

Abstract
Although gemcitabine has been approved as the first-line chemotherapeutic reagent for pancreatic cancer, its response rate is low and average survival duration is still only marginal. Because epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), and platelet-derived growth factor receptor (PDGFR) modulate tumor progression, we hypothesized that inhibition of phosphorylation of all three on tumor cells, tumor-associated endothelial cells, and stroma cells would improve the treatment efficacy of gemcitabine in an orthotopic pancreatic tumor model in nude mice and prolong survival. We implanted L3.6pl, a human pancreatic cancer cell, in the pancreas of nude mice. We found that tumor-associated endothelial cells in this model highly expressed phosphorylated EGFR, VEGFR, and PDGFR. Oral administration of AEE788, a dual tyrosine kinase inhibitor against EGFR and VEGFR, decreased phosphorylation of EGFR and VEGFR. PDGFR phosphorylation was inhibited by STI571. Although intraperitoneal (i.p.) injection of gemcitabine did not inhibit tumor growth, its combination with AEE788 and STI571 produced >80% inhibition of tumor growth and prolonged survival in parallel with increases in number of tumor cells and tumor-associated endothelial cell apoptosis, decreased microvascular density, decreased proliferation rate, and prolonged survival. STI571 treatment also decreased pericyte coverage on tumor-associated endothelial cells. Thus, inhibiting phosphorylation of EGFR, VEGFR, and PDGFR in combination with gemcitabine enhanced the efficacy of gemcitabine, resulting in inhibition of experimental human pancreatic cancer growth and significant prolongation of survival.
Keywords: AEE 788, STI571, EGFR, VEGFR, PDGFR
INTRODUCTION
Pancreatic adenocarcinoma remains one of the most aggressive malignancies and is the fourth leading cause of cancer-related death in the United States (1). Because of difficulties in early diagnosis, only 10–20% of pancreatic cancers can be surgically resected with curative intent at the time of diagnosis (2). Most patients develop local recurrence and metastatic disease. Although gemcitabine can prolong survival of patients, only less than 3% survive 5 years after the initial diagnosis, and the median survival duration is less than 6 mo (3, 4). Clearly, there is an urgent need to develop new treatment modalities for pancreatic cancer.
One general method under consideration is the modulation of cancer progression pathways and its interaction with the organ microenvironment. The epidermal growth factor (EGF) phosphorylates EGFR by binding to the EGFR and further stimulates multiple signaling pathways which are involved in cell proliferation (e.g., Ras/mitogen activated protein kinase [MAPK]), and anti-apoptosis (e.g., phosphatidylinositol 3-kinase(PI3k)/Akt, nuclear factor-kappa B [NF-κB]), and others (5–8). The overexpression of EGF and EGF-R by various type of malignancies has been shown to correlate with metastasis, apoptosis, resistance to chemotherapy and poor prognosis (9–11), indicating that inhibiting EGFR signaling is a good strategy for therapeutic intervention. Cetuximab (IMC C225, Erbitux, ImClone, New York, NY) is a monoclonal antibody to EGFR that inhibits binding of EGF to EGFR and stimulation of downstream signaling pathways (12). In locally advanced or pancreatic cancer expressing EGFR, Cetuximab in combination with gemcitabine produced a 12.2% partial response, and 63.4% of patients showed stable disease on a phase II clinical trial (13). Thus, inhibiting EGFR signaling in combination with gemcitabine for pancreatic cancer showed promising activity and has led to a phase III trial of Cetuximab plus gemcitabine.
Production of another growth modulator, vascular endothelial growth factor (VEGF), increased in most types of malignant tumors and is associated with angiogenesis and poor prognosis (14). VEGF is not only a proliferating and permeability factor but also an anti-apoptotic survival factor for vascular endothelial cells (15, 16). Inhibiting VEGFR signaling could have a therapeutic efficacy not only by preventing angiogenesis, but also by causing vascular endothelial cells in the tumor microenvironment to regress. Bevacizumab (Avastin, Genentech, Inc., South San Francisco, CA) is a recombinant humanized monoclonal antibody to VEGF that inhibits its binding to VEGFR and activation of downstream signaling (17). In stage IV advanced pancreatic cancer patients, Bevacizumab in combination with gemcitabine produced a median survival of 9.0 months and a 74% 6-month survival. The partial response rate was 21%, and 45% of patients achieved stable disease, which are encouraging results (18). A randomized phase III trial of Bevacizumab plus gemcitabine is ongoing.
PDGF and its receptor are expressed in many types of cancer, including prostate, lung, gastric, and pancreatic (19, 20). In our previous study, 29 of 31 human pancreatic cancer specimens expressed phosphorylated PDGFR (21). PDGFR signaling has been reported to increase proliferation of tumor cells in an autocrine manner (22,23) and to stimulate angiogenesis, recruit pericytes (which stabilize the tumor vasculature) (22, 24), and control the interstitial fluid pressure in stroma to influence transvascular transport of chemotherapeutic agents in a paracrine manner (25,26). Inhibition of PDGFR activity by tyrosine kinase inhibitor STI571 (Novartis Pharma, Basel, Switzerland) (27) in an orthotopic nude mouse model of pancreatic cancer decreased the growth of primary pancreatic tumors and decreased the incidence of peritoneal metastases when combined with gemcitabine (21).
The most recent data indicate that the biological heterogeneity of neoplasms includes expression of tyrosine kinase receptors (22). Indeed, dual immunohistochemistry of human pancreatic cancer cells growing in the pancreas of nude mice revealed that tumor cells express both EGFR and PDGFR (Fig. 1) and, thus, inhibition of one receptor’s signaling may not be sufficient to inhibit the progressive growth and spread of neoplasms. To overcome this heterogeneity and address the issue of redundancy in signaling pathways, we determined therapy of orthotopic human pancreatic cancer growing in nude mice by multiple PTK inhibitors. We examined whether the simultaneous inhibition of EGF-R, VEGF-R, and PDGF-R signaling pathway in pancreatic tumor cells, tumor-associated endothelial cells, and stroma cells would increase the therapeutic efficacy of gemcitabine against pancreatic cancer. AEE788 (Novartis Pharma) is a novel synthesized small molecule inhibitor of both EGF-R and VEGF-R tyrosine kinases (29), and STI571 is an inhibitor of PDGF-R, Bcr-abl, and c-kit tyrosine kinase (27). We determined whether the oral administrations of AEE788 and/or STI571 administered alone or combined with intraperitoneal injections of gemcitabine inhibited the progressive growth of human pancreatic cancer cells implanted into the pancreas of nude mice and prolonged survival.
Fig. 1.
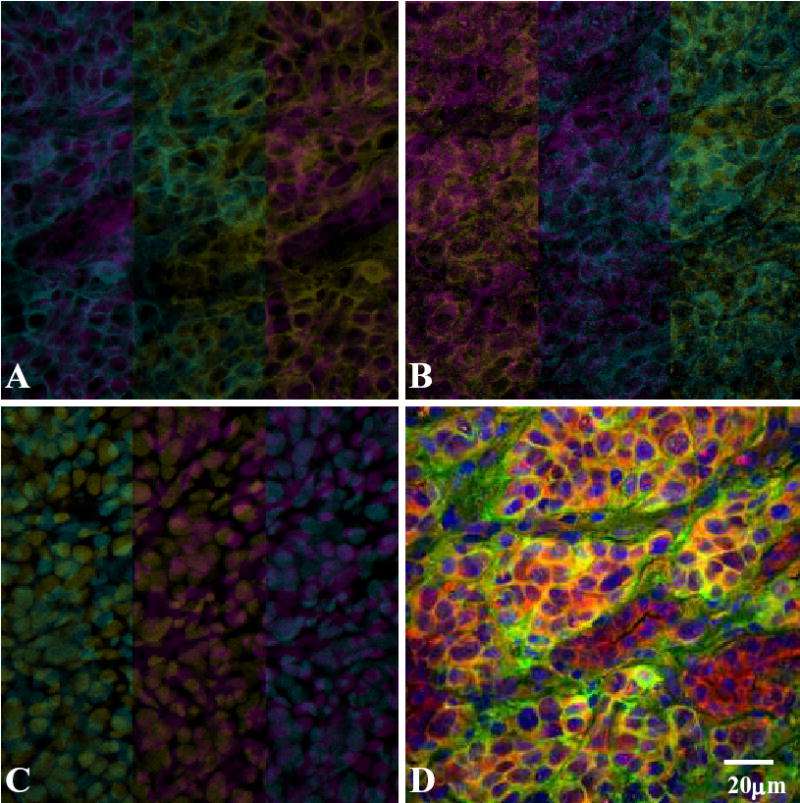
Double immunofluorescence staining for expression of EGFR and PDGFRβ in orthotopic L3.6pl tumor in nude mice. Samples were stained with anti-EGFR (A) and anti-PDGFRβ (B) antibodies as described in Materials and Methods. The nuclei were visualized by staining with Sytox green (C). Colocalization of EGFR and PDGFRβ appears as yellow fluorescence (D).
MATERIALS AND METHODS
Pancreatic Cancer Cell Line and Culture Condition
The human pancreatic cancer cell line L3.6pl was maintained in minimal essential medium supplemented with 10% fetal bovine serum (FBS), sodium pyruvate, nonessential amino acids, L-glutamine, a twofold vitamin solution (Life Technologies, Inc., Grand Island, NY), and a penicillin-streptomycin mixture (Flow Laboratories, Rockville, MD) as described previously (21).
Nucleotide Sequence Analysis of EGFR in Pancreatic Cancer L3.6pl Cell Line
Mutations in exons 18, 19, and 21 of the kinase domain of EGFR have been shown to correlate with response of patients to therapy with the tyrosine kinase inhibitor Iressa (30). To exclude the possibility that the response to AEE788 was associated with mutation of the EGFR, we assayed DNA extracted from log phase cultures of L3.6pl cells using the DNeasy Tissue Kit No. 69504 (Qiagen, Inc., Valencia, CA). Mutational analysis was performed by the Molecular Diagnostic Laboratory of the M. D. Anderson Cancer Center (Houston, TX). Nested PCR products of exons 18, 19, and 21 obtained using primers previously described (30) were directly sequenced in sense and antisense directions. All sequences were screened for the presence of mutations both manually and using the SeqScape software and confirmed by two independent PCR amplifications. The results indicated that the L3.6pl cells contain a wild-type EGFR.
Reagents
AEE 788 (Novartis Pharma), 7H-pyrrolo[2,3-d]pyrimidine lead scaffold, is a low molecular weight, ATP-competitive dual EGF-R and VEGF-R tyrosine kinase family inhibitor (29). STI571 (imatinib mesylate or Gleevec; Novartis Pharma) is a 2-phenylaminopyrimidine class protein-tyrosine kinase inhibitor of PDGFR, BCR-ABL, and c-Kit (27). For oral administration, AEE 788 was diluted in DMSO and STI571 was diluted in sterile water. Gemcitabine (Gemzar, Eli Lilly Co, Indianapolis, IN) was maintained at room temperature and dissolved in phosphate buffered saline (PBS) on the day of use. It was administered by i.p. injection.
Primary antibodies were purchased from the following manufacturers: rabbit anti-pVEGFR 2/3 (Flk-1) (Oncogene, Boston, MA); rabbit anti-human, mouse, rat VEGF-R(Flk-1)(C1158) (Santa Cruz Biotechnology, Santa Cruz, CA); rabbit anti-human pEGFR (Tyr 1173) (Biosource, Camarillo, CA); rabbit anti-human EGF and rabbit anti-human EGFR for paraffin samples (Santa Cruz Biotechnology); rabbit anti-human EGFR for frozen samples (Zymed, San Francisco, CA); rabbit anti-VEGF (A20) (Santa Cruz Biotechnology); polyclonal rabbit anti-PDGFR-β, polyclonal goat anti-phosphorylated PDGFR-β, and polyclonal rabbit anti-PDGF-β (all obtained from Santa Cruz Biotechnology, Santa Cruz, CA); rat anti-mouse CD31 (BD PharMingen, San Diego, CA); mouse anti-proliferating cell nuclear antigen (PCNA) clone PC 10 (Dako A/S, Copenhagen, Denmark); and rabbit anti-desmin (Dako A/S)(as a pericyte marker). The following secondary antibodies were used for colorimetric immunohistochemistry: peroxidase-conjugated goat anti-rabbit IgG; F(ab’)2 (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA); biotinylated goat anti-rabbit (Biocare Medical, Walnut Creek, CA); streptavidin horseradish peroxidase (Dako A/S); rat anti-mouse IgG2a horseradish peroxidase (Serotec, Harlan Bioproducts for Science, Inc., Indianapolis, IN); and goat anti-rat horseradish peroxidase (Jackson ImmunoResearch Laboratories, Inc.). The following fluorescent secondary antibodies were used: Alexa488 conjugated goat anti-rabbit IgG (Molecular Probes Inc., Eugene, OR) and Alexa 594 conjugated goat anti-rat IgG (Molecular Probes Inc.). Terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) staining was performed using a commercial apoptosis detection kit (Promega, Madison, WI) with modifications.
Animals and Orthotopic Implantation of Tumor Cells
Male athymic nude mice (NCI-nu) were purchased from the Animal Production Area of the National Cancer Institute Frederick Cancer Research and Development Center (Frederick, MD). The mice were housed and maintained under specific pathogen-free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the U.S. Department of Agriculture, U.S. Department of Health and Human Services, and the National Institutes of Health. The mice were used in accordance with institutional guidelines when they were 8 to 12 weeks old.
To produce pancreatic tumors, L3.6pl cells were harvested from subconfluent cultures by a brief exposure to 0.25% trypsin and 0.02% EDTA. Trypsinization was stopped with medium containing 10% FBS, and the cells were washed once in serum-free medium and resuspended in Hanks’ balanced salt solution (HBSS). Only suspensions consisting of single cells with greater than 90% viability were used for injection into the pancreas of nude mice as described previously (21).
Treatment of Established Human Pancreatic Carcinoma Tumors Growing in the Pancreas of Athymic Nude Mice
Twenty-one days after the intra-pancreatic injection of 0.5 × 106 viable L3.6pl cells in 50 μl HBSS, the pancreatic tumors reached the size of 5–6-mm. At that time, the mice were randomized to the following 8 treatments (n=10): (1) Control mice: administration of water diluted at 1:20 with DMSO–0.5% Tween 80 (diluent) by oral gavage 3 times per week, daily oral gavage with sterile water, and i.p. injections of PBS twice a week; (2) administration of diluent by oral gavage 3 times per week, daily oral gavage with sterile water, and twice weekly i.p. injections of gemcitabine (50 mg/kg); (3) oral gavage of AEE788 (50 mg/kg), 3 times per week, daily oral gavage with sterile water, and twice per week i.p. injections of PBS; (4) oral gavage of AEE788 (50 mg/kg) three times per week, daily oral gavage with sterile water, and twice per week i.p. injection of gemcitabine (50 mg/kg); (5) daily oral gavage of STI571 (50 mg/kg), diluent of AEE788 by oral gavage 3 times per week and i.p. injections of PBS twice per week; (6) daily oral STI571 (50 mg/kg), oral gavage of diluent for AEE788 3 times per week, and i.p. injections of gemcitabine (50 mg/kg) twice weekly; (7) combination of oral AEE788 (50 mg/kg) 3 times per week, daily STI571 (50 mg/kg), and twice per week i.p. injections of PBS; and (8) combination of oral AEE788 (50 mg/kg) 3 times per week, STI571 (50 mg/kg) 7 times per week, and twice per week i.p. injections of gemcitabine (50 mg/kg). All mice were treated for 4 weeks and killed on day 49 of the experiment.
For survival studies, 21 days after the intra-pancreatic injection of 1.0 × 106 tumor cells in 50 μl HBSS, at which time the tumors in the pancreas exceeded 6- to 8-mm in diameter, the mice were randomized (n=10) to one of the 8 treatment groups, as described above. The mice were killed and necropsied when they became moribund. Survival was evaluated by the Kaplan-Meier method. The study was repeated.
Necropsy Procedures and Histological Studies
In the first treatment study, the mice were killed on day 49 after tumor cell injection, weighted, and necropsied. Tumors growing in the pancreas were excised and weighed. For immunohistochemical staining procedures, one part of the tumor tissue was fixed in formalin and embedded in paraffin and the other was embedded in OCT compound (Miles, Inc., Elkhart, IN), rapidly frozen in liquid nitrogen, and stored at −70°C.
Immunohistochemical (IHC) Analysis to Detect EGF, VEGF, PDGF-BB, EGFR, VEGFR, PDGFRβ pEGFR, pVEGFR, pPDGF-R in Pancreatic Tumors
Paraffin-embedded pancreatic tumors of mice from all treatment groups were immunostained to evaluate the expression of EGF, VEGF, PDGF-BB, EGFR, VEGFR, PDGFRβ, phosphorylated (p)EGFR, pVEGFR, and pPDGFRβ. The sections were deparaffinized in xylene, dehydrated with alcohol and rehydrated in PBS. Endogenous peroxidase was blocked with 3% hydrogen peroxide in PBS. Samples were exposed to protein block (5% normal horse serum, 1% normal goat serum in PBS) and incubated overnight at 4°C with each primary antibody at the appropriate dilution. After 1 h incubation at room temperature with peroxidase-conjugated secondary antibody, positive reaction was detected by exposure to stable 3,3′-diaminobenzidine (DAB) (Phoenix Biotechnologies, Huntsville, AL). Slides were counterstained with Gill’s #3 hematoxylin. Sections stained for immunoperoxidase or hematoxylin and eosin were examined in a Nikon Microphot-FX microscope equipped with a three-chip-charged coupled device (CCD) color video camera (Model DXC990, Sony Corp., Tokyo, Japan). Digital images were captured using Optimas Image Analysis software (Media Cybernetics, Silver Spring, MD).
IHC Determination of Proliferating Cell Nuclear Antigen (PCNA), CD31/PECAM-1 (Endothelial Cells) and TUNEL (Apoptosis)
Paraffin-embedded tissues were used for IHC identification of proliferating cell nuclear antigen (PCNA). Frozen tissues used for identification of CD31/PECAM-1 were sectioned (8–10 μm), mounted on positively charged slides, and air-dried for 30 min. Frozen sections were fixed in cold acetone (5 min), in acetone/chloroform (v/v; 5 min), and again in acetone (5 min), and washed with PBS. IHC procedures were performed as described previously (21). Control samples exposed to a secondary antibody alone showed no specific staining. For the quantification of mean vessel density (MVD) in sections stained for CD31, 10 random 0.159-mm2 fields at X100 magnification were captured for each tumor, and microvessels were quantified. For quantification of PCNA expression, the number of positive cells was counted in 10 random 0.159-mm2 fields at X100 magnification.
Analysis of apoptotic cells was performed by using a commercially available TUNEL kit (Promega) with the following modifications: Samples were fixed and incubated with an equilibration buffer followed by a reaction buffer (containing nucleotide mix and terminal deoxynucleotidyl transferase enzyme). Immunofluorescence microscopy was performed in a Zeiss Axioplan microscope (Carl Zeiss, Inc., Thornwood, NY) equipped with an HBO 100 mercury lamp, narrow bandpass filters to individually select for green, red, and blue fluorescence (Chroma Technology Corp., Brattleboro, VT). Images were captured using a cooled CCD Hamamatsu Orca camera (Hamamatsu Corp., Bridgewater, NJ) and Image Pro Analysis software (Media Cybernetics, Silver Spring, MD). Photomontages were prepared using Adobe Photoshop software (Adobe Systems, Inc., San Jose, CA). The number of TUNEL-positive cells in 10 random 0.159-mm2 fields at ×100 magnification was used to quantify apoptosis.
Double Immunofluorescence Staining for CD31/PECAM-1 and EGFR, pEGFR, VEGFR, pVEGFR, PDGFRβ, pPDGFRβ, Pericytes (desmin-positive cells), and TUNEL
Frozen sections of pancreatic tumors were mounted on slides and fixed. Immunofluorescence for CD31 was performed using Alexa594 conjugated secondary antibody, and samples were again blocked briefly in a blocking solution (5% normal horse serum and 1% normal goat serum in PBS) as described above and incubated with antibody against human EGFR, pEGFR, VEGFR, pVEGFR, PDGFRβ, pPDGFRβ, or desmin at 4°C overnight. After washes and blocking with blocking solution, samples were incubated with Alexa488 conjugated secondary antibody. Endothelial cells were identified by red fluorescence, and EGF-R, pEGFR, VEGFR, pVEGFR, PDGFRβ, pPDGFRβ and desmin positive cells (pericytes) were identified by green fluorescence. The presence of growth factor receptors and phosphorylated receptors on endothelial cells were detected by colocalization of red and green fluorescence, which appeared yellow.
The coverage of pericytes on endothelial cells was determined by counting CD31 positive cells in direct contact with desmin-positive cells and CD31-positive cells without direct association with desmin-positive cells in five randomly selected microscopic fields (at ×200 magnification) (31–33).
TUNEL-positive apoptotic cells were detected by localized green fluorescence within cell nuclei, and endothelial cells were identified by red fluorescence. Apoptotic endothelial cells were identified by yellow fluorescence within the nuclei. Quantification of apoptotic endothelial cells was expressed as the ratio of apoptotic endothelial cells to the total number of endothelial cells in ten 0.159-mm2 fields at ×100 magnification.
Statistical Analysis
Body weight, tumor weight, PCNA-positive cells, mean vessel density (CD31/PECAM-1), and TUNEL-positive cells were compared using the Mann-Whitney U test. Survival analysis was computed by the Kaplan-Meier method and compared by the Log rank test.
RESULTS
Therapy of Human Pancreatic Cancer Growing in the Cecum of Nude Mice
In the first set of experiments, the effect of treatment with AEE788, STI571, and gemcitabine alone and in various combinations was determined against well-established (5–6 mm) pancreatic tumors. The mice were killed and necropsied on day 49 of the study (Table1). Tumor incidence in the pancreas was 100% in all treatment groups. None of the treatments significantly affected body weight, indicating no obvious side effects. Control mice had the largest tumors (0.77 g). Treatment with STI571 or gemcitabine alone did not inhibit tumor growth, but mice treated with AEE788 had significantly smaller tumors (0.33g: p<0.001). The combination of AEE788 and gemcitabine or AEE788 and STI571 (but not STI571 and gemcitabine) significantly decreased tumor weight in the pancreas (0.19 g, p<0.0001, 0.33 g; p<0.001 vs control, and 0.71 g, respectively). Combining AEE788, STI571, and gemcitabine for therapy produced the most significant inhibition of tumor growth (0.14 g, p<0.0001 versus control).
Table 1.
Therapy of L3.6pl human pancreatic cancer cells implanted in the pancreas of nude mice
| Body weight(g)
|
Tumor weight (g)
|
|||
|---|---|---|---|---|
| Treatment | Median | (Range) | Median | (Range) |
| Control | 24.8 | (18.8–27.8) | 0.77 | (0.48–1.80) |
| Gemcitabine | 25.7 | (20.0–28.1) | 0.78 | (0.36–1.23) |
| STI571 | 23.5 | (18.7–27.2) | 0.96 | (0.45–1.83) |
| STI571 + Gemcitabine | 25.0 | (21.1–28.1) | 0.71 | (0.42–1.35) |
| AEE788 | 26.2 | (21.3–28.5) | 0.33 | (0.08–0.44)a |
| AEE788 + Gemcitabine | 25.3 | (22.1–28.8) | 0.19 | (0.05–0.40)b |
| AEE788 + STI571 | 24.1 | (22.2–29.0) | 0.33 | (0.05–0.50)a |
| AEE788 + STI571 + Gemcitabine | 24.0 | (21.5–28.9) | 0.14 | (0.04–0.30)b,c |
L3.6pl cells (0.5 × 106) were injected into the pancreas of nude mice. Three weeks later, the mice were randomized (n=10) to receive the following regimens: (1) Control: oral and i.p. diluent only; (2) Gemcitabine: twice per week i.p. injection of gemcitabine (50 mg/kg); (3) STI571: daily oral gavage of STI571 (50 mg/kg); (4) STI571 and Gemcitabine: combination of oral STI571 (50 mg/kg) and i.p. injection of gemcitabine (50 mg/kg) twice weekly; (5) AEE788: oral gavage of AEE788 (50 mg/kg) 3 times per week; (6) AEE788 and Gemcitabine: Combination of oral AEE788 (50 mg/kg) and twice per week i.p. injection of gemcitabine (50 mg/kg); (7) AEE788 and STI571: Combination of oral AEE788 (50 mg/kg) 3 times per week and STI571 (50 mg/kg) daily; (8) AEE788, STI571, and Gemcitabine: Combination of oral AEE788 (50 mg/kg) 3 times per week, STI571 (50 mg/kg) daily, and i.p. injection of gemcitabine (50 mg/kg) twice weekly. All mice were treated for 4 wk and killed on day 49 of the study. Body weight, tumor incidence, and tumor weight were recorded. All mice had pancreatic tumors.
P<0.001 vs control.
P<0.0001 vs control.
P<0.05 vs AEE788 or AEE788 and STI571.
In the next survival study, treatment began 21 days after the intrapancreatic injection of 1.0 × 106 L3.6pl cells. The pancreatic tumors measured 6–8 mm in diameter and thus were well established. Treatment continued until the mice became moribund, at which time they were killed. Survival was analyzed using the Kaplan-Meier method as shown in Figure 2. All treatments other than STI571 alone or gemcitabine alone significantly prolonged survival as compared to the control treatment group. Mice treated with the combination of AEE788, STI571, and gemcitabine had the greatest prolongation of survival.
Fig. 2.
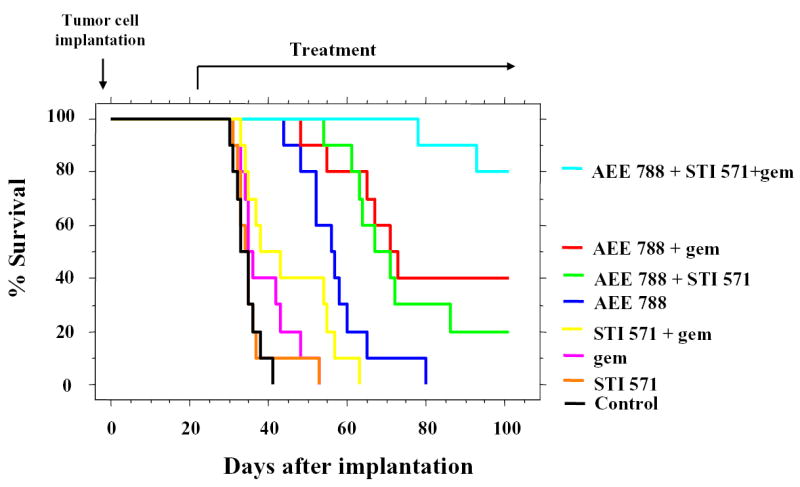
Therapeutic effects of AEE788, STI571, gemcitabine and their combinations on survival rate. Nude mice were injected with L3.6pl human pancreatic cancer cells (1 × 106) into the pancreas. Twenty-one days after the injection, the mice were randomized into 8 treatment groups (n=10) as detailed in Table 1. Mice were killed when got moribund. Survival analysis was done by the Kaplan-Meier method and compared by the Logrank test. AEE 788 + STI571 + Gemcitabine: p<0.0001 vs Control, STI, Gem, STI+Gem, p<0.001 vs AEE, p<0.01 vs AEE+STI, p<0.05 vs AEE+Gem. AEE788+Gemcitabine: p<0.0001 vs Control, STI, Gem, STI+Gem, p<0.05 vs AEE. AEE788+STI571: p<0.0001 vs Control, STI, Gem, STI+Gem, p<0.001 vs AEE. AEE788: P<0.0001 vs Control, p<0.01 vs STI, STI571+Gemcitabine: p<0.05 vs Control
Immunohistochemical Analysis of L3.6pl Pancreatic Tumors
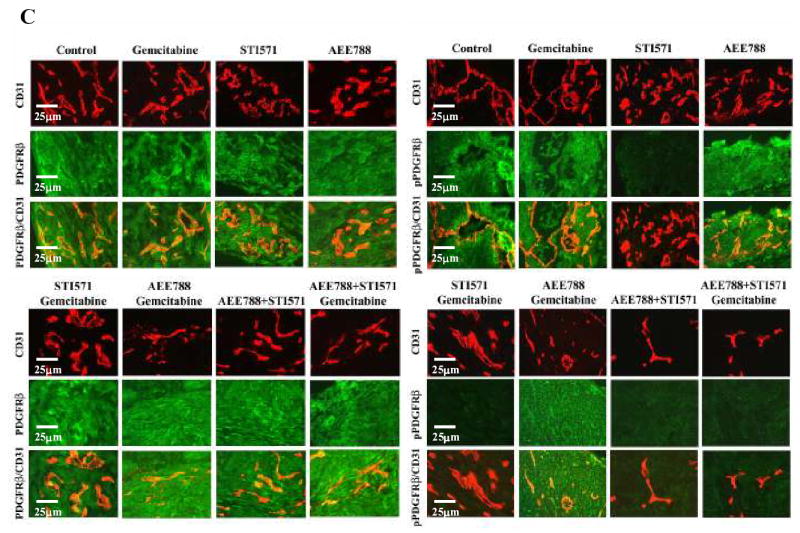
Tumor sections were analyzed immunohistochemically for the expression of EGF, EGFR, and pEGFR (Fig. 3A), VEGF, VEGFR, and pVEGFR (Fig. 3B), and PDGF-BB, PDGFRβ and pPDGFRβ (Fig. 3C). Treatment with AEE 788, STI571, gemcitabine, or any of the combination treatments did not alter the expression level of EGF, VEGF, PDGF-BB, EGFR, VEGFR, and PDGFRβ by the tumor cells or in the stroma cells. The phosphorylation of EGFR and VEGFR (but not PDGFR) was significantly reduced in tumors from mice treated with AEE788 alone or any combination therapy including AEE788 (Fig. 3A, 3B). In contrast, PDGFRβ (but not EGFR or VEGFR) phosphorylation was inhibited in tumors from mice treated with STI571 alone or combination therapy including STI571 (Fig. 3C). These data confirmed that at the concentration administered to mice, the PTK inhibitors produced specific inhibition of their respective target receptors. As expected, the combination therapies with AEE788 and STI571 and with AEE788, STI571, and gemcitabine inhibited phosphorylation of all three receptors.
Fig. 3.
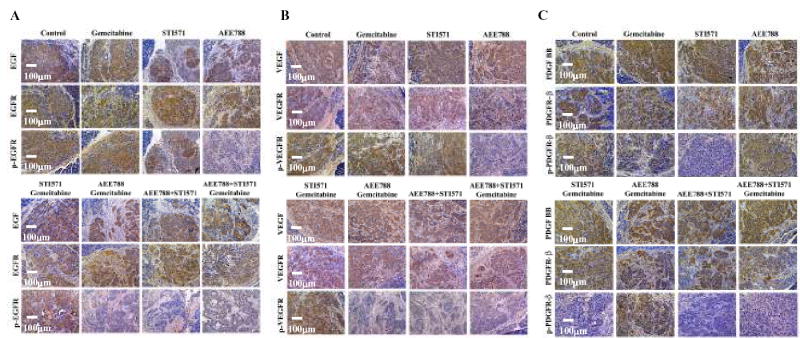
Immunohistochemical analysis of the expression of EGF, EGFR, pEGFR, VEGF, VEGFR, pVEGR and PDGF-BB, PDGFRβ and pPDGFRβ. L3.6pl human pancreatic cancer cells growing in the pancreas of nude mice were treated as described in Materials and Methods for 4 weeks, and all the mice were killed on day 28. Tumor tissue sections were stained for EGF, EGF-R, p-EGF-R (A), VEGF, VEGF-R, p-VEGR (B) and PDGF-BB, PDGFRβ, pPDGFRβ (C) as described in Material and Methods. Tumors from all treatment groups expressed similar levels of the ligands and receptors. Tumor from mice treated with AEE788 showed decreased phosphorylation of EGFR and VEGFR and tumor from mice treated with STI571 showed inhibition of phosphorylation of PDGFRβ. Combination of AEE788 and STI571 inhibited phosphorylation of EGFR, VEGFR, and PDGFRβ.
EGF-R, VEGFR, PDGFR, pEGFR, pVEGFR and pPDGFR on Tumor-associated Endothelial Cells
To determine whether tumor-associated endothelial cells expressed EGFR, VEGFR, PDGFRβ, pEGFR, pVEGFR, or pPDGFRβ, we used a double immunofluorescence staining technique. Tumor-associated endothelial cells from all treatment groups expressed similar levels of EGFR (Fig. 4A), VEGFR (Fig. 4B), and PDGFRβ (Fig. 4C). The phosphorylation of EGFR and VEGFR was diminished on endothelial cells from tumors of mice treated with AEE788 or combination treatments including AEE788 (Fig. 4A, 4B).
Fig. 4.
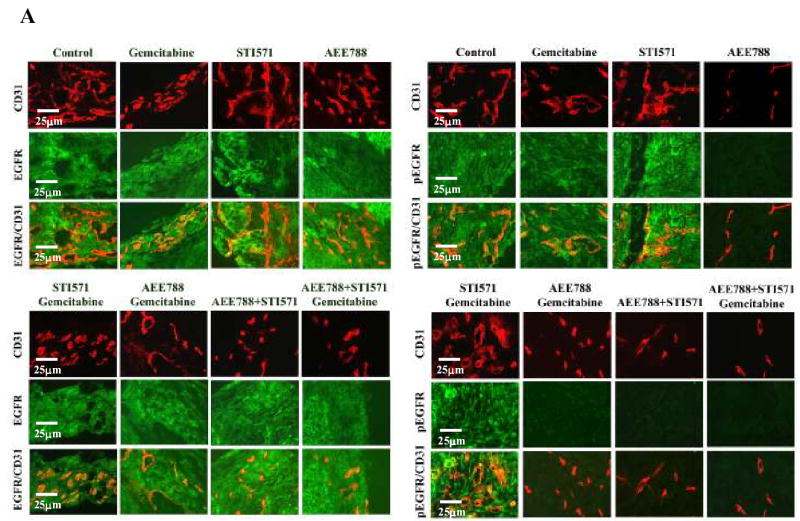
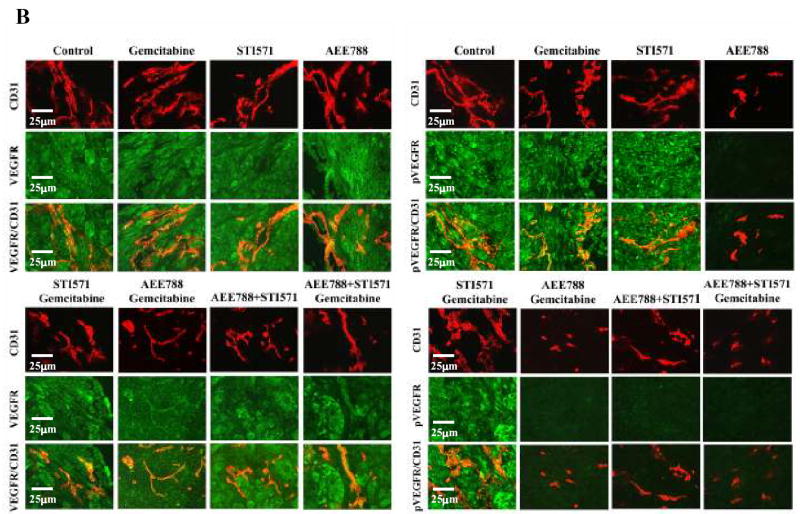
Double immunofluorescence staining for CD31/PECAM-1 and EGFR, pEGFR VEGFR, pVEGFR, PDGFRβ or pPDGFRβ in pancreatic tumors. Tumor sections were stained with anti-CD31/PECAM1 antibody (red) and anti-EGFR, pEGFR(A), VEGFR, pVEGR(B), PDGFRβ or pPDGFRβ (C) in green fluorescence as described in Materials and Methods. Colocalization of CD31 and EGF-R, pEGFR, VEGFR, pVEGR, PDGFRβ, or pPDGFRβ appears in yellow fluorescence. Expression of EGFR, VEGFR or PDGFRβ by tumor-associated endothelial cells was found in tumors from all treatment groups. Phosphorylation of EGF-R and VEGFR on endothelial cells was decreased by treatment with AEE 788 and phosphorylation of PDGFRβ on tumor-associated endothelial cells was decreased by treatment with STI571. Combination of AEE788 and STI571 inhibited phosphorylation of EGFR, VEGFR and PDGFRβ simultaneously.
Phosphorylation of the PDGFRβ was decreased on endothelial cells from tumors of mice treated with STI571 or combination treatments including STI571 (Fig. 4C). Administration of AEE788 and STI571 or AEE788, STI571, and gemcitabine inhibited phosphorylation of EGFR, VEGFR, and PDGFRβ on tumor-associated endothelial cells.
Cell Proliferation (PCNA), Apoptosis (TUNEL), and Mean Vessel Density (MVD)
Cell proliferation was evaluated by staining for PCNA (Fig. 5). In tumors from control mice, the median number of PCNA-positive cells was 371 ± 88. As shown in Table 2, treatment with gemcitabine alone or STI571 alone decreased the number of dividing PCNA-positive cells. A significant decrease of PCNA-positive cells was found in tumors from all other treatment groups, with the highest inhibition produced in tumors from mice treated with AEE788, STI571, and gemcitabine (155 ± 54, P<0.001).
Fig 5.
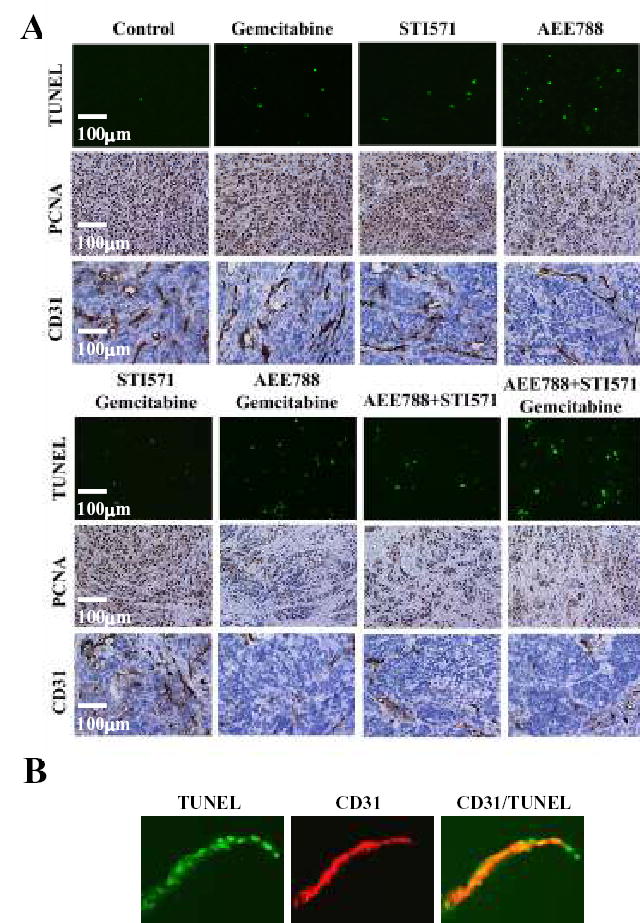
(A) Analysis of apoptosis (TUNEL), cell proliferation (PCNA), and microvessel density (CD31). Mice were treated with control, gemcitabine, AEE 788, STI571, or the combination of AEE 788 and gemcitabine, STI571 and gemcitabine, AEE788 and STI571, or AEE788 and STI571 and gemcitabine. Pancreatic tumors were resected and processed for immunohistochemical evaluation of PCNA, TUNEL, and CD31 as described in Materials and Methods. (B) Double immunofluorescence staining of CD31/PECAM-1 and TUNEL in pancreatic tumors from mice treated with the combination of AEE788, STI571, and gemcitabine. Endothelial cells (CD31+) stained red fluorescence and apoptotic cells (TUNEL+) stained green fluorescence. Colocalization of endothelial cells undergoing apoptosis yielded yellow fluorescence.
Table 2.
Immunohistochemical analysis of L3.6pl human pancreatic cancer cells growing in the pancreas of nude mice
| Tumor cells
|
Endothelial cells
|
|||
|---|---|---|---|---|
| Treatment | PCNAa,c | TUNELb,c | CD31c | TUNEL+ (%)c |
| Control | 371 ± 88 | 1 ± 1 | 46 ± 11 | 0 ± 0 |
| Gemcitabine | 305 ± 71 | 8 ± 3f | 38 ± 7 | 1 ± 1 |
| STI571 | 301 ± 49 | 6 ± 2 | 37 ± 7 | 0 ± 0 |
| STI571 + Gemcitabine | 254 ± 48e | 11 ± 4f | 34 ± 8d | 0 ± 1 |
| AEE788 | 233 ± 54e | 14 ± 4f | 25 ± 5d | 3 ± 3e |
| AEE788 + Gemcitabine | 187 ± 48f | 22 ± 7f,k | 28 ± 7f,k | 8 ± 6e |
| AEE788 + STI571 | 204 ± 69e | 18 ± 6f | 21 ± 5f | 5 ± 5e |
| AEE788 + STI571 + Gemcitabine | 155 ± 54f,h | 30 ± 10g,i,j | 16 ± 6f,I | 8 ± 5e |
PCNA, proliferating cellular nuclear antigen;
TUNEL, terminal deoxynucleotidyltransferase-mediated nick end labeling;
Median ± S.D.;
P<0.05 vs control;
P<0.01 vs control;
P<0.001 vs control;
P<0.001 vs control;
P<0.05 vs AEE788;
P<0.01 vs AEE788;
P<0.05 vs AEE788 + STI571;
P<0.05 vs AEE788.
The induction of apoptosis in the pancreatic tumors was evaluated by the TUNEL method (Table 2). In tumors from control-treated mice, the median number of apoptotic tumor cells was minimal (1 ± 1). The number of apoptotic cells in tumors from mice in all other treatment groups (except those treated with only STI571) increased, with the highest produced by therapy with the combination of AEE788, STI571, and gemcitabine (30 ± 10).
MVD in the tumors was determined by IHC staining with antibodies against CD31 (Table 2). The median number of CD31-positive tumor cells from control mice was 46 ± 11. Treatment with gemcitabine alone or STI571 alone did not decrease MVD. The number of CD31-positive cells was significantly decreased in tumors from all other treatment groups, with the largest decrease in MVD in tumors from mice treated with AEE 788, STI571, and gemcitabine (16 ± 6) (P<0.001).
Immunofluorescence Double Staining for CD31/PECAM-1 and TUNEL
Next, we determined whether therapy was associated with apoptosis of endothelial cells by using the CD31/TUNEL fluorescent double-labeling technique (Fig. 5B). Tumors from control mice had no apoptosis in tumor-associated endothelial cells. Treatment of mice with AEE788, STI571, and gemcitabine produced a median of 8 ± 5% apoptosis in tumor-associated endothelial cells (Table 2).
Pericyte coverage on tumor-associated endothelial cells
The effect of the different treatments on pericyte coverage on tumor-associated endothelial cells was evaluated using the double immunofluorescence staining technique with anti-CD31 antibody and anti-desmin antibody (Fig. 6A). Pericyte coverage rate in tumors from control-treated mice was 35.4 ± 9.8% (median ± S.D.). Treatment with STI571 alone or STI571 and gemcitabine produced a significant decrease in pericyte coverage (p<0.05, 18.8 ± 14.7%, 18.1 ± 10.3% respectively) (Fig. 6B). In contrast, treatment with gemcitabine alone, AEE788 alone, or treatment including AEE788 did not produce a measurable decrease in pericyte coverage. Thus, in this study, we did not find a correlation between inhibition of pericyte coverage of endothelial cells and a decrease in MVD.
Fig 6.
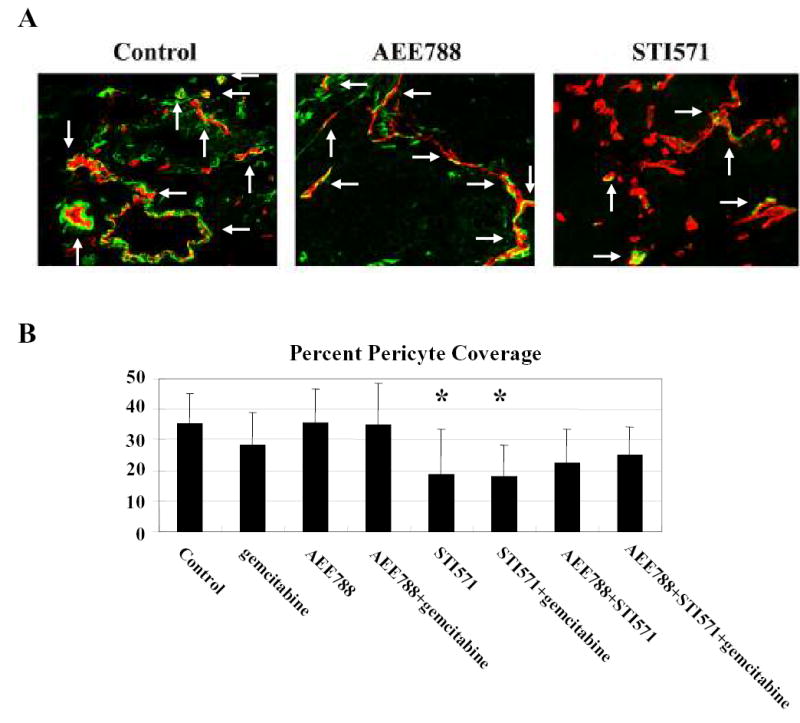
Pericyte coverage on tumor-associated endothelial cells in the pancreatic tumors. Tumor sections were stained with anti-CD31/PECAM1 antibody (red) and anti-desmin antibody (pericyte marker) in green fluorescence, and the pericyte coverage rate was determined as described in Materials and Methods. Representative photomicrographs of pericyte coverage from control, AEE788, and STI571 treatment groups. Arrowhead indicates pericyte coverage of tumor-associated endothelial cells (A). Pericyte coverage rate was significantly decreased by STI571 or combination with STI571 and gemcitabine treatment as compared to those in control (B). *: p<0.05 versus control.
DISCUSSION
The expression levels of EGF, VEGF, PDGF and their receptors have been reported to correlate with the progressive growth, metastasis, and resistance to chemotherapy of a variety of cancers (11, 20, 34, 35). We previously reported that the majority (29/31) of human pancreatic cancer clinical specimens expressed PDGFR and pPDGFR (21). We also found that more than 80% of pancreatic cancer clinical specimens expressed EGF, VEGF, EGFR, VEGFR, pEGFR and pVEGFR on tumor cells and tumor-associated endothelial cells (Yokoi et al., manuscript submitted). These data suggest that EGF-R, VEGF-R, and PDGF-R could be attractive targets for therapy of this cancer.
In the present study, human pancreatic cancer cells growing in the pancreas of nude mice expressed high levels of EGF, VEGF, PDGF-BB, and their receptors, and the receptors were phosphorylated. In addition to the tumor cells, tumor-associated endothelial cells also expressed these receptors, probably in response to specific ligands produced by tumor cells (19). Oral treatment with AEE788 inhibited the phosphorylation of EGFR and VEGFR (but not the expression of EGF, VEGF, EGFR and VEGFR) on pancreatic tumor cells and tumor-associated endothelial cells. Oral treatment with STI571 inhibited phosphorylation of PDGFR but did not alter PDGF-BB and PDGF-R expression levels. When AEE788 and STI571 were combined, phosphorylation of the EGFR, VEGFR, and PDGFR was inhibited on both the implanted human pancreatic cancer cells and the tumor-associated endothelial cells of the recipient mice.
L3.6pl cells growing in the pancreas of nude mice were resistant to treatment with gemcitabine (Fig. 1, Table 1). When combined with AEE788, however, gemcitabine reduced tumor growth by nearly 75% and significantly prolonged survival (p<0.0001). This therapeutic effect was significantly better than that from treatment with AEE788 alone (p<0.05). Indeed, the combination treatment using AEE788 and gemcitabine induced a significantly higher level of apoptosis in tumor and tumor-associated endothelial cells, decreased the number of proliferating cells, and a decreased MVD as compared to control. These data indicate that inhibition of both the EGFR and VEGFR signaling pathways on tumor cells and tumor-associated endothelial cells combined with a chemotherapeutic reagent is superior to either treatment administered alone.
STI571 as a single treatment had a limited effect on the inhibition of tumor growth and prolongation of survival. However, the combination of STI571 with AEE788 significantly lowered the number of PCNA-positive cells and the MVD and increased the number of apoptotic tumor cells and apoptotic endothelial cells; all these were associated with prolongation of survival. Similar data were produced by combining AEE788 with gemcitabine. The best therapy, however, was produced by combining AEE788 with STI571 and gemcitabine. This combination led to a decrease in tumor size, prolonged survival (P<0.0001), the fewest PCNA-positive tumor cells, the lowest MVD, and the highest number of apoptotic cells.
In our study, tumor-associated endothelial cells expressed not only EGFR and VEGFR, but also PDGFR, which would provide another target for inhibition of its signaling by STI571. PDGFR as well as EGFR and VEGFR signaling, which activates the anti-apoptotic protein Akt and bcl-2, acts like a survival factor for endothelial cells (36–38). With the inhibition of survival mechanisms by AEE788 and STI571, tumor-associated endothelial cells, whose proliferating frequency is 20–2000 times higher than that of endothelial cells in normal organs (39,40), would be more sensitive to anticycling chemotherapeutic treatment. Indeed, we found the largest number of apoptotic cells on tumor-associated endothelial cells (Table 2).
Until now, antiangiogenic therapy has focused mainly on endothelial cells. Recent studies, however, imply that pericyte can also play an important role in angiogenesis (22–24). Since pericyte recruitment and covering of endothelial cells for stabilization and maturation of vessel structure is dependent on PDGFRβ signaling (22), the inhibition of PDGFR signaling by a PTK inhibitor should inhibit pericyte recruitment and attachment to endothelial cells which would in turn confer resistance to VEGFR antagonists on endothelial cells (41,42). In agreement with other reports, we found that treatment with STI571 decreased pericyte coverage on tumor-associated endothelial cells, whereas AEE788 did not. However, administration of AEE788 seemed to reverse the effect of STI571, suggesting that AEE788 may target endothelial cells or targeted endothelial cells with relatively poor pericyte coverage.
The increased interstitial hyperpressure found in tumor stroma can decrease delivery of drugs. A number of studies reported that inhibition of PDGFR signaling can decrease this pressure and hence enhance the effects of chemotherapeutic reagents (25, 26). Increased vascular permeability is a major reason for increased interstitial high pressure (43, 44). Anti-VEGF mAb treatment can lower vascular permeability by normalization of vascular architecture and function (43). Taken together, these reports suggest that treatment with AEE788 and STI571 may decrease interstitial pressure as well as vascular permeability and, hence, increase delivery of gemcitabine to cancer cells.
In conclusion, pancreatic cancer cells produce EGF, VEGF, and PDGF. These ligands can activate their receptors on tumor cells by an autocrine manner and on tumor-associated endothelial cells by a paracrine manner. As a consequence, both tumor cells and tumor-associated endothelial cells have increased survival and resistance to chemotherapeutic agents (36). Inhibiting these signaling pathways by tyrosine kinase inhibitors combined with conventional chemotherapy induced a significant apoptosis in tumor-associated endothelial cells and tumor cells, resulting in decreased tumor size and significant prolongation of survival. The success of this multimodality therapy can be attributed to the heterogeneous nature of cancer. Targeting both tumor cells and tumor-associated endothelial cells can therefore be of great therapeutic benefit.
Acknowledgments
The authors thank Walter Pagel for critical editorial review and Lola López for expert assistance with the preparation of this manuscript.
Footnotes
Grant support: This work was supported in part by Cancer Center Support Grant CA16672, SPORE in Prostate Cancer Grant CA90270, and SPORE in Pancreatic Cancer Grant CA10193-06 from the National Cancer Institute, National Institutes of Health, and by a sponsored research agreement from Novartis Pharma, Basle, Switzerland.
References
- 1.Jemal A, Tiwari RC, Murray T, et al. Cancer Statistics, 2004. CA Cancer J Clin. 2004;54:8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- 2.Li D, Xie K, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. 2004;363:1049–57. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 3.Burris HA, 3rd, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–13. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 4.Abbruzzese JL. New application of gemcitabine and future directions in the management of pancreatic cancer. Cancer. 2002;95:941–5. doi: 10.1002/cncr.10753. [DOI] [PubMed] [Google Scholar]
- 5.Perugini RA, McDade TP, Vittimberga FJ, et al. Pancreatic cancer cell proliferation is phosphatidylinositol 3-kinase dependent. J Surg Res. 2000;90:29–44. doi: 10.1006/jsre.2000.5833. [DOI] [PubMed] [Google Scholar]
- 6.Nicholson KM, Anderson NG. The protein kinase B/Akt signaling pathway in human malignancy. Cell Signal. 2002;14:381–95. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 7.Wang W, Abbruzzese JL, Evans DB, et al. The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–27. [PubMed] [Google Scholar]
- 8.Douziech N, Calvo E, Laine J, et al. Activation of MAP kinases in growth responsive pancreatic cancer cells. Cell Signal. 1999;11:591–602. doi: 10.1016/s0898-6568(99)00030-3. [DOI] [PubMed] [Google Scholar]
- 9.Ghaneh P, Kawesha A, Evans JD, Neoptolemos JP. Molecular prognostic markers in pancreatic cancer. J Hepatobiliary Pancreat Surg. 2002;9:1–11. doi: 10.1007/s005340200000. 2002. [DOI] [PubMed] [Google Scholar]
- 10.Kuwahara K, Sasaki T, Kuwada Y, Murakami M, Yamasaki S, Chayama K. Expressions of angiogenic factors in pancreatic ductal carcinoma: a correlative study with clinicopathologic parameters and patient survival. Pancreas. 2003;26(4):344–9. doi: 10.1097/00006676-200305000-00006. [DOI] [PubMed] [Google Scholar]
- 11.Yamanaka Y, Friess H, Kobrin MS, Buchler M, Beger HG, Korc M. Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res. 1993;13:565–9. [PubMed] [Google Scholar]
- 12.Mendelsohn J. The epidermal growth factor receptor as a target for cancer therapy. Endocrinal Rel Cancer. 2001;8:3–9. doi: 10.1677/erc.0.0080003. [DOI] [PubMed] [Google Scholar]
- 13.Xiong HQ, Rosenberg A, LoBuglio A, et al. Cetuximab, a monoclonal antibody targeting the epidermal growth factor receptor, in combination with gemcitabine for advanced pancreatic cancer: a multicenter phase II trial. J Clin Oncol. 2004;22(13):2610–6. doi: 10.1200/JCO.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 14.Ferrara N, Alitalo K. Clinical applications of angiogenic growth factors and their inhibitors. Nat Med. 1999;5:1359–64. doi: 10.1038/70928. [DOI] [PubMed] [Google Scholar]
- 15.Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273:13313–6. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- 16.Tran J, Rak J, Sheehan C, et al. Marked induction of the IAP family antiapoptotic proteins survivin and XIAP by VEGF in vascular endothelial cells. Biochem Biophys Res Commun. 1999;264:781–8. doi: 10.1006/bbrc.1999.1589. [DOI] [PubMed] [Google Scholar]
- 17.Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 18.Kindler HL, Friberg G, Stadler WM, et al. Bevacizumab (B) plus gemcitabine (G) in patients (pts) with advanced pancreatic cancer (PC): updated results of a multicenter phase II trial (abstract) Proc ASCO Meeting. 2004;22:315. [Google Scholar]
- 19.Kim SJ, Uehara H, Yazici S, Langley RR, et al. Simultaneous blockade of platelet-derived growth factor-receptor and epidermal growth factor-receptor signaling and systemic administration of paclitaxel as therapy for human prostate cancer Metastasis in bone of nude mice. Cancer Res. 2004;64(12):4210–8. doi: 10.1158/0008-5472.CAN-03-3763. [DOI] [PubMed] [Google Scholar]
- 20.Ebert M, Yokoyama M, Friess H, Kobrin MS, Buchler MW, Korc M. Induction of platelet-derived growth factor A and B chains and over-expression of their receptors in human pancreatic cancer. Int J Cancer. 1995;62:529–35. doi: 10.1002/ijc.2910620507. [DOI] [PubMed] [Google Scholar]
- 21.Hwang RF, Yokoi K, Bucana CD, et al. Inhibition of platelet-derived growth factor receptor phosphorylation by STI571 (Gleevec) reduces growth and metastasis of human pancreatic carcinoma in an orthotopic nude mouse model. Clin Cancer Res. 2003;9:6534–44. [PubMed] [Google Scholar]
- 22.Ostman A. PDGF receptors-mediators of autocrine tumor growth and regulators of tumor vasculature and stroma. Cytokine Growth Factor Rev. 2004;15(4):275–86. doi: 10.1016/j.cytogfr.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Heldin C-H, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 199979:1283–316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 24.Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111(9):1287–95. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pietras K, Rubin K, Sjoblom T, et al. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002;62:5476–84. [PubMed] [Google Scholar]
- 26.Pietras K. Increasing tumor uptake of anticancer drugs with imatinib. Semin Oncol. 2004;31:18–23. doi: 10.1053/j.seminoncol.2004.03.036. [DOI] [PubMed] [Google Scholar]
- 27.Buchdunger E, Cioffi CL, Law N, et al. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-Kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–45. [PubMed] [Google Scholar]
- 28.Ciardiello F, Bianco R, Caputo R, et al. Antitumor activity of ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor, in human cancer cells with acquired resistance to anti-epidermal growth factor receptor therapy. Clin Cancer Res. 2004;10:784–93. doi: 10.1158/1078-0432.ccr-1100-03. [DOI] [PubMed] [Google Scholar]
- 29.Traxler P, Allegrini PR, Brandt R, et al. AEE788: a dual family epidermal growth factor receptor/ErbB2 and vascular endothelial growth factor receptor tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2004;64(14):4931–41. doi: 10.1158/0008-5472.CAN-03-3681. [DOI] [PubMed] [Google Scholar]
- 30.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 31.McCarty MF, Wey J, Stoeltzing O, et al. ZD6474, a vascular endothelial growth factor receptor tyrosine kinase inhibitor with additional activity against epidermal growth factor receptor tyrosine kinase, inhibits orthotopic growth and angiogenesis of gastric cancer. Mol. Cancer Ther. 2004;3(9):1041–8. [PubMed] [Google Scholar]
- 32.Stoeltzing O, McCarty MF, Jane S, et al. Role of hypoxia-inducible factor 1α in gastric cancer cell growth, angiogenesis, and vessel maturation. J Natl Cancer Inst. 2004;96(12):946–56. doi: 10.1093/jnci/djh168. [DOI] [PubMed] [Google Scholar]
- 33.Chan-Ling T, Page MP, Gardiner T, Baxter L, Rosinova E, Hughes S. Desmin ensheathment ratio as an indicator of vessel stability: evidence in normal development and in retinopathy of prematurity. Am J Pathol. 2004;165(4):1301–13. doi: 10.1016/S0002-9440(10)63389-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt M, Lichtner RB. EGF receptor targeting in therapy-resistant human tumors. Drug Resistance Update. 2002;5:11–18. doi: 10.1016/s1368-7646(02)00004-3. [DOI] [PubMed] [Google Scholar]
- 35.Ozawa F, Friess H, Tempia-Caliera A, Kleeff J, Buchler MW. Growth factors and their receptors in pancreatic cancer. Teratog Carcinog Mutagen 2002;21:27–44. Buchler P, Reber HA, Buchler MW, Friess H, Hines OJ. VEGF-RII influences the prognosis of pancreatic cancer. Ann Surg. 2001;236:738–49. doi: 10.1002/1520-6866(2001)21:1<27::aid-tcm4>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 36.Langley RR, Fan D, Tsan RZ, et al. Activation of the platelet-derived growth factor-receptor enhances survival of murine bone endothelial cells. Cancer Res. 2004;64:3727–30. doi: 10.1158/0008-5472.CAN-03-3863. [DOI] [PubMed] [Google Scholar]
- 37.Nicholson KM, Anderson NG. The protein kinase B/Akt signaling pathway in humanmalignancy. Cell Signal. 2002;14:381–95. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 38.Gerber HP, McMurtrey A, Kowalski J, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–43. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 39.Hobson B, Denekamp J. Endothelial proliferation in tumours and normal tissues: continuous labeling studies. Br J Cancer. 1984;49:405–13. doi: 10.1038/bjc.1984.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res. 2000;60:1388–93. [PubMed] [Google Scholar]
- 41.Erber R, Thurnher A, Katsen AD, et al. Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. FASEB J. 2004;18(2):338–40. doi: 10.1096/fj.03-0271fje. [DOI] [PubMed] [Google Scholar]
- 42.Pietras K, Hanahan D. A multi-targeted, metronomic, and maximum-tolerated dose “chemo-switch” regimen is antiangiogenic, producing objective responses and survival benefit in a mouse model of cancer. J Clin Oncol. 2005;23(5):939–52. doi: 10.1200/JCO.2005.07.093. [DOI] [PubMed] [Google Scholar]
- 43.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–6. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 44.Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–7. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]