

Abstract
Mapping markers from linkage data continues to be a task performed in many genetic epidemiological studies. Data collected in a study may be used to refine published map estimates and a study may use markers that do not appear in any published map. Furthermore, inaccuracies in meiotic maps can seriously bias linkage findings. To make best use of the available marker information, multilocus linkage analyses are performed. However, two computational issues greatly limit the number of markers currently mapped jointly; the number of candidate marker orders increases exponentially with marker number and computing exact multilocus likelihoods on general pedigrees is computationally demanding. In this article, a new Markov chain Monte Carlo (MCMC) approach that solves both these computational problems is presented. The MCMC approach allows many markers to be mapped jointly, using data observed on general pedigrees with unobserved individuals. The performance of the new mapping procedure is demonstrated through the analysis of simulated and real data. The MCMC procedure performs extremely well, even when there are millions of candidate orders, and gives results superior to those of CRI-MAP.
GENETIC (or meiotic) maps of polymorphic DNA markers are an important resource in human genetics. Information from genetic maps is used in linkage analysis to identify disease-predisposing genes. Genetic maps, however, are not known with certainty. Comparison studies of sequence-based physical maps and genetic maps have revealed discrepancies in the order of some markers (Matise et al. 2002; Nievergelt et al. 2004). Differences in marker order and marker location between published genetic maps have also been found (Nievergelt et al. 2004). These inaccuracies can seriously bias linkage findings from genetic epidemiologic studies (Buetow 1991; Halpern and Whittemore 1999; Daw et al. 2000). Inaccuracies in genetic maps are due to a number of factors. The number of meioses studied, marker informativeness, genotyping error, and missing data all contribute to map misspecification. Map accuracy is also affected by the statistical procedure used to construct the map.
Genetic maps are most accurately constructed using multilocus linkage methods. Two-locus procedures are easy to implement, computationally inexpensive, and may give results less sensitive to departures from the assumed genetic model and genotyping errors (Buetow 1991; Shields et al. 1991). However, multilocus procedures make better use of available information leading to increased accuracy (Lathrop et al. 1984, 1985; Thompson 1984), especially if the data are relatively error free and there are missing data. Two issues limit the number of loci mapped jointly. First, for data observed on L markers, there are L!/2 candidate orders. Even for a moderate number of markers, the set of candidate orders is extremely large and increases exponentially with marker number. Second, exact calculation of multilocus likelihoods on pedigree data can be computationally demanding. For a given marker map, pedigree-peeling algorithms (Elston and Stewart 1971; Cannings et al. 1978) and the Lander-Green algorithm (Lander and Green 1987) are efficient procedures for calculating multilocus likelihoods on pedigrees. However, pedigree-peeling computations are exponential in marker number. Lander-Green computations are exponential in family size. Furthermore, mapping multiple loci jointly requires calculating a multilocus likelihood under many different parameter values under each candidate order.
Marker loci are typically mapped using maximum-likelihood estimation. For a given marker order and set of marker allele frequencies, recombination fractions are estimated by finding values that maximize the likelihood. The “best” marker order is then the order with the largest maximized likelihood. Maximum-likelihood estimates are often found using the expectation-maximization (EM) algorithm (Dempster et al. 1977). However, obtaining maximum-likelihood estimates under each candidate order from multilocus likelihoods can be computationally challenging. Approaches to limit the set of candidate orders and reduce the computational burden include preliminary ranking procedures (Weeks and Lange 1987; Wilson 1988; Weeks 1991; Falk 1992), constructing the map stepwise (Lathrop et al. 1984), and combining the EM algorithm with optimization techniques such as branch-and-bound (Lathrop et al. 1985) and simulated annealing (Thompson 1984; Weeks and Lange 1987; Stam 1993). Even by reducing the set of candidate orders, multilocus maximum-likelihood procedures are often limited to the analysis of data on few markers jointly.
Little attention has been given to the development of Bayesian multilocus linkage approaches for mapping genetic markers. This is despite Bayesian linkage techniques showing promise for the detection and localization of putative trait loci influencing genetically complex diseases (Bartlett et al. 2002; Gagnon et al. 2003; Logue et al. 2003; Badzioch et al. 2004; Wijsman et al. 2004). There are also several advantages to using Bayesian marker mapping procedures over maximum-likelihood approaches. First, prior information can be correctly incorporated into the analysis. Using Bayes' theorem, prior information is combined with observed information to form the posterior distribution of the model variables. Bayesian inference is then based on this posterior distribution. Second, evidence for a particular order is measured on the familiar probability scale. Third, uncertainty about nuisance parameters such as marker allele frequencies are taken into account instead of being assumed known.
Bayesian procedures are computationally demanding. Bayesian inference requires integration of the joint posterior distribution, often over many variables. Integrating joint probability distributions over large multidimensional parameter spaces is extremely challenging. Monte Carlo procedures are an invaluable tool for approximating integrals. Several Bayesian marker-mapping methods have been implemented using Monte Carlo. Stephens and Smith (1993) obtained Monte Carlo estimates of the posterior probability of a marker order and marker position using two-locus data. George et al. (1999) and Rosa et al. (2002) developed a Monte Carlo strategy for the analysis of data observed from experimental designs. A Monte Carlo approach capable of ordering many markers was developed by Heath (1997a) for radiation hybridization mapping data. These implementations, however, restrict the Bayesian analysis to certain types of data and/or pedigree structure.
In this article, a Bayesian multilocus linkage approach for mapping many genetic markers simultaneously is presented. Bayesian quantities such as posterior probabilities and posterior means are approximated using Markov chain Monte Carlo (MCMC). MCMC makes feasible the analysis of multilocus data observed on general pedigrees containing possibly consanguineous marriages and missing information. The performance characteristics of the MCMC procedure are improved by combining Monte Carlo sampling with exact computation. The methodology is demonstrated through its application to the analysis of simulated data and real data originating from the Framingham Heart Study.
MATERIALS AND METHODS
Notation and assumptions:
The following notation and assumptions are used in this article. Suppose marker data Y are observed on L arbitrarily ordered genetic marker loci {Mj: j = 1, 2, … , L} on families of arbitrary size and complexity. Codominant multiallelic loci are assumed with allele frequencies p = (p1, p2, … , pL), where at locus Mj the set of allele frequencies is denoted by pj. Marker loci are in linkage equilibrium within the population. Markers are ordered 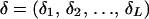 , where δk is the kth element in the ordered list of marker indexes. Let
, where δk is the kth element in the ordered list of marker indexes. Let 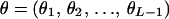 be the vector of (sex-averaged) recombination fractions between pairs of adjacent loci, where θk is the recombination fraction between
be the vector of (sex-averaged) recombination fractions between pairs of adjacent loci, where θk is the recombination fraction between 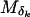 and
and 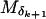 . To demonstrate, suppose data are observed on five markers {Mj: j = 1, 2, … , 5} ordered M1 M4 M5 M2 M3. Then
. To demonstrate, suppose data are observed on five markers {Mj: j = 1, 2, … , 5} ordered M1 M4 M5 M2 M3. Then 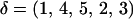 and
and 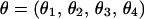 , where θ1, θ2, θ3, and θ4 are the recombination fractions between markers M1 and M4, M4 and M5, M5 and M2, and M2 and M3, respectively.
, where θ1, θ2, θ3, and θ4 are the recombination fractions between markers M1 and M4, M4 and M5, M5 and M2, and M2 and M3, respectively.
Meiosis indicators are used to trace the passage of unobservable founder alleles (or identical-by-descent genes) through a pedigree. Founder alleles are latent because marker data observed on families are incomplete. Data on large families may be only sparsely observed with many individuals unavailable for sampling. Also the parental origin of an observed allele is unknown and cannot always be inferred from parental information. Let S denote the array of meiosis indicators Sij for i = 1, 2, … , m and j = 1, 2, … , L, where m is the total number of meioses in the families. Here, Sij is 0 or 1 if the copied founder allele is the parent's maternal or paternal founder allele, respectively. The set of meiosis indicators can be partitioned on loci S = (S·1, S·2, … , S·L) or partitioned on meioses S = (S1·, S2·, … , Sm·), where S·j is the set of meiosis indicators at Mj and Si· is the set of meiosis indicators at meiosis i.
Bayesian model:
A Bayesian probability model for mapping many markers jointly is presented. The probability model defines the joint posterior distribution of the model variables 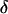 ,
,  conditioned on the observed marker data Y. In presenting the probability model, first the functional form of the joint prior distribution is given and prior distributions are specified. Second, the probability of the data given the variables (i.e., the likelihood) is constructed and its calculation is discussed. Third, the likelihood and the priors are combined to form the joint posterior distribution.
conditioned on the observed marker data Y. In presenting the probability model, first the functional form of the joint prior distribution is given and prior distributions are specified. Second, the probability of the data given the variables (i.e., the likelihood) is constructed and its calculation is discussed. Third, the likelihood and the priors are combined to form the joint posterior distribution.
The Bayesian paradigm provides opportunity for the inclusion of prior information through the specification of prior distributions on the model variables. The joint prior distribution on 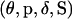 is
is
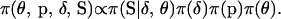 |
Here, the meiosis indicators are assumed to be conditionally independent of the marker allele frequencies given the marker order and recombination fractions. That is, the probability of S depends only on 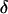 and
and 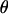 . Marker order, recombination fractions, and marker allele frequencies are assumed independent a priori. The prior distribution on S, assuming meioses are independent, is
. Marker order, recombination fractions, and marker allele frequencies are assumed independent a priori. The prior distribution on S, assuming meioses are independent, is 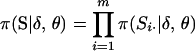 , where Si· is the vector of meiosis indicators at meiosis i across loci. Assuming independence of recombination events in disjoint marker intervals (i.e., no interference), the prior distribution on Si· is
, where Si· is the vector of meiosis indicators at meiosis i across loci. Assuming independence of recombination events in disjoint marker intervals (i.e., no interference), the prior distribution on Si· is 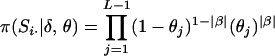 , where
, where 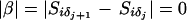 if no recombination event has occurred between the j + 1th and jth ordered locus and 1 if a recombination event has taken place. Equal prior probability is assigned to each candidate marker order by placing a uniform prior on
if no recombination event has occurred between the j + 1th and jth ordered locus and 1 if a recombination event has taken place. Equal prior probability is assigned to each candidate marker order by placing a uniform prior on 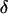 such that
such that 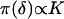 , where K is a constant. The marker allele frequencies, under the assumption of linkage equilibrium, are independent at different loci such that
, where K is a constant. The marker allele frequencies, under the assumption of linkage equilibrium, are independent at different loci such that 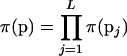 . The Dirichlet distribution, a multivariate generalization of the beta distribution, is placed on pj. For analyses conducted in this article, the parameters of the Dirichlet distribution, which can be thought of as counts, are set to 1. Equal prior probability is assigned to all combinations of allele frequencies at a marker locus. Recombination fractions, assuming no interference, are independent such that the prior distribution on
. The Dirichlet distribution, a multivariate generalization of the beta distribution, is placed on pj. For analyses conducted in this article, the parameters of the Dirichlet distribution, which can be thought of as counts, are set to 1. Equal prior probability is assigned to all combinations of allele frequencies at a marker locus. Recombination fractions, assuming no interference, are independent such that the prior distribution on 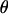 is
is 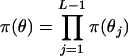 . A truncated beta distribution B(aj, bj) is placed on θj over the interval [0, 0.5]. A special case of the beta distribution, used here, is when aj = bj = 1. The beta distribution then becomes a uniform distribution.
. A truncated beta distribution B(aj, bj) is placed on θj over the interval [0, 0.5]. A special case of the beta distribution, used here, is when aj = bj = 1. The beta distribution then becomes a uniform distribution.
The likelihood is the joint probability of the observed marker data Y given the marker allele frequencies p and meiosis indicators S, where
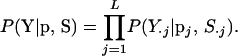 |
The joint probability of Y is the product of L single-locus probabilities since the data Y·j depend only on the allele frequencies and descent of founder alleles at Mj. Calculation of the single-locus probability P(Y·j|pj, S·j) is due to Thompson (1974) and requires identifying all possible assignments of marker allelic type to founder alleles that appear in observed individuals. Kruglyak et al. (1996) present an efficient algorithm for identifying all valid assignments. The probability of a particular assignment, under Hardy-Weinberg and linkage equilibrium, is then the product of the appropriate marker allele frequencies. The value of P(Y·j|pj, S·j) is obtained by summing these probabilities over all possible assignments (Thompson and Heath 1999).
The joint posterior distribution of 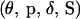 conditional on Y is
conditional on Y is
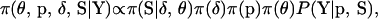 |
(1) |
where each term on the right-hand side has been discussed above. From (1) it is clear that the joint posterior distribution combines prior knowledge with information from the observed data. Also note that for given values of the model variables, calculation of (1) is computationally inexpensive, a feature exploited in MCMC.
MCMC procedure:
Bayesian inference requires integrating over (1), a high-dimensional probability distribution. Here, analytic solution is not possible. An alternative is Monte Carlo integration via MCMC. MCMC, namely the Metropolis-Hastings (M-H) algorithm (Hastings 1970) and the Gibbs sampler (Geman and Geman 1984), is a procedure for drawing dependent realizations of the model variables from high-dimensional probability distributions. These dependent realizations form a Markov chain with the distribution of interest as its stationary distribution. Bayesian quantities are then estimated via averages of the dependent realizations.
The MCMC procedure begins by generating an initial configuration 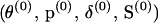 . Starting values for
. Starting values for 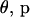 , and
, and 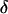 are easily drawn from their prior distributions. However, generating an initial set of meiosis indicators is challenging. If S is initialized using its prior, many starting configurations are rejected before a set of meiosis indicators consistent with the observed data is generated. Instead, S(0), consistent with the observed data and Mendelian inheritance, is generated using sequential imputation (Kong et al. 1993, 1994). Sequential imputation is an importance-sampling technique that can be used to impute latent genetic data. Loci are processed in sequence where for a given locus j, S·j is generated conditional on the observed data Y·j and previously processed loci. The dependence between loci is only partially captured since only data for loci to one side of a given locus contribute to imputation at that locus. However, for the purpose of initializing the MCMC procedure, sequential imputation leads to adequate starting configurations.
are easily drawn from their prior distributions. However, generating an initial set of meiosis indicators is challenging. If S is initialized using its prior, many starting configurations are rejected before a set of meiosis indicators consistent with the observed data is generated. Instead, S(0), consistent with the observed data and Mendelian inheritance, is generated using sequential imputation (Kong et al. 1993, 1994). Sequential imputation is an importance-sampling technique that can be used to impute latent genetic data. Loci are processed in sequence where for a given locus j, S·j is generated conditional on the observed data Y·j and previously processed loci. The dependence between loci is only partially captured since only data for loci to one side of a given locus contribute to imputation at that locus. However, for the purpose of initializing the MCMC procedure, sequential imputation leads to adequate starting configurations.
To draw 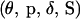 from the joint posterior distribution
from the joint posterior distribution 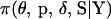 such that a Markov chain
such that a Markov chain 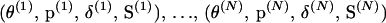 with equilibrium distribution
with equilibrium distribution 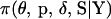 is constructed, the following update steps are performed:
is constructed, the following update steps are performed:
Step 1. Meiosis indicators are updated either across loci or across meioses using a joint Gibbs sampler.
Step 2. Marker allele frequencies are updated for a randomly chosen locus using the M-H algorithm.
Step 3. Marker order and recombination fractions are jointly updated using the M-H algorithm with an integrated acceptance probability. To complete the move, meiosis indicators of select loci are also updated.
These steps can be performed in any order. After completion of these three steps, a new sample is realized and the process is repeated. Each step is now discussed.
In step 1, S is updated via one of two randomly chosen strategies: a block update of all meiosis indicators at each locus (in random order) using the whole-locus Gibbs sampler (L-sampler) or a block update of the meiosis indicators for all loci in each meiosis (in random order) using the whole-meiosis Gibbs sampler (M-sampler). The L-sampler (Heath 1997b), using single-locus pedigree peeling, jointly updates the complete set of meiosis indicators at a locus conditional on the observed data at that locus and current values of the locus order, recombination fractions, and meiosis indicators at neighboring loci. The M-sampler (Thompson and Heath 1999; Thompson 2000), using the forward-backward Baum algorithm (Baum et al. 1970), jointly updates the complete set of meiosis indicators in a meiosis conditional on the observed data and current values of the locus order, recombination fractions, and meiosis indicators at other meioses. A combination of L- and M-sampler steps is used here to improve the performance characteristics of the MCMC procedure (Heath and Thompson 1997).
In step 2, for a randomly chosen marker locus Mj, marker allele frequencies pj are updated using a M-H step. A proposed set of allele frequencies for Mj, p′j, is drawn from a Dirichlet distribution with parameters set to 1 and accepted with M-H probability 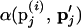 . Here,
. Here, 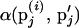 is the M-H acceptance probability of the Markov chain moving from the current state
is the M-H acceptance probability of the Markov chain moving from the current state 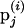 to a proposed state p′j. The acceptance probability is
to a proposed state p′j. The acceptance probability is
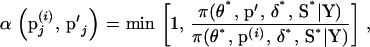 |
where * denotes realization (i) or (i + 1) depending on the update order and π(·) is the joint posterior distribution (1) evaluated at the model variable values. If the move is accepted, the proposed set of marker allele frequencies becomes the current state where 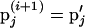 . If the move is rejected,
. If the move is rejected, 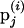 becomes the current state where
becomes the current state where 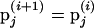 .
.
In step 3, 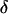 and
and 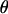 are jointly updated as follows: First, a block of k adjacent markers is randomly chosen from the currently ordered marker loci. These markers are referred to as selected markers and markers not included in the block are referred to as unselected markers. Second, the block of markers is moved to a new chromosomal position and positioned using a uniform distribution on [0, 0.5]. Current values of the recombination fractions are preserved between adjacent selected markers and, where possible, between adjacent unselected markers. This proposal mechanism results in a new
are jointly updated as follows: First, a block of k adjacent markers is randomly chosen from the currently ordered marker loci. These markers are referred to as selected markers and markers not included in the block are referred to as unselected markers. Second, the block of markers is moved to a new chromosomal position and positioned using a uniform distribution on [0, 0.5]. Current values of the recombination fractions are preserved between adjacent selected markers and, where possible, between adjacent unselected markers. This proposal mechanism results in a new 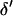 and
and 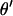 . Third, the proposed values are accepted with M-H integrated acceptance probability
. Third, the proposed values are accepted with M-H integrated acceptance probability 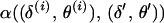 . The acceptance probability is based on a probability distribution where the set of meiosis indicators at either or both end block markers is integrated out of
. The acceptance probability is based on a probability distribution where the set of meiosis indicators at either or both end block markers is integrated out of 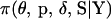 via single-locus pedigree peeling. This updated M-H promotes good mixing and has previously been used in updating the position of a disease locus (George and Thompson 2003) and a quantitative trait locus (Heath 1997b) relative to a fixed marker map. If
via single-locus pedigree peeling. This updated M-H promotes good mixing and has previously been used in updating the position of a disease locus (George and Thompson 2003) and a quantitative trait locus (Heath 1997b) relative to a fixed marker map. If 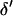 and
and 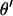 are accepted, the move is completed by using the L-sampler to sample the meiosis indicators at either or both end block markers. Further details are given in the appendix.
are accepted, the move is completed by using the L-sampler to sample the meiosis indicators at either or both end block markers. Further details are given in the appendix.
Description of data and analyses:
Simulated data and analysis:
Multilocus data are generated on 11 extended families originating from the Framingham Heart Study (Dawber et al. 1951; Feinleib et al. 1975). These families are three- and four-generation pedigrees having multiple founding couples and ranging in size from 26 to 47 individuals. A total of 396 meioses are contained in the pedigree data. Data are first generated assuming all individuals are observed. Marker data on randomly chosen individuals are then removed and a new set of data created. The probability of an individual being unobserved changes with generation number (Table 1), resulting in ∼50% of the individuals being unobserved. These probabilities are based on patterns of missing data observed in the Framingham study.
TABLE 1.
The probability distribution used in the simulation study to generate families with missing data
| Generation | Pr(unobs|gen) |
|---|---|
| Last | 0.0 |
| 2nd last | 0.50 |
| 3rd last | 0.90 |
| 4th last | 0.98 |
| ≥5th last | 1.0 |
Pr(unobs|gen), the conditional probability of an individual being unobserved given the individual's generation.
Data are generated under two different marker maps, an 8-cM map and a 1-cM map. The 8-cM map has eight approximately evenly spaced microsatellite markers along a 78-cM chromosomal segment with an average intermarker distance of 8 cM. Each marker has between 6 and 13 possible alleles although only a subset of these is observed in any given family. Marker positions are derived from the Marshfield map for chromosome 9. Marker allele frequencies are based on previous estimates obtained from the Framingham Heart Study. The 1-cM map is based on the same eight microsatellite markers but the markers span an 8-cM chromosomal segment with an average intermarker distance of 1 cM. Hence, this simulation study is composed of four data sets: an 8-cM map and full data (8-F), an 8-cM map and missing data (8-M), a 1-cM map and full data (1-F), and a 1-cM map and missing data (1-M). The simulated marker order is M1 M2 M3 M4 M5 M6 M7 M8 and for notational convenience, δS = (1, 2, 3, 4, 5, 6, 7, 8). There are 8!/2 = 20,160 candidate orders and each marker set is replicated 100 times.
MCMC analyses of the data are performed as follows. Using the same starting configuration, the run length is gradually increased until the posterior probability of a marker order stabilizes across visited marker orders. Four repeated MCMC analyses of each data replicate are then performed using different randomly generated starting configurations to access the variation in Bayesian estimates. The analysis is concluded by examining plots of the sequence of realizations of the model variables. Systematic patterns of values in these plots may suggest poor MCMC performance and a run length that is too short.
For comparison, maximum-likelihood analyses via the software package CRI-MAP (Lander and Green 1987) are also performed. CRI-MAP contains several interactive and automated routines for constructing and refining genetic maps. Here, markers are first ordered using the “build” routine. This routine uses maximum-likelihood estimation for the stepwise construction of marker maps, where markers are added in decreasing order of informativeness. The integrity of this initial order is then tested via the “flips” routine. The flips routine reverses the order between a pair of markers and recalculates the likelihood under the new order. If this results in an increase in the likelihood, then the initial order is replaced by this new order. Analysis via the flips routine is repeated until the likelihood no longer increases with a change in marker order.
The simulation study is concluded by examining the mixing characteristics of the MCMC procedure. MCMC can suffer mixing problems if markers are tightly linked (Thompson and Heath 1999). Tightly linked markers, the pattern of observed and unobserved individuals, and the laws of Mendelian inheritance constrain the model space. A MCMC sampler can become “trapped” in a local part of the model space. To investigate this, exact multilocus likelihoods are calculated on all eight markers jointly, under each candidate order, for replicates one and two from 1-F. This data set was chosen because the markers are tightly linked and likelihood computations are tractable. Calculating a single eight-locus likelihood on replicates from 1-M or 8-M was estimated to take many months. Exact likelihoods are computed using SUPERLINK V1.4 (Fishelson and Geiger 2002). Each replicate is generated such that the number of crossover events between any two loci is known. The true recombination fractions are then easily calculated from the simulated data and used as the parameter values in the likelihood calculation.
Real data and analysis:
Real data observed on the families used in the simulation study are also analyzed. Multilocus data are available on 12 linked microsatellite markers on chromosome 9. These markers span a chromosomal segment of 123 cM with an average intermarker distance of 10 cM. As in the simulation study, each marker has between 6 and 13 possible alleles. Approximately 50% of the individuals are unobserved. Markers are ordered 1, 2, … , 12 (on the basis of the Marshfield map), where for clarity of exposition markers are referenced by their marker indexes. MCMC and CRI-MAP analyses of these data are performed as described above. There are over 200 million candidate marker orders. All analyses are performed on a Linux-based workstation using a single AMD Athlon 2800+ processor.
RESULTS
Simulation study:
Results are presented from the analysis of data sets 8-F, 8-M, 1-F, and 1-M. A single MCMC analysis of a replicate from 8-F or 1-F consists of 3 × 105 iterations and takes ∼2 hr. A single MCMC analysis of a replicate from 8-M or 1-M consists of 6 × 105 iterations and takes ∼4 hr. There is little variation in Bayesian estimates across repeated analyses. Hence, only results from a single MCMC analysis of a replicate are reported below.
The estimated posterior probabilities of the marker orders for each replicate are plotted in Figure 1. These estimates are obtained by normalizing the number of times a marker order is sampled within a MCMC run. To help visualize the departure of a sampled order from the simulated marker order, 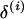 are converted into distances using the measure
are converted into distances using the measure 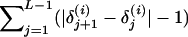 . Here, the larger the distance, the greater the departure of the sampled order from
. Here, the larger the distance, the greater the departure of the sampled order from 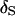 . For example, for sample orders (1, 2, 3, 4, 5, 6, 7, 8), (1, 2, 3, 4, 5, 8, 7, 6), and (8, 7, 6, 3, 2, 1, 4, 5), the distances are 0, 2, and 4, respectively.
. For example, for sample orders (1, 2, 3, 4, 5, 6, 7, 8), (1, 2, 3, 4, 5, 8, 7, 6), and (8, 7, 6, 3, 2, 1, 4, 5), the distances are 0, 2, and 4, respectively.
Figure 1.

The estimated posterior probabilities of a marker order from the Bayesian analysis of data sets (A) 8-F, (B) 8-M, (C) 1-F, and (D) 1-M. The numbers are distances measuring the discrepancy between the sample order and the simulated marker order. For clarity, results are shown only from the analysis of the first 50 replicates. The simulated marker order has a distance of 0.
From Figure 1, the impact of tightly linked markers is obvious. Under an 8-cM map, the posterior probability of 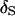 is near one across replicates when data are observed on all individuals (Figure 1A). Even for analyses of families containing unobserved individuals, the average posterior probability of
is near one across replicates when data are observed on all individuals (Figure 1A). Even for analyses of families containing unobserved individuals, the average posterior probability of 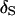 is 85% and all but one of the MCMC analyses result in
is 85% and all but one of the MCMC analyses result in 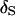 having the highest posterior probability (Figure 1B). Under a 1-cM map, several marker orders may be supported. In Figure 1D, the average estimated posterior probability of
having the highest posterior probability (Figure 1B). Under a 1-cM map, several marker orders may be supported. In Figure 1D, the average estimated posterior probability of 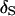 is 24% and in only 38 replicates is
is 24% and in only 38 replicates is 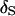 assigned the highest posterior probability. Also, by using a probability scale to measure the strength of evidence, a better appreciation of the uncertainty associated with a marker order is obtained. For example, consider the analysis of replicate 9 from 1-M. The estimated posterior probabilities of orders (1, 2, 3, 4, 5, 6, 7, 8), (1, 2, 3, 5, 4, 6, 7, 8), (1, 3, 2, 4, 5, 6, 7, 8), and (1, 3, 2, 5, 4, 6, 7, 8) are 0.25, 0.20, 0.31, and 0.24, respectively. Here, the simulated marker order is not assigned the highest posterior probability but it is clear that a single unique ordering of the markers is not supported by the data.
assigned the highest posterior probability. Also, by using a probability scale to measure the strength of evidence, a better appreciation of the uncertainty associated with a marker order is obtained. For example, consider the analysis of replicate 9 from 1-M. The estimated posterior probabilities of orders (1, 2, 3, 4, 5, 6, 7, 8), (1, 2, 3, 5, 4, 6, 7, 8), (1, 3, 2, 4, 5, 6, 7, 8), and (1, 3, 2, 5, 4, 6, 7, 8) are 0.25, 0.20, 0.31, and 0.24, respectively. Here, the simulated marker order is not assigned the highest posterior probability but it is clear that a single unique ordering of the markers is not supported by the data.
In Figure 2, the marker ordering capabilities of the MCMC procedure and CRI-MAP are compared. The marker order with the highest estimated posterior probability is converted into a distance and plotted against replicate number. For clarity, only results from the analysis of the first 50 replicates are shown. The marker order (converted into distances) with the largest maximized likelihood obtained via CRI-MAP is also plotted against replicate number. Here, the benefits of using MCMC for marker ordering are evident. By making good use of the available data, it is possible to obtain clear evidence in favor of the simulated marker order despite substantial missing data (Figure 2B). Also, MCMC generally identifies marker orders closer to the true simulated marker order.
Figure 2.

The marker order with the highest estimated posterior probability is converted into a distance and plotted against replicate number for data sets (A) 8-F, (B) 8-M, (C) 1-F, and (D) 1-M. Also, the marker order (converted into a distance) with the largest maximized likelihood obtained via CRI-MAP is plotted against replicate number. For clarity, results are shown only for the first 50 replicates. x, a result obtained using the MCMC procedure; *, a result obtained using CRI-MAP. The simulated marker order has a distance of 0.
The MCMC procedure allows a variety of Bayesian quantities to be calculated on the model variables including posterior means, posterior modes, standard deviations, and credible intervals. These quantities are easily derived from averages of the MCMC samples. Table 2 reports the posterior means and posterior modes of 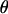 under the simulated marker order averaged over analyses. The accuracy of the estimator is measured via the mean square error (MSE). For comparison, maximum-likelihood estimates obtained via CRI-MAP of
under the simulated marker order averaged over analyses. The accuracy of the estimator is measured via the mean square error (MSE). For comparison, maximum-likelihood estimates obtained via CRI-MAP of 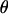 under the simulated marker order and their MSEs are also reported.
under the simulated marker order and their MSEs are also reported.
TABLE 2.
The estimated posterior means (Mean) and modes (Mode) of the recombination fractions (θ1, … , θ7), under the simulated marker order, from the Bayesian analysis of data sets 8-F, 8-M, 1-F, and 1-M
| Recombination fractions
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Data set | Implementation | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| 8-F | MCMC | True | 0.15 | 0.11 | 0.12 | 0.07 | 0.09 | 0.04 | 0.11 |
| Mean | 0.15 (6.3) | 0.11 (3.8) | 0.12 (4.2) | 0.08 (2.5) | 0.09 (2.8) | 0.04 (1.7) | 0.11 (3.5) | ||
| CRI-MAP | Mode | 0.15 (6.4) | 0.11 (3.8) | 0.12 (4.2) | 0.07 (2.5) | 0.09 (2.9) | 0.04 (1.6) | 0.11 (3.6) | |
| MLE | 0.13 (6.9) | 0.10 (3.4) | 0.11 (4.3) | 0.07 (2.0) | 0.08 (2.8) | 0.04 (1.3) | 0.10 (3.5) | ||
| 8-M | MCMC | True | 0.15 | 0.11 | 0.12 | 0.07 | 0.09 | 0.04 | 0.11 |
| Mean | 0.16 (15.9) | 0.12 (8.2) | 0.13 (8.6) | 0.08 (5.4) | 0.10 (5.3) | 0.05 (3.7) | 0.11 (6.5) | ||
| CRI-MAP | Mode | 0.16 (13.5) | 0.11 (7.5) | 0.13 (7.9) | 0.08 (5.0) | 0.09 (5.0) | 0.04 (3.4) | 0.11 (6.2) | |
| MLE | 0.12 (23.9) | 0.08 (13.2) | 0.10 (14.5) | 0.06 (7.5) | 0.07 (10.1) | 0.03 (4.7) | 0.08 (11.9) | ||
| 1-F | MCMC | True | 0.02 | 0.01 | 0.01 | 0.01 | 0.01 | 0.00 | 0.01 |
| Mean | 0.02 (0.7) | 0.01 (0.4) | 0.02 (0.5) | 0.01 (0.3) | 0.01 (0.4) | 0.01 (0.2) | 0.01 (0.4) | ||
| CRI-MAP | Mode | 0.02 (0.7) | 0.01 (0.4) | 0.01 (0.5) | 0.01 (0.2) | 0.01 (0.3) | 0.00 (0.1) | 0.01 (0.3) | |
| MLE | 0.02 (0.6) | 0.01 (0.3) | 0.01 (0.4) | 0.01 (0.2) | 0.01 (0.3) | 0.00 (0.1) | 0.01 (0.4) | ||
| 1-M | MCMC | True | 0.02 | 0.01 | 0.01 | 0.01 | 0.01 | 0.00 | 0.01 |
| Mean | 0.02 (1.7) | 0.02 (1.1) | 0.02 (3.1) | 0.01 (2.5) | 0.02 (9.4) | 0.01 (1.3) | 0.02 (0.7) | ||
| CRI-MAP | Mode | 0.01 (1.3) | 0.01 (0.7) | 0.02 (2.1) | 0.01 (1.7) | 0.01 (7.4) | 0.01 (0.7) | 0.01 (0.6) | |
| MLE | 0.01 (1.8) | 0.01 (1.0) | 0.01 (1.0) | 0.01 (0.5) | 0.01 (0.7) | 0.00 (0.3) | 0.01 (0.3) | ||
For comparison, average maximum-likelihood estimates (MLEs) obtained via CRI-MAP are also given. The accuracy of the estimator is measured via the mean square error (MSE × 106).
From Table 2, the average posterior means and modes agree closely with the true values under which the data are generated. As expected, an estimator's MSE increases if the data contain missing information. MCMC and CRI-MAP give similar results but other statistics such as standard errors and confidence intervals are not easily obtained via CRI-MAP. In contrast, MCMC does offer opportunity for a more detailed investigation of the data since the entire joint distribution of the model variables is realized.
The six marker orders with the largest exact multilocus log likelihoods from replicates 1 and 2 in 1-F are given in Table 3. The estimated posterior probabilities of these orders are also given. The results from Table 3 suggest that the sampler is performing well. In the analysis of replicate 1, the three marker orders with the largest log likelihoods are visited most often by the MCMC sampler. The two marker orders not sampled have log likelihoods up to 5 log units smaller than the log likelihood under the simulated marker order. In the analysis of replicate 2, the four marker orders with the highest log likelihoods are close in value. This is mirrored by the estimated posterior probabilities for these orders, which are also close in value. The two marker orders not sampled have log-likelihood values up to 12 log units smaller than the log-likelihood value under the simulated marker order. These results suggest that marker orders with relatively high likelihoods are being sampled by the MCMC procedure.
TABLE 3.
The estimated posterior probability of 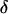 for the six marker orders with the largest exact log likelihoods (log L) for replicates 1 and 2 from 1-F
for the six marker orders with the largest exact log likelihoods (log L) for replicates 1 and 2 from 1-F
| Replicate 1
|
Replicate 2
|
||||
|---|---|---|---|---|---|
| Marker order | log L | Posterior prob | Marker order | log L | Posterior prob |
| 1 2 3 4 5 6 7 8 | −1903.19 | 0.967 | 1 2 3 4 5 6 7 8 | −1888.35 | 0.284 |
| 2 1 3 4 5 6 7 8 | −1905.52 | 0.018 | 1 2 3 4 5 7 6 8 | −1888.35 | 0.264 |
| 3 1 2 4 5 6 7 8 | −1906.67 | 0.015 | 1 3 2 4 5 7 6 8 | −1888.47 | 0.217 |
| 1 2 3 5 4 6 7 8 | −1907.42 | NS | 1 3 2 4 5 7 6 8 | −1888.47 | 0.235 |
| 1 2 3 4 5 6 8 7 | −1908.17 | NS | 1 2 3 4 6 5 7 8 | −1892.91 | NS |
| 3 2 1 4 5 6 7 8 | −1909.60 | 0.001 | 1 3 2 4 6 5 7 8 | −1893.10 | NS |
NS, a marker order not sampled by the MCMC procedure.
Real data:
The estimated posterior probabilities of a marker order from the Bayesian analysis of the Framingham data are presented in Table 4. Only the five marker orders sampled with the highest frequency are given, although many more orders are sampled. Each MCMC run consists of 5 × 105 iterations and takes ∼20 hr. This run length is excessive but it does give the MCMC sampler opportunity to visit marker orders of low posterior probability. The published marker order (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12) has the highest posterior probability, ranging between 80 and 86% across the repeated MCMC analyses. The three marker orders with the highest estimated posterior probabilities are the same across the repeated MCMC analyses and are visited with approximately equal frequency.
TABLE 4.
Results from the Bayesian analysis of chromosome 9 data from the Framingham Heart Study
| MCMC analysis
|
||||
|---|---|---|---|---|
| Marker order | 1 | 2 | 3 | 4 |
| 1 2 3 4 5 6 7 8 9 10 11 12 | 0.8008 | 0.8247 | 0.8146 | 0.8635 |
| 12 1 2 3 4 5 6 7 8 9 10 11 | 0.1187 | 0.1011 | 0.1121 | 0.1133 |
| 1 2 3 4 5 7 6 8 9 10 11 12 | 0.0497 | 0.0352 | 0.0259 | 0.0113 |
| 1 2 3 4 5 6 7 8 9 10 12 11 | 0.0126 | 0.0035 | ||
| 1 2 3 4 5 6 7 8 10 9 11 12 | 0.0124 | 0.0282 | 0.0378 | 0.0058 |
| 12 1 2 3 4 5 7 6 8 9 10 11 | 0.0067 | |||
| 12 1 2 3 4 5 6 7 8 10 9 11 | 0.0041 | |||
The five marker orders with the highest posterior probability are shown. Four MCMC analyses with different starting configurations are shown. The published marker order is 1, 2, … , 12, where markers are referenced by their marker index. Markers ordered differently from the published order are underlined.
The markers could be only partially ordered using CRI-MAP. The best marker order was (1, 2, 3, 4, 5, 6, 8, 9), where the remaining markers could not be uniquely positioned with certainty. This is in contrast to the Bayesian analysis where there is strong a posteriori evidence in favor of the published marker order.
DISCUSSION
In this article, a new MCMC procedure has been developed, implementing a Bayesian multilocus linkage approach for ordering many markers jointly on general pedigrees. These pedigrees may be large, have complex structures, and contain unobserved individuals. Ordering multiple markers simultaneously is challenging because the number of candidate orders increases exponentially with marker number. Furthermore, calculating exact multilocus likelihoods on general pedigrees is often computationally intractable. The MCMC procedure presented here circumvents these problems by using Monte Carlo sampling to form a Markov chain with 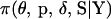 as its stationary distribution. Bayesian quantities are then formed from averages of the dependent realizations.
as its stationary distribution. Bayesian quantities are then formed from averages of the dependent realizations.
The MCMC procedure is demonstrated through the analysis of simulated and real data on 11 extended pedigrees. These pedigrees are large, contain half-sibships, and have multiple founding couples. The simulation study focuses on the analysis of multilocus data generated under two different genetic maps: one map with an average intermarker distance of 8 cM (8-cM map) and the other with an average intermarker distance of 1 cM (1-cM map). It was possible to unambiguously order markers from data generated under an 8-cM map using the estimated posterior probabilities of a marker order. However, there was insufficient information in the data generated under a 1-cM map to unambiguously order markers. The MCMC procedure was superior to CRI-MAP for identifying the correct marker order when the data contained unobserved individuals. Real data on 12 microsatellite markers on chromosome 9 were also analyzed. With >200 million possible orders, the MCMC procedure predominantly sampled the published marker order, resulting in a posterior probability of ∼80%. Here, CRI-MAP could only partially order the markers in the published map. It should be noted that results presented in this article should not be construed as a general failure of the CRI-MAP software. CRI-MAP is an excellent package for the rapid construction of multilocus linkage maps from data on nuclear families or CEPH-like pedigrees. Even data on extended pedigrees can be analyzed. However, as warned in the user documentation, likelihoods on extended pedigrees with unobserved individuals are approximated with the accuracy of the approximation yet to be tested.
The Bayesian probability model presented in this article can be extended in several ways. First, the Bayesian probability model can be extended to accommodate genotyping errors. Aberrant marker observations can bias linkage findings where map lengths are inflated and even the marker order is misspecified (Buetow 1991; Goldstein et al. 1997). Accounting for aberrant data, however, does increase the computational complexity of the analysis since any marker phenotype is now potentially consistent with any latent marker genotype. The calculation of P(Y·j|pj, S·j) now requires a procedure analogous to pedigree peeling (Thompson and Heath 1999). Bayesian quantities can still be estimated using the MCMC procedure presented in this article. Second, the Bayesian probability model can be extended to make use of known information. Given detailed physical and genetic maps, the partial order of some markers, their relative distances, and marker allele frequencies may be known with a high degree of certainty. This information is not easily incorporated into a maximum-likelihood-based linkage analysis. However, by placing subjective priors on the model variables, this information can be incorporated into a Bayesian analysis. Again, Bayesian quantities can be estimated using the MCMC procedure described in this article. Third, the Bayesian probability model can accommodate genetic interference by replacing 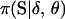 , the transmission probabilities of S under no interference, by
, the transmission probabilities of S under no interference, by 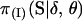 , the transmission probabilities of S under interference. Again, the MCMC procedure previously discussed can also be used here. However, the first-order Markov property of the indicators S·j over the loci j, upon which the M-sampler is based, is no longer true under interference. Hence, the whole-meiosis Gibbs update is replaced by a M-H step. A new set of indicators at meiosis i, Si·, is proposed, using the M-sampler assuming no interference. But instead of immediately accepting these values as before, the proposed indicators are accepted with M-H probability
, the transmission probabilities of S under interference. Again, the MCMC procedure previously discussed can also be used here. However, the first-order Markov property of the indicators S·j over the loci j, upon which the M-sampler is based, is no longer true under interference. Hence, the whole-meiosis Gibbs update is replaced by a M-H step. A new set of indicators at meiosis i, Si·, is proposed, using the M-sampler assuming no interference. But instead of immediately accepting these values as before, the proposed indicators are accepted with M-H probability
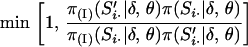 |
See Thompson (2000) for further details. Fourth, the Bayesian probability model can be extended to accommodate differences in male and female recombination fractions. The joint posterior distribution of the model variables conditioned on Y is 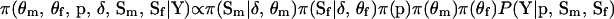 , where the subscripts m and f denote male and female, respectively, and it is assumed that sex-specific recombination fractions and paternal and maternal founder alleles are independent a priori. The same MCMC procedure previously described can be used here except that step 3 is modified to use the M-H algorithm with an integrated acceptance probability to jointly sample
, where the subscripts m and f denote male and female, respectively, and it is assumed that sex-specific recombination fractions and paternal and maternal founder alleles are independent a priori. The same MCMC procedure previously described can be used here except that step 3 is modified to use the M-H algorithm with an integrated acceptance probability to jointly sample 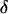 ,
, 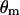 , and
, and 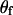 . It should be noted that the challenge in implementing these extensions lies in the development of efficient computer code.
. It should be noted that the challenge in implementing these extensions lies in the development of efficient computer code.
A computer software package implementing the Bayesian marker-ordering methodology and some of the extensions discussed above is currently under development. This software will allow users to construct and refine genetic maps from multilocus data on general pedigrees using MCMC. Options will include a facility to map a single marker relative to a fixed marker map, to map a group of markers relative to a fixed marker map, and to map many markers simultaneously. The software will accept linkage-formatted files and results will be reported as text and graphically. A version of this software will also be distributed with the MORGAN package for Monte Carlo genetic analysis (http://www.stat.washington.edu/thompson/Genepi/MORGAN/Morgan.shtml).
Acknowledgments
I am grateful to the University of Iowa's Center for Statistical Genetics Research for the use of their computational resources. This work was funded by a University of Iowa Internal Funding Initiative (85022911).
APPENDIX: JOINT M-H UPDATING OF δ AND θ
The joint updating of 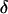 and
and 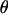 via the M-H algorithm consists of three steps. First, new values of
via the M-H algorithm consists of three steps. First, new values of 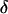 and
and 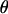 are proposed as follows: A block of k adjacent loci is randomly selected where markers are ordered
are proposed as follows: A block of k adjacent loci is randomly selected where markers are ordered 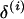 . Loci within the marker block are referred to as selected markers and loci outside the marker block are referred to as unselected markers. At each iteration, k is drawn from a uniform distribution on [1, 3, 4, … , L − 1]. Here, to simplify the calculation of the integrated acceptance probability, moves involving blocks of only two loci are not considered. If the block consists of three or more markers, with 50% probability, the order is reversed. A marker interval I ∈ {0, 1, … , L − k} is then chosen, where 0 is the interval to the left of the first unselected locus, 2, … , L − k − 1, are intervals between adjacent unselected loci, and L − k is the interval to the right of the last unselected locus. The marker block is positioned within a randomly chosen interval by sampling the recombination fraction between neighboring unselected and end block markers from a uniform distribution on [0, 0.5].
. Loci within the marker block are referred to as selected markers and loci outside the marker block are referred to as unselected markers. At each iteration, k is drawn from a uniform distribution on [1, 3, 4, … , L − 1]. Here, to simplify the calculation of the integrated acceptance probability, moves involving blocks of only two loci are not considered. If the block consists of three or more markers, with 50% probability, the order is reversed. A marker interval I ∈ {0, 1, … , L − k} is then chosen, where 0 is the interval to the left of the first unselected locus, 2, … , L − k − 1, are intervals between adjacent unselected loci, and L − k is the interval to the right of the last unselected locus. The marker block is positioned within a randomly chosen interval by sampling the recombination fraction between neighboring unselected and end block markers from a uniform distribution on [0, 0.5].
To illustrate this proposal mechanism, suppose data are available on markers M1, M2, M3, M4, M5, and M6. Markers are currently ordered 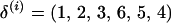 and positioned
and positioned 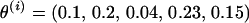 . Suppose a block of three markers is chosen, say M2, M3, and M6. Here, the selected markers are M2, M3, and M6, and the unselected markers are M1, M4, and M5. In Figure A1A, the marker block is moved to the left of the unselected markers, resulting in a proposed order
. Suppose a block of three markers is chosen, say M2, M3, and M6. Here, the selected markers are M2, M3, and M6, and the unselected markers are M1, M4, and M5. In Figure A1A, the marker block is moved to the left of the unselected markers, resulting in a proposed order 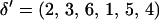 . To position the marker block, θ3 is drawn from a uniform distribution where θ3 is the recombination fraction between M6 and M1. The recombination fractions between the other pairs of markers are calculated or obtained directly from
. To position the marker block, θ3 is drawn from a uniform distribution where θ3 is the recombination fraction between M6 and M1. The recombination fractions between the other pairs of markers are calculated or obtained directly from 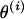 . The proposed set of recombination fractions then becomes
. The proposed set of recombination fractions then becomes 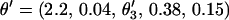 . In Figure A1B, the marker block is placed between M5 and M4, resulting in a new order
. In Figure A1B, the marker block is placed between M5 and M4, resulting in a new order 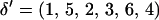 . To position the marker block relative to the unselected markers, θ2 and θ5 are drawn from a uniform distribution where θ2 is the recombination fraction between M5 and M2, and θ5 is the recombination fraction between M6 and M4. From
. To position the marker block relative to the unselected markers, θ2 and θ5 are drawn from a uniform distribution where θ2 is the recombination fraction between M5 and M2, and θ5 is the recombination fraction between M6 and M4. From 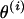 and the sampled recombination fractions, the new set of recombination fractions is
and the sampled recombination fractions, the new set of recombination fractions is  .
.
Figure A1.

The proposal mechanism for generating a new marker order and set of recombination fractions. A block of markers (M2, M3, and M6) is moved to a new position, resulting in a new marker order and set of recombination fractions. (A) The marker block is moved to the left of marker M1. (B) The marker block is moved between markers M5 and M4. Marker locations are denoted by vertical lines. For the proposed state, a solid horizontal line denotes a chromosomal distance that does not differ from the current state, and a cross-hatched horizontal line denotes a chromosomal distance that is sampled from a uniform distribution.
Second, the proposal is accepted with probability 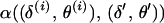 , where α(·) is the (integrated) M-H probability of the Markov chain moving from the current state
, where α(·) is the (integrated) M-H probability of the Markov chain moving from the current state 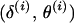 to the proposed state
to the proposed state 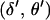 . The M-H acceptance probability is
. The M-H acceptance probability is
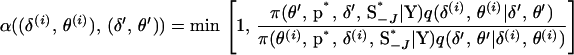 |
(A1) |
Here J is the set of indexes of block markers adjacent to unselected markers. For example, in Figure A1A where the marker block is moved to the left of the unselected markers, J = 6 since M6 is adjacent to an unselected marker. In Figure A1B where the marker block is moved between unselected markers, J = (2, 6) since M2 and M6 are now adjacent to unselected markers. Also, S−J is the set of meiosis indicators across loci excluding meiosis indicators at the locus or loci referenced in J, 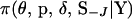 is the joint probability distribution of the model variables
is the joint probability distribution of the model variables 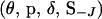 with SJ integrated out of the joint posterior distribution
with SJ integrated out of the joint posterior distribution 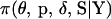 , and q(·) is the proposal distribution.
, and q(·) is the proposal distribution.
Since block size, marker interval, and block position are sampled from a uniform distribution and with 50% probability the order of the selected markers is reversed, the proposal probability is 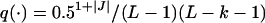 , where |J| = 1 or 2 depending on the number of elements in J. Furthermore,
, where |J| = 1 or 2 depending on the number of elements in J. Furthermore,
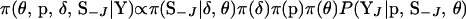 |
and the acceptance probability (A1) simplifies to
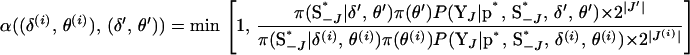 |
If J references a single end marker, the probabilities 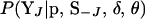 are obtained by single-locus peeling over MJ at positions
are obtained by single-locus peeling over MJ at positions 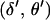 and
and 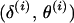 . If J references both end markers, the joint conditional probability of
. If J references both end markers, the joint conditional probability of 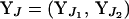 is
is
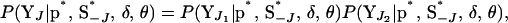 |
which is obtained by independently peeling over 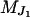 and
and 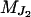 . This joint conditional probability can be factorized in this way since moves involving only two neighboring loci are not considered and due to the assumed conditional independence structure between Y and S.
. This joint conditional probability can be factorized in this way since moves involving only two neighboring loci are not considered and due to the assumed conditional independence structure between Y and S.
Third, if the proposal is accepted, the move is completed by updating SJ via the L-sampler. The (i + 1)th state of the Markov chain then becomes 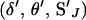 . If the proposal is rejected, the (i + 1)th state of the Markov chain becomes
. If the proposal is rejected, the (i + 1)th state of the Markov chain becomes 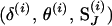 .
.
References
- Badzioch, M. D., R. P. Igo, F. Gagnon, J. D. Brunzell, R. M. Krauss et al., 2004. Low-density lipoprotein particle size loci in familial combined hyperlipidemia—evidence for multiple loci from a genome scan. Arterioscler. Thromb. Vasc. Biol. 24: 1942–1950. [DOI] [PubMed] [Google Scholar]
- Bartlett, C. W., J. F. Flax, M. W. Logue, V. J. Vieland, A. S. Bassett et al., 2002. A major susceptibility locus for specific language impairment is located on 13q21. Am. J. Hum. Genet. 71: 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, L. E., T. Petrie, G. Soules and N. Weiss, 1970. A maximization technique occurring in the statistical analysis of probabilistic functions on Markov chains. Ann. Math. Stat. 41: 164–171. [Google Scholar]
- Buetow, K. H., 1991. Influence of aberrant observations on high-resolution linkage analysis outcomes. Am. J. Hum. Genet. 49: 985–994. [PMC free article] [PubMed] [Google Scholar]
- Cannings, C., E. A. Thompson and M. H. Skolnick, 1978. Probability functions on complex pedigrees. Adv. Appl. Probab. 10: 26–61. [Google Scholar]
- Daw, E. W., E. A. Thompson and E. M. Wijsman, 2000. Bias in multipoint linkage analysis arising from map misspecification. Genet. Epidemiol. 19: 366–380. [DOI] [PubMed] [Google Scholar]
- Dawber, T. R., G. F. Meadors and F. E. Moore, 1951. Epidemiologic approaches to heart disease: the Framingham study. Am. J. Public Health 41: 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster, A. P., N. M. Laird and D. B. Rubin, 1977. Maximum likelihood from incomplete data via the EM algorithm (with discussion). J. R. Stat. Soc. B 39: 1–37. [Google Scholar]
- Elston, R. C., and J. Stewart, 1971. A general model for the analysis of pedigree data. Hum. Hered. 21: 523–542. [DOI] [PubMed] [Google Scholar]
- Falk, C. T., 1992. Preliminary ordering of multiple linked loci using pairwise linkage data. Genet. Epidemiol. 9: 367–375. [DOI] [PubMed] [Google Scholar]
- Feinleib, M., W. B. Kannel and R. J. Garrison, 1975. The Framingham offspring study. Design and preliminary data. Prev. Med. 4: 518–525. [DOI] [PubMed] [Google Scholar]
- Fishelson, M., and D. Geiger, 2002. Exact genetic linkage computations for general pedigrees. Bioinformatics 18: S189–S198. [DOI] [PubMed] [Google Scholar]
- Gagnon, F., G. P. Jarvik, A. G. Motulsky, S. S. Deeb, J. D. Brunzell et al., 2003. Evidence of linkage of HDL level variation to APOC3 in two samples with different ascertainment. Hum. Genet. 113: 522–533. [DOI] [PubMed] [Google Scholar]
- Geman, S., and D. Geman, 1984. Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images. IEEE Trans. Patt. Anal. Machine Intell. 6: 721–741. [DOI] [PubMed] [Google Scholar]
- George, A. W., and E. A. Thompson, 2003. Discovering disease genes: multipoint linkage analysis via a new Markov chain Monte Carlo approach. Stat. Sci. 18: 515–531. [Google Scholar]
- George, A. W., K. L. Mengersen and G. P. Davis, 1999. A Bayesian approach to ordering gene markers. Biometrics 55: 419–429. [DOI] [PubMed] [Google Scholar]
- Goldstein, D. R., H. Y. Zhao and T. P. Speed, 1997. The effects of genotyping errors and interference on estimation of genetic distance. Hum. Hered. 47: 86–100. [DOI] [PubMed] [Google Scholar]
- Halpern, J., and A. S. Whittemore, 1999. Multipoint linkage analysis: a cautionary note. Hum. Hered. 49: 194–196. [DOI] [PubMed] [Google Scholar]
- Hastings, W. K., 1970. Monte Carlo sampling methods using Markov chains and their applications. Biometrika 57: 97–109. [Google Scholar]
- Heath, S. C., 1997. a Markov chain Monte Carlo methods for radiation hybrid mapping. J. Comput. Biol. 4: 505–515. [DOI] [PubMed] [Google Scholar]
- Heath, S. C., 1997. b Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am. J. Hum. Genet. 61: 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath, S. C., and E. A. Thompson, 1997. MCMC samplers for multilocus analyses on complex pedigrees. Am. J. Hum. Genet. 61: A278. [Google Scholar]
- Kong, A., N. Cox, M. Frigge and M. Irwin, 1993. Sequential imputations and multipoint linkage analysis. Genet. Epidemiol. 10: 483–488. [DOI] [PubMed] [Google Scholar]
- Kong, A., J. S. Liu and W. H. Wong, 1994. Sequential imputations and Bayesian missing data problems. J. Am. Stat. Assoc. 89: 278–288. [Google Scholar]
- Kruglyak, L., M. J. Daly, M. P. Reeve-Daly and E. S. Lander, 1996. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am. J. Hum. Genet. 58: 1347–1363. [PMC free article] [PubMed] [Google Scholar]
- Lander, E. S., and P. Green, 1987. Construction of multilocus genetic linkage maps in humans. Proc. Natl. Acad. Sci. USA 84 (8): 2363–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop, G. M., J. M. Lalouel, C. Julier and J. Ott, 1984. Strategies for multilocus linkage analysis in humans. Proc. Natl. Acad. Sci. USA 81: 3443–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop, G. M., J. M. Lalouel, C. Julier and J. Ott, 1985. Multilocus linkage analysis in humans: detection of linkage and estimation of recombination. Am. J. Hum. Genet. 37: 482–498. [PMC free article] [PubMed] [Google Scholar]
- Logue, M. W., V. J. Vieland, R. J. Goedken and R. R. Crowe, 2003. Bayesian analysis of a previously published genome screen for panic disorder reveals compelling evidence for linkage to chromosome 7. Am. J. Med. Genet. Neuropsychiatr. Genet. 121B: 95–99. [DOI] [PubMed] [Google Scholar]
- Matise, T. C., C. J. Porter, S. Buyske, A. J. Cuttichia, E. P. Sulman et al., 2002. Systematic evaluation of map quality: human chromosome 22. Am. J. Hum. Genet. 70: 1398–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nievergelt, C. M., D. W. Smith, J. B. Kohlenberg and N. J. Schork, 2004. Large-scale integration of human genetic and physical maps. Genome Res. 14: 1199–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa, G. J. M., B. S. Yandell and D. Gianola, 2002. A Bayesian approach for constructing genetic maps when markers are miscoded. Genet. Sel. Evol. 34: 353–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields, D. C., A. Collins, K. H. Buetow, N. E. Morton, E. P. Sulman et al., 1991. Error filtration, interference, and the human linkage map. Proc. Natl. Acad. Sci. USA 88: 6501–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam, P., 1993. Construction of integrated genetic linkage maps by means of a new computer package: JOINMAP. Plant J. 3: 739–744. [Google Scholar]
- Stephens, D. A., and A. F. M. Smith, 1993. Bayesian inference in multipoint gene mapping. Ann. Hum. Genet. 57: 65–82. [DOI] [PubMed] [Google Scholar]
- Thompson, E. A., 1974. Gene identities and multiple relationships. Biometrics 30: 667–680. [PubMed] [Google Scholar]
- Thompson, E. A., 1984. Information gain in joint linkage analysis. IMA J. Math. Appl. Med. Biol. 1: 31–49. [DOI] [PubMed] [Google Scholar]
- Thompson, E. A., 2000. MCMC estimation of multi-locus genome sharing and multipoint gene location scores. Int. Stat. Rev. 68: 53–73. [Google Scholar]
- Thompson, E. A., and S. C. Heath, 1999. Estimation of conditional multilocus gene identity among relatives, pp. 95–113 in Statistics in Molecular Biology and Genetics: Selected Proceedings of a 1997 Joint AMS-IMS-SIAM Summer Conference on Statistics in Molecular Biology (IMS Lecture Note–Monograph Series, Vol. 33), edited by F. Seillier-Moiseiwitsch. Institute of Mathematical Statistics, Hayward, CA.
- Weeks, D. E., 1991. Human linkage analysis: strategies for locus ordering, pp. 297–330 in Advanced Techniques in Chromosome Research, edited by K. W. Adolph. Marcel Dekker, New York.
- Weeks, D. E., and K. Lange, 1987. Preliminary ranking procedures for multilocus ordering. Genomics 1: 236–242. [DOI] [PubMed] [Google Scholar]
- Wijsman, E. M., E. W. Daw, C. E. Yu, H. Payami, E. J. Steinbart et al., 2004. Evidence for a novel late-onset Alzheimer disease locus on chromosome 19p13.2. Am. J. Hum. Genet. 75: 398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, S. R., 1988. A major simplification in the preliminary ordering of linked loci. Genet. Epidemiol. 5: 75–80. [DOI] [PubMed] [Google Scholar]