

Abstract
Bayesian segregation analyses were used to investigate the mode of inheritance of osteochondral lesions (osteochondrosis, OC) in pigs. Data consisted of 1163 animals with OC and their pedigrees included 2891 animals. Mixed-inheritance threshold models (MITM) and several variants of MITM, in conjunction with Markov chain Monte Carlo methods, were developed for the analysis of these (categorical) data. Results showed major genes with significant and substantially higher variances (range 1.384–37.81), compared to the polygenic variance (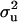 ). Consequently, heritabilities for a mixed inheritance (range 0.65–0.90) were much higher than the heritabilities from the polygenes. Disease allele frequencies range was 0.38–0.88. Additional analyses estimating the transmission probabilities of the major gene showed clear evidence for Mendelian segregation of a major gene affecting osteochondrosis. The variants, MITM with informative prior on
). Consequently, heritabilities for a mixed inheritance (range 0.65–0.90) were much higher than the heritabilities from the polygenes. Disease allele frequencies range was 0.38–0.88. Additional analyses estimating the transmission probabilities of the major gene showed clear evidence for Mendelian segregation of a major gene affecting osteochondrosis. The variants, MITM with informative prior on 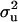 , showed significant improvement in marginal distributions and accuracy of parameters. MITM with a “reduced polygenic model” for parameterization of polygenic effects avoided convergence problems and poor mixing encountered in an “individual polygenic model.” In all cases, “shrinkage estimators” for fixed effects avoided unidentifiability for these parameters. The mixed-inheritance linear model (MILM) was also applied to all OC lesions and compared with the MITM. This is the first study to report evidence of major genes for osteochondral lesions in pigs; these results may also form a basis for underpinning the genetic inheritance of this disease in other animals as well as in humans.
, showed significant improvement in marginal distributions and accuracy of parameters. MITM with a “reduced polygenic model” for parameterization of polygenic effects avoided convergence problems and poor mixing encountered in an “individual polygenic model.” In all cases, “shrinkage estimators” for fixed effects avoided unidentifiability for these parameters. The mixed-inheritance linear model (MILM) was also applied to all OC lesions and compared with the MITM. This is the first study to report evidence of major genes for osteochondral lesions in pigs; these results may also form a basis for underpinning the genetic inheritance of this disease in other animals as well as in humans.
OSTEOCHONDROSIS (OC) is a term used to describe a variety of joint diseases, all of which involve abnormal cartilage and/or bone development. These diseases can affect the shoulder, elbow, knee, or hock joints in animals. Degenerative osteoarthritis and necrosis will be a resulting outcome as abnormalities within the joint lead to further wear and tear and joint degeneration. Osteochondrosis is thought to have a multifactorial etiology but genetic factors that affect weight gain, growth rate, and body conformation/type are undoubtedly involved in the etiology of osteochondrosis (e.g., Kadarmideen et al. 2004). By virtue of its etiology, this disease affects all food animals such as cattle (e.g., Trostle et al. 1997), pigs (Kadarmideen et al. 2004), and deer (Audige et al. 1995) and companion animals such as dogs (e.g., Neàs et al. 1999) and horses (e.g., Barneveld and van Weeren 1999), as well as humans (e.g., Seidler et al. 2001).
Modern food production programs from pigs need to consider osteochondrosis as recent findings suggest that selection for high growth and meat yield has antagonistic/unfavorable genetic effects on susceptibility to this disease in pigs (Kadarmideen et al. 2004). However, breeding pigs for resistance to osteochondrosis would be difficult by conventional breeding programs due to low (polygenic) heritability and the high cost of data recording on the farm or station. Identification and use of quantitative trait loci (QTL) for OC disease may help in breeding for disease resistance but would need costly resource populations and QTL mapping experiments. Before such experiments, it is worthwhile to conduct a statistical analysis to test existing phenotypic data on OC disease for evidence of a segregating major gene. Segregation analysis is the most powerful statistical method to identify a major gene, using only phenotypic data and without DNA marker information (e.g., Guo and Thompson 1992; Janss et al. 1995, 1997; Thaller et al. 1996). Inbreeding and marriage loops in a typical segregation analysis using pedigreed populations make the exact computations of likelihoods or marginal densities impossible. This problem has been overcome by the development of a Bayesian method implemented via Gibbs sampling, a Monte Carlo Markov chain (MCMC) methodology (Guo and Thompson 1992), and the application of this methodology to livestock populations (Janss et al. 1997; Hagger et al. 2004; Ilahi and Kadarmideen 2004) as well as to companion animals (Cargill et al. 2004).
No investigations have been carried out so far to determine the mode of inheritance of osteochondrosis in animals (in particular, pigs) by a complex segregation analysis. In addition, the development and application of a “liability or threshold model,” in the context of segregation analysis of polygenic diseases in complex animal pedigrees with MCMC methods, have not been explored extensively.
The main objective of this study was to investigate whether inheritance of osteochondral diseases in pigs is determined as a mixed inheritance (of polygenes and a major gene) by the mixed inheritance threshold or liability models (MITM). The emphasis is on the development of the MITM method and investigation of its variants for segregation analysis of polygenic diseases observed as binary traits.
MATERIALS AND METHODS
Osteochondral disease:
The company SUISAG provides herd book, field, and station tests and artificial insemination and conducts a national pig-breeding program in Switzerland. Detailed descriptions of breeding program and station-tested traits in SUISAG were given by Kadarmideen et al. (2004). Trained personnel in SUISAG conducted pathological-anatomical examination of front and hind leg bones of slaughtered pigs and recorded OC lesions with a score of 1 equaling “normal” and 4 or 5 or 6 equaling “severely affected,” depending on the lesion. Figure 1 (from Kadarmideen et al. 2004) shows positions on the bones on which different lesions are scored: head of humerus (HK), condylus medialis humeri (CMH), condylus lateralis humeri (CLH), radius and ulna proximal (RUP), distal epiphyseal cartilage of ulna (DEU), head of femur (FK), and condylus medialis femoris (CMF). More details and description of OC lesions were given in Kadarmideen et al. (2004). Disease data were available on 1163 station-tested pigs born to 194 boars and 697 dams. Data could be characterized by 23 year-months of testing, 59 stable periods, four breeds (Swiss Large White, Swiss Large White-sire line, Swiss Landrace, and Duroc), and two sexes (male and female). As seen in Figure 2, 70–98% of observations of all OC lesions (except DEU) had a score of 1 (normal). For this reason and to investigate the threshold model segregation analysis of binary data, all lesions (except DEU) were recoded such that “healthy” animals with a score of 1 received 0 and “diseased” animals with a score of 2 and above received a score of 1, creating a 0 or 1 binary trait. Pedigrees were traced as far back as possible, which included 2891 animals. The description of the data used for segregation analyses is given in Table 1.
Figure 1.

Illustration of positions of different osteochondral lesions found on bones in pigs. Lesions are scored from 1 to 6 with score 1 showing normal and scores 2–6 showing from mild to severe lesions. HK, head of humerus (1–4); CMH, condylus medialis humeri (1–4); CLH, condylus lateralis humeri (1–4); RUP, radius and ulna proximal (1–4); DEU, distal epiphyseal cartilage of ulna (1–6); FK, head of femur (1–4); CMF, condylus medialis femoris (1–6) (from Kadarmideen et al. 2004).
Figure 2.

Distribution of nine osteochondral (OC) lesions scored on 1163 station-tested pigs.
TABLE 1.
Descriptive statistics for OC lesions from 1163 station-tested pigs
| Traits | Units | Abbreviations of traits | Mean | SD | Min | Max | Incidencea |
|---|---|---|---|---|---|---|---|
| Weight at end of test | kg | WET | 103.53 | 3.50 | 90.41 | 115.21 | — |
| Age at end of test | Days | Age | 168 | 12 | 136 | 215 | — |
| Head of humerusa | 1–4 | HK | 1.02 | 0.15 | 1 | 3 | 1.4 |
| Condylus medialis humeria | 1–4 | CMH | 1.11 | 0.37 | 1 | 3 | 10.0 |
| Condylus lateralis humeria | 1–4 | CLH | 1.02 | 0.14 | 1 | 3 | 2.0 |
| Radius and ulna proximala | 1–4 | RUP | 1.04 | 0.20 | 1 | 3 | 4.0 |
| Distal epiphyseal cartilage of ulna | 1–6 | DEU | 2.30 | 0.92 | 1 | 5 | — |
| Condylus medialis femorisa | 1–6 | CMF | 1.33 | 0.52 | 1 | 4 | 31.0 |
| Total front leg | 1–6 | TFL | 6.49 | 1.04 | 5 | 10 | — |
| Total hind leg | 1–4 | THL | 1.33 | 0.52 | 1 | 4 | — |
OC lesions were transformed to two categories (binary data) as healthy (original score =1) and diseased (original score >1); “incidence” was calculated as percentage of diseased pigs.
Two more “new” traits were created: all OC lesions within front legs were summed to give “total front leg” lesions (TFL) as TFL = CMH + CLH + RUP + HK + DEU and those within hind legs were summed to give “total hind leg” lesions (THL) as THL = CMF + FK scores. This summing of scores was performed on the basis of the assumption that all OC lesions within the front or the hind leg bones were likely controlled by similar genes. The TFL resulted in six categories with 175, 483, 318, 135, 48, and 4 animals in categories 1–6, respectively. Since the incidence of FK was nearly zero (Figure 2; only 2 animals were affected with a score of 2), THL was basically equal to CMF, except that there was an increase in the number of animals in category 1 of 1289 and in category 2 of 2 animals. The distribution of animals within each category of TFL and THL is also given in Figure 2.
MITM:
A genetic model describing polygenetic and monogenetic factors for the inheritance of the osteochondral disease (so-called mixed-inheritance model) was used to detect the presence of a major gene in the inheritance of OC in pigs.
Threshold-liability models for analysis of binary data were first proposed by Wright (1934) and were later adapted for the analysis of a mixed inheritance by Morton and MacLean (1974) and to QTL detection and mapping in backcross and F2 populations (e.g., Rebai 1997) and in outbred populations on a within-family basis (Kadarmideen et al. 2000a; Kadarmideen and Dekkers 2001). These models assume the presence of an underlying continuous random variable, called liability, λ = {λi}. Under the liability scale, the MITM model has the form
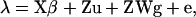 |
(1) |
where λN×1 is the vector of liability to OC lesions and β is a vector of nongenetic fixed effects that included: stable period, which is a time period nested within stable in the testing station; year and month of performance test; breed, consisting of Swiss Large White, Swiss Landrace, Swiss Large White sire line, and Duroc breed; Sex, being a male and female; and weight at end of test as a covariate. Polygenetic effects are modeled by u, while major gene effects are modeled by Wg, composed of a matrix W assigning one of three genotypes to each individual and a vector g to parameterize the genetic effects of the major gene (see details below). The X and Z are incidence matrices, connecting the unknowns in β, u, and Wg to the observations. Finally, e is a vector of random residuals for λ.
The “liability,” λ in model (1) is such that the observed binary (disease) responses (y = {yi}) are the result of the relationship
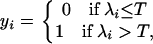 |
(2) |
where T is a fixed threshold. Here, yi = 0 denotes a healthy animal and yi = 1 denotes a diseased animal, depending on whether their liability, λi, exceeded the “threshold point,” T, for manifestation of the disease.
Note that λi's are not observed; however, they are used to simplify the likelihood and the construction of the MCMC. Often, liabilities are assumed to be normally distributed with mean vector μ and covariance matrix 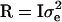 . Further, as a consequence of the well-known identification problem, the threshold and the dispersion parameter can be set equal to some arbitrary values (here, T = 0 and
. Further, as a consequence of the well-known identification problem, the threshold and the dispersion parameter can be set equal to some arbitrary values (here, T = 0 and 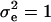 ).
).
Assumptions:
Prior distribution for the “fixed” effects β was N(0, 10). This normal distribution with relatively large variance (residual variance is set to 1) will apply a mild shrinkage to these fixed effects to avoid unidentifiability and poor convergence of their solutions on the liability scale. Distributional assumptions for polygenic effects were 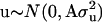 , where A is the numerator relationship matrix accounting for the additive genetic relationship between individuals with data and their parental generations (Henderson 1984) and where, in our standard implementation of the MITM, the prior distribution for polygenic variance
, where A is the numerator relationship matrix accounting for the additive genetic relationship between individuals with data and their parental generations (Henderson 1984) and where, in our standard implementation of the MITM, the prior distribution for polygenic variance 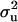 was a uniform on [0, 2].
was a uniform on [0, 2].
The major gene was modeled as an autosomal biallelic (alleles A and B) locus, initially, with Mendelian transmission probabilities. The allele A is defined to decrease the phenotypic value (lower the OC scores, i.e., associated with healthy phenotypes), and the allele B is defined to increase the phenotypic value (increase the OC scores, i.e., associated with diseased phenotypes). The prior distribution for genotypes can be specified as founder genotypes to have prior probabilities of (f−)2, 2f−f+, and (f+)2 for genotypes AA, AB/BA, and BB, respectively, and as nonfounder genotypes to have prior probabilities conditional on parental genotypes given Mendelian transmission. Hence, this parameterization assumes a single population and Hardy-Weinberg equilibrium for prior probabilities in founders, although actually multiple subpopulations were present within the founders. However, this prior distribution in founders generally has only a small influence, being largely overruled by the data, which is also reflected by a generally low accuracy to estimate founder allele frequency; hence we consider that this parameterization is also adequate for the data being analyzed here. For founder allele frequency, only 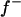 is estimated (
is estimated (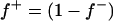 ), and a flat prior distribution on [0, 1] was assigned for
), and a flat prior distribution on [0, 1] was assigned for 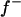 . Effects of the three genotypes AA, AB/BA, and BB were modeled as g′ = (−a, 0, a), assuming no dominance, because in our experience the estimation of dominance on a liability scale is difficult, often leading to implausible estimates with large overdominance. The prior distribution for the additive effect a was taken as N(0, 5), which, similarly as for fixed effects, applies a mild shrinkage to a major gene effect. For the model (1), the matrix W and the vector g are given as
. Effects of the three genotypes AA, AB/BA, and BB were modeled as g′ = (−a, 0, a), assuming no dominance, because in our experience the estimation of dominance on a liability scale is difficult, often leading to implausible estimates with large overdominance. The prior distribution for the additive effect a was taken as N(0, 5), which, similarly as for fixed effects, applies a mild shrinkage to a major gene effect. For the model (1), the matrix W and the vector g are given as
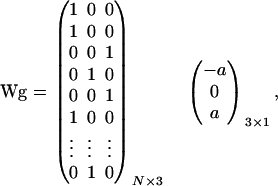 |
where W selects, with a 1 in column 1, 2, or 3, depending on whether each individual has genotype AA, AB/BA, or BB, respectively, and g specifies the effect of the genotype. Note that W and g are unknown and have to be estimated by the segregation analysis; that is, the actual genotype of the N individuals and its corresponding genotypic effects are unknowns. Note further that, in the MCMC implementation, W indeed contains 0's and 1's, not probabilities; probabilities can be obtained by averaging multiple realizations of W from the Markov chain.
The above assumed Mendelian transmission of alleles from parents to offspring can be relaxed, by modeling the prior probability of nonfounder genotypes using transmission probabilities τA|AA, τA|AB, and τA|BB, which represent the probabilities of receiving an A allele from a parent with genotype AA, AB (and BA), and BB, respectively (Elston and Stewart 1971; Lynch and Walsh 1998, pp. 366–369). The Mendelian case is expressed as τA|AA = 1, τA|AB = 0.5, and τA|BB = 0. A general expression for the transmission probability of a progeny genotype given parental genotypes then is
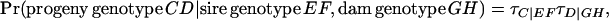 |
(3) |
where C, D, E, F, G, and H represent any of the two alleles A or B. The above general expression distinguishes between the two heterozygotes, and terms τB|.. arising are 1 − τA|... By estimation of the transmission probabilities, extra safeguards against detection of false-positive major genes can be made: as suggested by Elston and Stewart (1971) a robust indication for the segregation of a major gene is obtained only when estimated transmission probabilities do not deviate significantly from Mendelian, but do differ significantly from equal transmission probabilities, in conjunction with overall significance of the major gene component in the model. Estimation of transmission probabilities was done by fixing obtained major gene parameters and polygenic variance to estimated values, allowing the estimation of transmission probabilities to indicate whether the inferred major gene might have been caused by other sources.
Variants of MITM:
The informative prior model:
Polygenic variance of OC lesions under threshold models previously estimated with a pure polygenic model by restricted maximum likelihood and a generalized linear mixed model (REML/GLMM) methodology by Kadarmideen et al. (2004) was used as a prior for 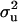 [the informative prior model (MITM-IP)]. This can be seen as a conservative approach to inference for a mixed inheritance, because this prior suggests that all genetic variance should be polygenic. In conjunction with the prior for
[the informative prior model (MITM-IP)]. This can be seen as a conservative approach to inference for a mixed inheritance, because this prior suggests that all genetic variance should be polygenic. In conjunction with the prior for 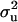 a weighting factor is also specified; with higher weights attached to this prior for
a weighting factor is also specified; with higher weights attached to this prior for 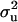 , the sample (and the marginal posterior distribution) is kept closer to the prior value for
, the sample (and the marginal posterior distribution) is kept closer to the prior value for 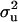 , but with low weights it is allowed to still deviate considerably.
, but with low weights it is allowed to still deviate considerably.
The reduced polygenic model:
A “reduced polygenic model” (MITM-RM) was used instead of an “individual polygenic model,” estimating only polygenic effects of parents in the form of “transmitting abilities” (TA), or half breeding values. This reduced-model approach was shown to be particularly useful for threshold models (as shown in Janss and Bolder 2000). In model (1), u is then replaced by v, the subset of u that pertains to parents, and the design matrix is altered so that the record of an individual i is linked to the TAs of the two parents of i. This model is equivalent to the previous model as long as parents do not have records, but would become approximate otherwise. Relationships between parents were taken into account, by taking the prior for v ∼ N(0, Av 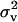 ), where Av is the numerator relationship matrix specifying the relationships between parents in the analysis. The variance component for TAs expresses one-quarter of polygenic variance, and with every record modeled with two TAs, the total variance fitted is
), where Av is the numerator relationship matrix specifying the relationships between parents in the analysis. The variance component for TAs expresses one-quarter of polygenic variance, and with every record modeled with two TAs, the total variance fitted is 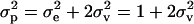 . In this model an unbounded flat prior for
. In this model an unbounded flat prior for 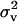 was used.
was used.
One representative lesion each from front and hind leg bones that had reasonable incidence was chosen. These were CMH and CMF (10 and 31%, respectively) and the above model variants were applied only to these lesions to be able to draw reasonable conclusions.
The Gibbs sampler:
Gibbs sampling (Geman and Geman 1984) was used to obtain marginal posterior distributions of model parameters. The Gibbs sampling strategy with blocking of parents and progeny developed by Janss et al. (1995) was used. For liability-based genetic analysis of polygenic diseases in livestock, Bayesian methods have been used by, e.g., Kadarmideen et al. (2001). The MaGGic 4.1. software package, developed by Janss (1998), was used to derive marginal posterior distributions of model parameters in (1) for each OC lesion. OC lesion FK was dropped in the final analysis; FK had only 0.1% incidence and zero variance. The addition of FK to CMF (THL) did not make any difference for the reason mentioned above and hence THL was not analyzed. The generation of samples in the MCMC scheme proceeded as follows.
Liabilities:
The liabilities λi are resampled every MCMC cycle according to Equation 1 by resampling the error term, conditioning on current values of all model parameters β, u, W, and g. According to the model definition, errors come from a N(0, 1) distribution. A straightforward rejection sampler was implemented to sample from a truncated normal such that the conditions from Equation 2 are met; i.e., for yi = 0, errors are sampled until λi ≤ T and for yi = 1, errors are sampled until λi > T. Conditional on sampled liabilities, estimation of all other model parameters can proceed as if continuous normal data are available. For a general linear model with normal errors this has been described by Wang et al. (1994) and for a mixed-inheritance model with normal errors by Janss et al. (1995).
Fixed effects:
Fixed effects are handled per effect, separating β into β1, … , β5 with corresponding incidence matrices (or covariate vector) X1, … , X5 for the mentioned effects of stable, year-month, breed, sex, and weight. Then, using corrected liability data 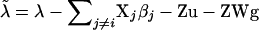 , new βi are sampled from a normal distribution with mean
, new βi are sampled from a normal distribution with mean 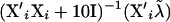 and variance
and variance 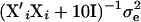 , where the 10I originates from the normal “shrinkage” prior used for fixed effects, and here
, where the 10I originates from the normal “shrinkage” prior used for fixed effects, and here 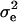 = 1. Because
= 1. Because 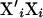 has a simple diagonal structure, the sampling can be simplified to scalar sampling of each element of
has a simple diagonal structure, the sampling can be simplified to scalar sampling of each element of 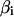 , leading to the sampling given by Wang et al. (1994).
, leading to the sampling given by Wang et al. (1994).
Polygenic effects:
Polygenic effects are equally sampled from normal distributions (Wang et al. 1994), using corrected liability data as 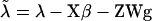 and mean and variance of u as
and mean and variance of u as 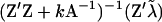 and
and 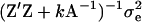 , respectively, where
, respectively, where 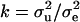 , and
, and 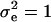 . The elements of u are sampled scalar-wise, correcting for each ui the off-diagonals arising from A−1, which involves only correction terms from parents, progeny, and mates. A scalar equation for ui is given in Janss et al. (1995) and also follows from Wang et al. (1994). For the model with reduced parameterization of polygenic effects (MITM-RM) sampling follows analogously, with the principal difference being the differently defined incidence matrix Z.
. The elements of u are sampled scalar-wise, correcting for each ui the off-diagonals arising from A−1, which involves only correction terms from parents, progeny, and mates. A scalar equation for ui is given in Janss et al. (1995) and also follows from Wang et al. (1994). For the model with reduced parameterization of polygenic effects (MITM-RM) sampling follows analogously, with the principal difference being the differently defined incidence matrix Z.
Genotypes:
For genotype sampling, the blocking strategy described by Janss et al. (1995) was used to avoid poor mixing due to “lock-up” in large families. The most general equation is the equation to compute probabilities for the genotype wi for a sire i,
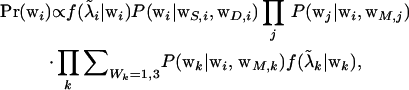 |
(4) |
where 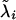 are corrected liabilities; in this step
are corrected liabilities; in this step 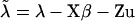 , the first term is the likelihood or penetrance of the corrected liability of individual i with normal kernel
, the first term is the likelihood or penetrance of the corrected liability of individual i with normal kernel 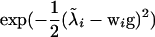 , the second term is the transmission probability for the genotype of individual i conditional on its parental genotypes, the third term is a product over all progeny j of i that are also parents themselves (nonfinal progeny of i), computing the product of transmission probabilities for progeny genotypes wj conditional on wi and the genotype of the other parent of j, mate of i, and the last term is a similar product over all progeny k of i but for progeny that are not parents themselves (final progeny of i), making a weighted sum of the transmission probabilities for progeny genotypes wk conditional on wi and the genotype of the other parent of k, mate of i, with the weight being the likelihood of the corrected liability of individual k (see above). The last term arises from the blocked sampling strategy to jointly sample sires and final progeny, giving as a first step this reduced conditional for the sire genotype probabilities; after the sire genotype has been sampled, the blocked sampling proceeds by sampling all genotypes of final progeny. For dams the above equation is simplified by considering all progeny within the third term, for final progeny the above equation reduces to the first two terms only, and for founder individuals the second term is replaced by the genotype probabilities in founders
, the second term is the transmission probability for the genotype of individual i conditional on its parental genotypes, the third term is a product over all progeny j of i that are also parents themselves (nonfinal progeny of i), computing the product of transmission probabilities for progeny genotypes wj conditional on wi and the genotype of the other parent of j, mate of i, and the last term is a similar product over all progeny k of i but for progeny that are not parents themselves (final progeny of i), making a weighted sum of the transmission probabilities for progeny genotypes wk conditional on wi and the genotype of the other parent of k, mate of i, with the weight being the likelihood of the corrected liability of individual k (see above). The last term arises from the blocked sampling strategy to jointly sample sires and final progeny, giving as a first step this reduced conditional for the sire genotype probabilities; after the sire genotype has been sampled, the blocked sampling proceeds by sampling all genotypes of final progeny. For dams the above equation is simplified by considering all progeny within the third term, for final progeny the above equation reduces to the first two terms only, and for founder individuals the second term is replaced by the genotype probabilities in founders 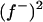 ,
, 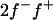 , and
, and 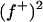 . The actual sampling of a genotype within the MCMC scheme proceeds by selecting one genotype for the individual i according to these probabilities, after normalization, by drawing a random uniform deviate.
. The actual sampling of a genotype within the MCMC scheme proceeds by selecting one genotype for the individual i according to these probabilities, after normalization, by drawing a random uniform deviate.
Polygenic variance:
Using an informative prior as in MITM-IP, 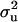 , conditional on all parameters and data, is taken to be distributed as
, conditional on all parameters and data, is taken to be distributed as 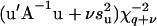 (Wang et al. 1994), where q is the number of levels in u,
(Wang et al. 1994), where q is the number of levels in u, 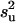 is the prior value used for
is the prior value used for 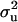 , and ν is the weight attached to that prior value, expressed in degrees of freedom. The quadratic
, and ν is the weight attached to that prior value, expressed in degrees of freedom. The quadratic 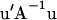 can be computed in a scalar form as exemplified in Janss et al. (1995). For a flat prior, as in our standard MITM,
can be computed in a scalar form as exemplified in Janss et al. (1995). For a flat prior, as in our standard MITM, 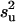 = 0 and ν = −2 (Wang et al. 1994). To implement a bounded flat prior on [0, 2] in MITM,
= 0 and ν = −2 (Wang et al. 1994). To implement a bounded flat prior on [0, 2] in MITM, 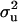 was sampled until a sample was obtained in the required interval.
was sampled until a sample was obtained in the required interval.
Additive allele effect:
Conditional on 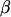 , u, and W the model for liabilities can be rewritten as
, u, and W the model for liabilities can be rewritten as 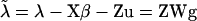 , which shows a structure for solving and sampling of g similar to that for fixed effects, considering ZW now as a composite known design matrix. Hence, g is also normally distributed with mean
, which shows a structure for solving and sampling of g similar to that for fixed effects, considering ZW now as a composite known design matrix. Hence, g is also normally distributed with mean 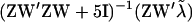 and variance
and variance 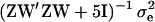 , where the 5I originates from the normal shrinkage prior used for the allele effect, and here
, where the 5I originates from the normal shrinkage prior used for the allele effect, and here 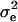 = 1. The allele effect was constrained to be positive so that the AA genotype remained identifiable as the genotype to lower the liabilities, by rejecting and repeating the sampling in case a negative allele effect was sampled.
= 1. The allele effect was constrained to be positive so that the AA genotype remained identifiable as the genotype to lower the liabilities, by rejecting and repeating the sampling in case a negative allele effect was sampled.
Founder allele frequency:
These frequencies were sampled from a beta distribution on the basis of counts nA and nB of alleles A and B in the founder individuals using 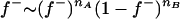 . An acceptance-rejection technique was used to sample a new
. An acceptance-rejection technique was used to sample a new 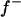 from this beta distribution as described in Janss et al. (1995), computing the required terms on a log scale.
from this beta distribution as described in Janss et al. (1995), computing the required terms on a log scale.
Transmission probabilities:
These were similarly sampled from beta distributions, on the basis of counts of allele transmissions from parents to progeny. Because our basic parameterization of genotypes did not distinguish between the two heterozygotes, in a first step this distinction was made for all heterozygotes in the pedigree; e.g., a heterozygote from a BB sire and an AA dam was marked to be “BA” to indicate that it inherited the B allele from its sire and the A allele from its dam. When both heterozygotes were possible, one of the two heterozygotes was selected randomly. Subsequently, starting with the transmission probability τA|AA, the number of A alleles nA and the number of B alleles nB to be transmitted from an AA parent were counted, after which τA|AA was sampled using 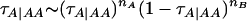 with a technique similar to that for sampling of allele frequency. The same counts and sampling were subsequently performed for the other transmission probabilities. The transmission probabilities were constrained so that τA|AA > τA|AB > τA|BB to prevent the Markov chain from switching to insensible mirror configurations where A alleles become predominantly transmitted by BB genotypes. This constraint was implemented by repeating the sampling of each transmission probability until the condition was met.
with a technique similar to that for sampling of allele frequency. The same counts and sampling were subsequently performed for the other transmission probabilities. The transmission probabilities were constrained so that τA|AA > τA|AB > τA|BB to prevent the Markov chain from switching to insensible mirror configurations where A alleles become predominantly transmitted by BB genotypes. This constraint was implemented by repeating the sampling of each transmission probability until the condition was met.
MCMC, post-MCMC analyses, and inferences:
For each OC lesion, three replicated Markov chains of 50,000 cycles were run. A spacing of 50 cycles was used to obtain 1000 samples per chain and 3000 samples in total for each lesion. A burn-in period of 1000 cycles was used to allow the Markov chains to reach the equilibrium. The following parameters were saved from the MCMC cycles: polygenic variance component 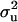 (or
(or 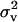 for MITM-RM), additive effects at the major gene a, and allele frequency
for MITM-RM), additive effects at the major gene a, and allele frequency 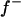 . The following functions of model parameters were derived from each Gibbs cycle to also allow inference on these parameters:
. The following functions of model parameters were derived from each Gibbs cycle to also allow inference on these parameters:
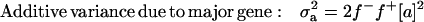 |
 |
 |
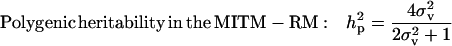 |
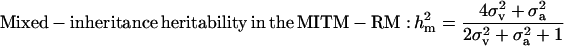 |
For the post-Gibbs analysis of the samples, an analysis of variance was used to check for equality of multiple chains. This test also yields information about the effective number of samples by comparing within-chain and between-chain variances (Janss et al. 1997).
The following summary statistics for model parameters were made on the basis of the samples: the marginal posterior means were used as point estimators; marginal posterior standard deviations were computed as measures of inaccuracy; nonparametric density estimates were constructed using the average shifted histogram method of Scott (1992); and on the basis of the latter, highest posterior density regions (HPDR) were determined. The HPDR is constructed in such a way that it includes the part (1 − α) of the probability mass about the smallest possible region of sampled parameter values (Box and Tiao 1973). The HPDR (as opposed to simple quantiles) can include the boundary of a parameter space, e.g., zero in the case of a variance component.
Hypothesis testing was based on HPDRs, using 1 − α = 0.95 and denoted as HPDR95%. For testing significance of the major gene component in the mixed inheritance, it was determined whether the HPDR95% of 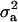 (major gene variance) excluded zero. Testing whether transmission probabilities were not significant from Mendelian was done by determining whether the HPDR95% of τA|AA, τA|AB, and τA|BB included the Mendelian values of 1, 0.5, and 0, respectively. Testing whether these transmission probabilities were significantly different from an equal transmission model was done by testing whether the HPDR95% of these parameters did not overlap.
(major gene variance) excluded zero. Testing whether transmission probabilities were not significant from Mendelian was done by determining whether the HPDR95% of τA|AA, τA|AB, and τA|BB included the Mendelian values of 1, 0.5, and 0, respectively. Testing whether these transmission probabilities were significantly different from an equal transmission model was done by testing whether the HPDR95% of these parameters did not overlap.
Mixed-inheritance linear model:
The mixed-inheritance linear model (MILM) was applied to DEU, which was the only OC lesion, with many observations in different categories; lesions were distributed into all five of the six possible classes (Figure 2). The MILM was also used to analyze the newly created trait, TFL. The statistical model was basically the same as that in Equation 1 except that the MILM was applied to the original observed data (y) instead of to liability (λ). MILM was also applied to some OC lesions to compare it with regular MITM (without its variants, MITM-IP or MITM-RM).
RESULTS
MITM:
The results of segregation analyses of osteochondrosis (CMH, CLH, RUP, CMF, and HK), using threshold models and our basic parameterization with polygenic effects for all individuals (MITM), are given in Table 2. Table 2 shows the posterior means and standard deviations of marginal distributions of all parameters. All results (except allele frequencies) from MITM relate to the underlying liability to have the disease. The existence of the major gene for any OC lesion was confirmed by magnitude and significance of genetic variance contributed by the major gene (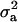 ) in the mixed model; with the use of average shifted histograms, density estimates were made to supply an HPDR95% to formally test whether these estimates were significantly different from zero at 95% probability.
) in the mixed model; with the use of average shifted histograms, density estimates were made to supply an HPDR95% to formally test whether these estimates were significantly different from zero at 95% probability.
TABLE 2.
Marginal posterior means and standard deviations of model and derived parameters for osteochondral diseases (transformed to healthy or diseased) in pigs by Bayesian mixed-inheritance threshold models modeling polygenic effects for all individuals in the pedigree and using a flat bounded prior for polygenic variance (MITM)
| CLH
|
CMH
|
RUP
|
CMF
|
HMK
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameters | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD |
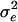 |
1.013 | 0.086 | 1.015 | 0.082 | 1.021 | 0.087 | 1.052 | 0.083 | 1.008 | 0.075 |
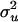 |
1.074 | 0.523 | 1.301 | 0.438 | 1.286 | 0.493 | 0.731a | 0.496 | 1.168b | 0.514 |
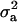 |
1.384a | 1.834 | 37.815 | 13.137 | 10.287a | 7.567 | 17.993a,b | 20.254 | 12.505 | 8.274 |
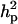 |
0.480 | 0.150 | 0.556 | 0.098 | 0.533 | 0.124 | 0.364a | 0.175 | 0.506b | 0.138 |
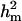 |
0.647 | 0.142 | 0.969 | 0.033 | 0.852 | 0.139 | 0.688 | 0.275 | 0.905b | 0.058 |
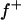 |
0.407 | 0.173 | 0.604 | 0.032 | 0.668 | 0.109 | 0.385 | 0.086 | 0.885 | 0.052 |
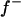 |
0.593 | 0.173 | 0.396 | 0.032 | 0.332 | 0.109 | 0.615 | 0.081 | 0.115 | 0.019 |
| a | 1.445a | 0.993 | 8.745 | 1.724 | 4.490 | 2.400 | 4.358a | 4.238 | 7.991 | 3.364 |
Results are based on 10,000 Gibbs samples from three replicated chains.
The 95% highest probability density region of this parameter (HPDR95%) included 0.0.
The 95% highest probability density region of this parameter (HPDR95%) included 2 HPDRs.
CLH:
Polygenic variance was lower than major gene variance (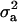 ) and
) and 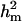 (0.65) was higher than
(0.65) was higher than 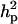 (0.48). Frequency of the positive allele that increases the genotypic value of the disease (from healthy to diseased or from score 1 to 4), f+, was (0.41) lower than that of the one that decreases it. The marginal posterior mean of additive major gene effect was 1.445. However, the major gene is not significant as the HPDR95% of its variance component (
(0.48). Frequency of the positive allele that increases the genotypic value of the disease (from healthy to diseased or from score 1 to 4), f+, was (0.41) lower than that of the one that decreases it. The marginal posterior mean of additive major gene effect was 1.445. However, the major gene is not significant as the HPDR95% of its variance component (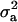 ) includes zero.
) includes zero.
CMH:
Polygenic variance was substantially lower than the additive major gene variance, resulting in higher 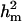 (0.96) than
(0.96) than 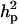 (0.55). Frequency of the disease allele, f+, was higher than that of the one that decreases it. The marginal posterior mean of additive major gene effect of 8.745 OC score, together with the high frequency of the disease allele, makes the
(0.55). Frequency of the disease allele, f+, was higher than that of the one that decreases it. The marginal posterior mean of additive major gene effect of 8.745 OC score, together with the high frequency of the disease allele, makes the 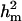 much higher than
much higher than 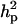 . The HPDR95% for parameters of CMH did not include zero for the variance components (including
. The HPDR95% for parameters of CMH did not include zero for the variance components (including 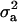 ), as well as for the corresponding derived parameters,
), as well as for the corresponding derived parameters, 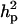 or
or 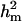 . Hence contributions of both a major gene component and additional polygenic background genes are statistically significant.
. Hence contributions of both a major gene component and additional polygenic background genes are statistically significant.
RUP:
Major gene variance (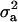 ) was substantially higher than polygenic variance resulting in
) was substantially higher than polygenic variance resulting in 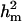 of 0.85 vs.
of 0.85 vs. 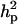 of 0.53. The frequency of the disease allele, f+, was higher than that of the one that decreases the incidence. However, the major gene component, judged by its variance, was not significant as its HPDR95% included zero; note that the allele effect for the major gene would be tested to be different from zero, but this is generally not a reliable guide for testing a major gene effect.
of 0.53. The frequency of the disease allele, f+, was higher than that of the one that decreases the incidence. However, the major gene component, judged by its variance, was not significant as its HPDR95% included zero; note that the allele effect for the major gene would be tested to be different from zero, but this is generally not a reliable guide for testing a major gene effect.
CMF:
Point estimates of 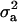 were ≫
were ≫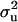 , resulting in
, resulting in 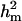 of 0.69 and
of 0.69 and 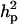 of 0.36. The additive effect was 4.35 OC liability units. The frequency of the disease allele, f+, was (0.39) lower than that of the one that decreases the incidence (f+ = 0.62). In general, however, estimates of genetic components for CMF were rather inaccurate, showing zero to be included in the HPDR95% for both the polygenic variance and the major gene variance, as well as the derived polygenic heritability. This does not necessarily mean that there is no genetic component for CMF; rather, it appears difficult to distinguish between these two genetic components, rendering both of them inaccurate when modeled jointly.
of 0.36. The additive effect was 4.35 OC liability units. The frequency of the disease allele, f+, was (0.39) lower than that of the one that decreases the incidence (f+ = 0.62). In general, however, estimates of genetic components for CMF were rather inaccurate, showing zero to be included in the HPDR95% for both the polygenic variance and the major gene variance, as well as the derived polygenic heritability. This does not necessarily mean that there is no genetic component for CMF; rather, it appears difficult to distinguish between these two genetic components, rendering both of them inaccurate when modeled jointly.
HK:
Major gene variance (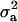 ) was substantially higher than polygenic variance, resulting in
) was substantially higher than polygenic variance, resulting in 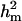 of 0.91 vs.
of 0.91 vs. 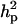 of 0.51. The frequency of the disease allele, f+, was (0.89) higher than that of the one that decreases the incidence. The HPDR95% for all parameters of HK did not include zero, which shows genetic components as well the presence of a major gene for this lesion.
of 0.51. The frequency of the disease allele, f+, was (0.89) higher than that of the one that decreases the incidence. The HPDR95% for all parameters of HK did not include zero, which shows genetic components as well the presence of a major gene for this lesion.
The MITM-RM:
With the MITM-RM, two OC lesions (CMH and CMF) were reanalyzed. Results are given in Table 3 and are compared with those from standard MITM without reduced polygenic parameterization (in Table 2). For CMH, the posterior mean of 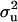 increased substantially from 1.30 to 10.89, and
increased substantially from 1.30 to 10.89, and 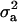 increased relatively less from 37.82 to 45.57 with a slight increase in additive effect from 8.75 to 10.21. Consequently, both
increased relatively less from 37.82 to 45.57 with a slight increase in additive effect from 8.75 to 10.21. Consequently, both 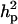 and
and 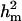 increased to 0.97 and 0.98, respectively. These results show the difficulties of estimating polygenic variance, where this reduced parameterization is considered to provide the more reliable estimates. Clear evidence for a major gene remain confirmed for CMH. For CMF, the posterior mean of
increased to 0.97 and 0.98, respectively. These results show the difficulties of estimating polygenic variance, where this reduced parameterization is considered to provide the more reliable estimates. Clear evidence for a major gene remain confirmed for CMH. For CMF, the posterior mean of 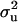 decreased to 0.56 (from 0.73) while
decreased to 0.56 (from 0.73) while 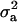 increased to 31.18 (from 17.99). There was a slight increase in additive effect from 4.36 to 6.36. Consequently, both
increased to 31.18 (from 17.99). There was a slight increase in additive effect from 4.36 to 6.36. Consequently, both 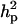 and
and 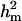 increased to 0.55 and 0.72, respectively. However, both genetic components remained to include zero in their HPDR95% and therefore indicated no significant contribution from either component. Hence, also with the reduced polygenic parameterization it appears difficult to distinguish between the two genetic components for CMF.
increased to 0.55 and 0.72, respectively. However, both genetic components remained to include zero in their HPDR95% and therefore indicated no significant contribution from either component. Hence, also with the reduced polygenic parameterization it appears difficult to distinguish between the two genetic components for CMF.
TABLE 3.
Marginal posterior means and standard deviations of model and derived parameters for osteochondral diseases in pigs by Bayesian mixed-inheritance threshold models using a reduced parameterization for polygenic effects with parental effects only (MITM-RM) or using an informative prior for polygenic variance suggesting a priori a pure polygenic inheritance (MITM-IP)
| MITM-RM
|
MITM-IP
|
|||||||
|---|---|---|---|---|---|---|---|---|
| CMH
|
CMF
|
CMH
|
CMF
|
|||||
| Parameters | Mean | SD | Mean | SD | Mean | SD | Mean | SD |
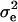 |
1.007 | 0.073 | 1.039 | 0.073 | 1.020 | 0.074 | 1.041 | 0.076 |
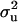 |
10.889b | 3.885 | 0.562ab | 0.516 | 0.844 | 0.426 | 0.712 | 0.453b |
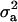 |
45.573b | 13.805 | 31.182a,b | 29.399 | 28.923 | 8.051 | 37.281 | 21.46ab |
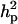 |
0.974 | 0.012 | 0.545b | 0.253 | 0.426 | 0.122 | 0.371 | 0.146 |
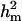 |
0.986b | 0.005 | 0.715b | 0.312 | 0.962 | 0.034 | 0.852 | 0.253 |
| f+ | 0.7192 | 0.026 | 0.603 | 0.103 | 0.621b | 0.055 | 0.674 | 0.057 |
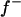 |
0.281 | 0.026 | 0.397 | 0.103 | 0.379b | 0.055 | 0.326 | 0.057 |
| a | 10.217b | 1.692 | 6.358ab | 5.252 | 7.813 | 1.250 | 8.244 | 4.111ab |
Results are based on 10,000 Gibbs samples from three replicated chains.
The 95% highest probability density region of this parameter (HPDR95%) included 0.0.
The 95% highest probability density region of this parameter (HPDR95%) included 2 HPDRs.
The MITM-IP:
With the MITM-IP, two OC lesions (CMH and CMF) were reanalyzed. Results are given in Table 3 and are compared with those from standard MITM without informative prior for polygenic variance (in Table 2).
For CMH, the posterior mean of 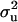 decreased from 1.30 to 0.84 and
decreased from 1.30 to 0.84 and 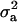 also decreased from 37.81 to 28.92. The
also decreased from 37.81 to 28.92. The 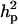 and
and 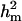 decreased from 0.56 to 0.43 and from 0.97 to 0.96, respectively. There was also a slight decrease in additive effect. Overall, the prior for polygenic variance therefore had a tempering effect on all genetic components, but did not affect the general conclusion about presence of a major gene, whose variance component remained significant.
decreased from 0.56 to 0.43 and from 0.97 to 0.96, respectively. There was also a slight decrease in additive effect. Overall, the prior for polygenic variance therefore had a tempering effect on all genetic components, but did not affect the general conclusion about presence of a major gene, whose variance component remained significant.
For CMF, the posterior mean of 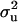 did not change much but
did not change much but 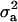 more than doubled (from 17.99 to 37.28), becoming comparable to the estimate from MITM-RM (31.18). There was also a twofold increase in additive effect (from 4.35 to 8.24). Consequently, the
more than doubled (from 17.99 to 37.28), becoming comparable to the estimate from MITM-RM (31.18). There was also a twofold increase in additive effect (from 4.35 to 8.24). Consequently, the 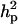 did not change much but
did not change much but 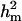 increased from 0.69 to 0.85. Now, accuracy of polygenic variance was improved, although this cannot be taken as an appropriate measure of accuracy because it includes prior information. Most importantly, accuracy of major gene variance remained low and major gene variance remained not significantly different from zero.
increased from 0.69 to 0.85. Now, accuracy of polygenic variance was improved, although this cannot be taken as an appropriate measure of accuracy because it includes prior information. Most importantly, accuracy of major gene variance remained low and major gene variance remained not significantly different from zero.
Tests for Mendelian segregation:
Representative OC lesions, one each from front and hind leg bones that had reasonable incidence, were chosen (as per Figure 1 and Table 1). These were CMH and CMF (with incidence 10 and 31%, respectively) and the additional tests for presence of a major gene were applied only to these lesions.
Mendelian transmission probabilities for CMH and CMF were estimated as follows. First, the variance components, allele affect, and founder allele frequency from the basic model (MITM) were fixed, and then the model was rerun while estimating jointly fixed effects, polygenic effects, genotypes, and transmission probabilities. Results for estimated transmission probabilities are in Table 4. For CMH, Mendelian transmission probabilities (1, 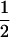 , or 0) were not rejected, while the test for equal transmission probabilities was rejected, as confirmed by HPDR95% (Table 4). This confirms evidence of a segregating Mendelian major gene for this OC lesion.
, or 0) were not rejected, while the test for equal transmission probabilities was rejected, as confirmed by HPDR95% (Table 4). This confirms evidence of a segregating Mendelian major gene for this OC lesion.
TABLE 4.
Estimated Mendelian transmission probabilities, expressed as the probability of receiving an A allele from a parent with genotype AA, AB, or BB, for osteochondral diseases CMF and CMH in pigs by Bayesian mixed-inheritance threshold models (MITM)
| Transmission probability
|
CMF
|
CMH
|
||||
|---|---|---|---|---|---|---|
| Mean | SD | HPDR95% | Mean | SD | HPDR95% | |
| Prob(A|AA) | 0.890 | 0.065 | 0.988–0.761 | 0.967 | 0.025 | 1–0.922 |
| Prob(A|AB) | 0.552 | 0.063 | 0.668–0.442 | 0.538 | 0.033 | 0.600–0.475 |
| Prob(A|BB) | 0.243 | 0.110 | 0.455–0.051 | 0.028 | 0.026 | 0.081–0 |
For CMF, the test for Mendelian transmission probabilities (1, 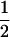 , or 0) rejected Mendelian segregation, but also the test for equal transmission probabilities was rejected as two transmission probabilities did not have overlapping HPDR95% (Table 4). This confirms evidence of some segregating genetic factor for this OC lesion, although it is likely not Mendelian.
, or 0) rejected Mendelian segregation, but also the test for equal transmission probabilities was rejected as two transmission probabilities did not have overlapping HPDR95% (Table 4). This confirms evidence of some segregating genetic factor for this OC lesion, although it is likely not Mendelian.
Posterior distributions:
For each parameter, a uni- or bimodal density was found (graphs not shown for all diseases and parameters). In all cases (models and diseases), mode was very close to the corresponding marginal posterior mean (Tables 2 and 3). In cases of bimodal densities with two peaks, two corresponding HPDR95% were available. For showing the differences between models with and without informative prior (MITM vs. MITM-IP) and between full vs. reduced polygenic models (MITM vs. MITM-RM), CMF was chosen as a representative disease as the population incidence was 31%. The densities of 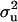 and
and 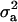 for OC lesions in DEU and TFL were compared with those in CMF, to show the impact of different phenotypic distributions on the posterior distribution of two important model parameters,
for OC lesions in DEU and TFL were compared with those in CMF, to show the impact of different phenotypic distributions on the posterior distribution of two important model parameters, 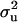 and
and 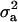 .
.
The posterior densities of 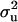 and
and 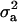 for CMF from MITM and its two variants MITM-IP and MITM-RM are plotted in Figure 3a through Figure 4c. Analyses of liability to CMF by MITM produced asymmetric distributions (bimodal), showing possibly the poor mixing of the Gibbs chains and/or convergence. However, comparing this with segregation analysis with the informative prior for
for CMF from MITM and its two variants MITM-IP and MITM-RM are plotted in Figure 3a through Figure 4c. Analyses of liability to CMF by MITM produced asymmetric distributions (bimodal), showing possibly the poor mixing of the Gibbs chains and/or convergence. However, comparing this with segregation analysis with the informative prior for 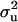 (MILM-IP), there is a marked difference. For example, distributions of
(MILM-IP), there is a marked difference. For example, distributions of 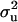 in Figure 3a vs. Figure 3b and of
in Figure 3a vs. Figure 3b and of 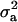 in Figure 4a vs. Figure 4b show the bimodal distribution becoming unimodel as well as more area under the curve for both
in Figure 4a vs. Figure 4b show the bimodal distribution becoming unimodel as well as more area under the curve for both 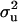 and
and 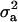 . The corresponding HPDR95% also changed from being nonsignificant to significant (including zero to not including zero) for
. The corresponding HPDR95% also changed from being nonsignificant to significant (including zero to not including zero) for 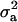 (Table 2 vs. Table 3).
(Table 2 vs. Table 3).
Figure 3.



Marginal posterior distributions of polygenic variance from the mixed-inheritance threshold models: (a) without informative prior (MITM), (b) with informative prior (MITM-IP), and (c) under the reduced polygenic model (MITM-RM) for the lesion condylus medialis femoris scored on a binary scale (CMF).
Figure 4.



Marginal posterior distributions of major gene variance from the mixed-inheritance threshold models: (a) without informative prior (MITM), (b) with informative prior (MITM-IP), and (c) under the reduced polygenic model (MITM-RM) for the lesion condylus medialis femoris scored on a binary scale (CMF).
In the case of the reduced polygenic model, MITM-RM, sampling of polygenic values or transmitting abilities was for parents only. The density distributions are given in Figure 3c for 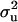 and in Figure 4c for
and in Figure 4c for 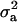 . Comparing Figure 3a and Figure 3c, the posterior mode is confirmed by MITM-RM to correspond to the first peak in Figure 3a although the density of the second peak became much smaller in MITM-RM. For
. Comparing Figure 3a and Figure 3c, the posterior mode is confirmed by MITM-RM to correspond to the first peak in Figure 3a although the density of the second peak became much smaller in MITM-RM. For 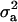 , there was not much difference between shapes of the densities but the area under the curve was larger under MITM-RM than under MITM (Figure 4a vs. Figure 4c).
, there was not much difference between shapes of the densities but the area under the curve was larger under MITM-RM than under MITM (Figure 4a vs. Figure 4c).
MILM analyses:
DEU:
With the linear model, all estimates were on the original (observed) scale. Point estimates of 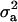 were approximately two times more than that of
were approximately two times more than that of 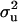 , resulting in
, resulting in 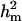 of 0.22 vs.
of 0.22 vs. 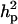 of 0.12. The additive gene effect was 0.73 OC units and dominance effect was −0.98 OC units. The HPDR95% for all parameters of DEU did not include zero, except
of 0.12. The additive gene effect was 0.73 OC units and dominance effect was −0.98 OC units. The HPDR95% for all parameters of DEU did not include zero, except 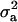 . These results show genetic components as well the presence of a major gene for this lesion.
. These results show genetic components as well the presence of a major gene for this lesion.
TFL:
With the linear model, all estimates were on the original (observed) scale. The 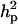 was 0.07 whereas
was 0.07 whereas 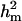 was 0.11. The major gene variance was much larger than polygenic variance. The frequency of the disease allele, f+, was (0.65) higher than that of the one that decreases the incidence, like many other OC lesions. As a result of combining different (genetically correlated) lesions in the front leg into one, the polygenic variances as well as major gene variances (
was 0.11. The major gene variance was much larger than polygenic variance. The frequency of the disease allele, f+, was (0.65) higher than that of the one that decreases the incidence, like many other OC lesions. As a result of combining different (genetically correlated) lesions in the front leg into one, the polygenic variances as well as major gene variances (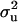 and
and 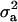 ) were much higher than those for other individual lesions in front legs (results not shown). Despite such high genetic variances, the heritabilities (both
) were much higher than those for other individual lesions in front legs (results not shown). Despite such high genetic variances, the heritabilities (both 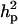 and
and 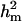 ) decreased, due to substantially larger residual variances,
) decreased, due to substantially larger residual variances, 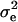 , relative to that of other lesions in the front leg. The impact of large residual variance was such that most of the model parameters were nonsignificant and inaccurate; the HPDR95% included zero for all but
, relative to that of other lesions in the front leg. The impact of large residual variance was such that most of the model parameters were nonsignificant and inaccurate; the HPDR95% included zero for all but 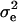 .
.
DISCUSSION
The results reported here clearly showed familial transmission and evidence for a segregating major gene for an important disease, osteochondrosis. Results for analysis of the CMH showed significant major gene variance in a mixed-inheritance model and passed the tests for Mendelian transmission. This is the first investigation to report the results from segregation or mixed-inheritance model analysis of osteochondrosis in pigs or in any other animal species. Hence none of the results from our study could be compared with the literature, except for polygenic parameters for osteochondrosis (e.g., Kadarmideen et al. 2004). In addition, only a few studies have investigated complex segregation analysis for ordinal or binary data (e.g., Thaller et al. 1996; Cargill et al. 2004) but this is the first study to extensively investigate a liability or threshold model method for identifying major genes for binary diseases in complex pedigrees with inbreeding and marriage loops. The basic theory and algorithms developed in this study could also be used in humans and companion animals (e.g., horses, cats, and dogs) where (poly- or oligogenic) diseases/disorders are observed in categorical form. While reporting evidence for major genes for osteochondrosis, this study also investigated and compared different approaches that can be adopted in the segregation analysis of categorical disease data, using threshold or liability models.
The magnitude and significance of genetic variance at the major gene (based on HPDR95%) was used to confirm the presence of a segregating major gene. This criterion is appropriate because it collectively represents the magnitude and significance of individual major gene parameters of the model and has been used by several studies (e.g., Janss et al. 1997; Ilahi and Kadarmideen 2004). General transmission models (Elston and Stewart 1971; Lynch and Walsh 1998, pp. 366–369) are usually not considered in animal populations, arguing that there are usually no nongenetic relationships or environmental resemblances between parents and progeny and that families are dispersed and linked through multiple marriages. Nevertheless, we have also fitted the general transmission model to further substantiate the presence of a Mendelian segregation. For CMH, results showed that the mixed-inheritance models gave a significant fit (as confirmed by HPDR of the major gene component) and that a model of equal transmission was rejected along with a failure to reject Mendelian transmission probabilities (Table 4). Thus, for CMH, all classical tests for providing evidence of a segregating major gene are passed. Results of the general transmission model for CMF were less clear, but showed a potential other use of this general model. Here, Mendelian segregation was rejected, but also an equal transmission model was rejected, concluding that something genetic, but not Mendelian, might be present, which may point to, e.g., an imprinted, X-linked, or digenic (epistatic) model.
When binary data have very low incidences, fitting several cross-classified “fixed” effects in a model may cause convergence problems and produce illogical estimates (e.g., due to all healthy or diseased phenotypes in a given class). This problem has been encountered in the genetic analysis of binary data (e.g., Kadarmideen et al. 2000b, 2001). We solved this problem by using shrinkage estimators for fixed effects. Apart from fixed effects, polygenic effects with an individual polygenic model parameterization also cause problems in the discrete data analyses with linear or threshold models (as shown by Kadarmideen et al. 2000b, 2004). One alternative is to fit a “sire” model but in MITM analysis, the genotype probabilities would still have to be estimated at an individual level. In an alternative parameterization we considered modeling only polygenic values of the parents. With this approach, the estimation of polygenic variance was similar to that used in the reduced polygenic model. A few OC lesions were reanalyzed by this reduced polygenic model with no restrictions on polygenic variances and other model parameters; however, this model still resulted in a much larger posterior mean for polygenic variances (Table 3 and Figure 3c). This result shows the impact of very small family sizes in the data (with the larger families, this approximation usually works well). In principle, the reduced parameterization can also be developed for genotypes, but this model is less straightforward to develop, because a heterogeneity of variance within families, dependent on parental genotypes, would result. We retained the full parameterization with individual genotypes and also applied a shrinkage estimator for the allele effect of the major gene, which appeared to work well.
The other approach investigated was the use of an informative prior for polygenic variance and the study of its impact on model parameters and their significance. Results (Table 3 and Figures 3b and 4b) showed significant improvement in point estimates and distributional properties of polygenic variance as well as major gene parameters. This shows the advantage of Bayesian methods over likelihood-based methods in segregation analysis with respect to the ability to include and update an informative prior.
The DEU lesion was the only lesion with many observations in different categories; lesions were distributed into all five of six possible classes (Figure 2). Hence, this trait by definition should mimic a continuous trait and give more reliable estimates/information than those traits with fewer categories and/or skewed distributions (e.g., binary traits with very high/low incidence). For this reason, this trait was analyzed by the MILM. It has been shown that there is a loss of information from continuous or ordinal data to binary data and it severely reduces power and accuracy in QTL mapping (Rebai 1997; Kadarmideen et al. 2000a). The distributional properties or advantages of DEU were reflected in good mixing of chains, convergence, and hence more reliable parameter (point) estimates (as seen in posterior distributions in Figure 5, a and b) than those traits with extreme distributions.
Figure 5.


Marginal posterior distributions of polygenic variance (a) and major gene variance (b) from the mixed-inheritance linear model (MILM) for the lesion, distal epiphyseal cartilage of ulna (DEU).
The TFL was defined as the sum of all OC lesions in front legs, which was meant to treat all the OC lesions in the front leg as one OC lesion; this also resulted in a trait that created more categories with a sufficient number of records to be analyzed by the MILM (Figure 6). The “clustering” of front leg lesions was based on our earlier findings (Kadarmideen et al. 2004) that they were genetically correlated traits (genetic correlation ranging from 0.57 to 0.69). The results based on the TFL lesion should be more reliable than those based on individual lesions, as they created six categories instead of only four (except for DEU) as well as ensured a reasonable number of animals in each category (Figure 2). The variances due to polygene and major gene did increase (as expected) but simultaneously there was a drastic increase in residual variance also, as there were very high environmental/error correlations among front leg lesions. In general, this approach shows how one can perform clustering of genetically related diseases in segregation analysis, when individual diseases have very low incidences.
Figure 6.


Marginal posterior distributions of polygenic variance (a) and major gene variance (b) from the mixed-inheritance linear model (MILM) for the lesion, total front leg (TFL).
We also applied MILMs to most OC lesions on the original scales (scores from 1 to 6 depending on the lesions) to make a comparison with the standard liability model (MITM) analysis of the binary form of the same OC data. Comparing results (not shown), the point estimates on the observed vs. liability scales, the major gene parameter 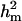 increased from 0.40 to 0.65, 0.40 to 0.96, 0.44 to 0.85, and 0.64 to 0.69 for CLH, CMH, RUP, and CMF, respectively. The frequency of the allele that increases the liability to the disease (f+) actually decreased when estimated with MITM; the f+ reduced from 0.83 to 0.41, 0.73 to 0.61, 0.80 to 0.67, and 0.47 to 0.39 for CLH, CMH, RUP, and CMF, respectively. Hence, on the discrete observed scale, major gene variances were generally higher, but must also be considered to be less reliable. The analysis of discrete traits by the linear model may have introduced some biases because it tries to match disease genotype (BB) directly to OC scores (1, 2, 3, … , etc) which may give a better fit but also causes an upward bias. The threshold model, in contrast, fits the genotypes to data on a continuous liability scale (rather than on a “one score-to-one genotype” fit in linear models). Hence, the genotypic values and frequencies are more reliable under liability models. This difference explains probably why the disease allele frequencies were overestimated by the linear model. Linear models typically are more sensitive/less robust to extreme (too low/high) incidence of diseases than threshold models (e.g., Kadarmideen et al. 2000a,b, 2001; Kadarmideen and Dekkers 2001). In this study, MILM results for CLH and RUP OC lesions (with incidence 2 and 4%, respectively) produced illogical polygenic and major gene parameter estimates. The advantages of MITM would be that gene-environment interactions could be handled more easily on the underlying scale than on the observed scale (Kadarmideen et al. 2004).
increased from 0.40 to 0.65, 0.40 to 0.96, 0.44 to 0.85, and 0.64 to 0.69 for CLH, CMH, RUP, and CMF, respectively. The frequency of the allele that increases the liability to the disease (f+) actually decreased when estimated with MITM; the f+ reduced from 0.83 to 0.41, 0.73 to 0.61, 0.80 to 0.67, and 0.47 to 0.39 for CLH, CMH, RUP, and CMF, respectively. Hence, on the discrete observed scale, major gene variances were generally higher, but must also be considered to be less reliable. The analysis of discrete traits by the linear model may have introduced some biases because it tries to match disease genotype (BB) directly to OC scores (1, 2, 3, … , etc) which may give a better fit but also causes an upward bias. The threshold model, in contrast, fits the genotypes to data on a continuous liability scale (rather than on a “one score-to-one genotype” fit in linear models). Hence, the genotypic values and frequencies are more reliable under liability models. This difference explains probably why the disease allele frequencies were overestimated by the linear model. Linear models typically are more sensitive/less robust to extreme (too low/high) incidence of diseases than threshold models (e.g., Kadarmideen et al. 2000a,b, 2001; Kadarmideen and Dekkers 2001). In this study, MILM results for CLH and RUP OC lesions (with incidence 2 and 4%, respectively) produced illogical polygenic and major gene parameter estimates. The advantages of MITM would be that gene-environment interactions could be handled more easily on the underlying scale than on the observed scale (Kadarmideen et al. 2004).
All results (except allele frequencies) from MITM were on the underlying liability (to have the disease) scale and on this scale one would expect the magnitude to be larger than that on the observed scale. These higher magnitudes of estimates from MITM (e.g., major gene heritabilities of liability to disease; Tables 2 and 3) should contribute to larger and faster genetic progress in selection for resistance/tolerance to osteochondrosis in pigs. Regardless of the method, very low incidence rates for certain lesions (as low as 1%) created problems in mixing of the chains and convergence. Similar problems were also encountered by other studies (e.g., Thaller et al. 1996). According to the central limit theorem, more power and accuracy in analysis of categorical/ordinal data could be obtained by increasing the population size and by intermediate incidences (Kadarmideen et al. 2000a, 2001). From a practical point of view, it may be financially prohibitive to collect data on diseases that require manual or anatomical-pathological examinations.
The results presented here open up a new possibility for investigating the presence of major genes in the inheritance of osteochondral lesions in other species such as humans, companion animals (e.g., horses, dogs, and cats), and other farm animals, as these species are known to have osteochondrosis and may also have similar patterns of mixed inheritance. Andersson-Eklund et al. (2000) reported very large QTL for osteochondrosis on chromosome 5 as well as on chromosomes 13 and 15. Lee et al. (2003) conducted QTL mapping in Large White × Meishan crossbred pigs and found no QTL for osteochondrosis but found a QTL for Physis, another closely related bone abnormality, in pig chromosome 7. The evidence collected from the current segregation analysis in Swiss pigs and from QTL for Physis and osteochondrosis in the United Kingdom and Swedish pig populations justifies that a resource/commercial population of pigs could be genotyped for these candidate chromosomes to fine map QTL for osteochondrosis.
Conclusion
The research conducted in this study is the first of its kind in that no study has so far investigated osteochondrosis in pigs or other animals for the evidence of a major gene by complex segregation analyses, accounting for inbreeding and marriage loops, commonly found in animal pedigrees. In the analysis, novel approaches such as Bayesian threshold mixed-inheritance models were introduced for the analysis of diseases observed as binary data, healthy or diseased, and compared with different alternative approaches as well as with linear mixed-inheritance models. All models provided significant evidence that osteochondrosis is not only under a polygenic mode of inheritance but also importantly affected by a major gene with Mendelian transmission (at least for osteochondrosis in the condylus medialis humeri). Models with an informative prior for polygenic variance and/or sampling only parental/grand-parental polygenic values resulted in higher efficiency of Gibbs sampling algorithms and corresponding accuracy of model parameters. Marginal distributions of major gene and polygene parameters in threshold models were for underlying liability of the animals to develop this disease while linear models provided the estimates for the disease episode; by virtue of the scale, the liabilities were higher in magnitude than observed scales. These results, in general, should be confirmed by molecular genome mapping for the disease loci in pig populations via linkage analysis and quantitative trait loci mapping methods. Since osteochondrosis is a health and welfare problem and has unfavorable (genetic) effects on many traits that are economically and functionally important, the evidence of a segregating major gene from this study provides an opportunity to select against this disease. The results from this study not only are useful for pig breeders but also may function as a “pig model” for underpinning genetic inheritance of this disease in other animals and in humans.
Acknowledgments
The authors thank A. Hofer and D. Schwörer from SUISAG for supplying data. This research was completed as part of an ongoing collaboration between the Federal Institute of Technology of Switzerland (ETH Zurich) and Wageningen University and Research Centre, Lelystad, The Netherlands.
References
- Andersson-Eklund, L., H. Uhlhorn, N. Lundeheim, G. Dalin and L. Andersson, 2000. Mapping quantitative trait loci for principal components of bone measurements and osteochondrosis scores in a wild boar × Large White intercross. Genet. Res. 75: 223–230. [DOI] [PubMed] [Google Scholar]
- Audige, L., P. R. Wilson, R. S. Morris and G. W. Davidson, 1995. Osteochondrosis, skeletal abnormalities and enzootic ataxia associated with copper deficiency in a farmed red deer (cervus-elaphus) herd. New Zealand Vet. J. 43: 70–76. [DOI] [PubMed] [Google Scholar]
- Barneveld, A., and R. van Weeren, 1999. Conclusions regarding the influence of exercise on the development of the equine musculoskeletal system with special reference to osteochondrosis. Equine Vet J. (Suppl.) 31: 112–119. [DOI] [PubMed] [Google Scholar]
- Box, G. E. P., and G. Tiao, 1973. Bayesian Inference in Statistical Analysis. Addison-Wesley, Reading, MA.
- Cargill, E. J., T. R. Famula, G. M. Strain and K. E. Murphy, 2004. Heritability and segregation analysis of deafness in U.S. dalmatians. Genetics 166: 1385–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston, R. C., and J. Stewart, 1971. A general model for the analysis of pedigree data. Hum. Hered. 21: 523–542. [DOI] [PubMed] [Google Scholar]
- Geman, S., and D. Geman, 1984. Stochastic relaxation, Gibbs distributions and Bayesian restoration of images. IEEE Trans. Patt. Anal. Mach. Intell. 6: 721–741. [DOI] [PubMed] [Google Scholar]
- Guo, S. W., and E. A. Thompson, 1992. Monte Carlo method for combined segregation and linkage analysis. Am. J. Hum. Genet. 51: 1111–1126. [PMC free article] [PubMed] [Google Scholar]
- Hagger, C., L. L. G. Janss, H. N. Kadarmideen and G. Stranzinger, 2004. Bayesian inference on major loci in related multi generation selection lines of laying hens. Poultry Sci. 83: 1932–1939. [DOI] [PubMed] [Google Scholar]
- Henderson, C. R., 1984. Applications of Linear Models in Animal Breeding. University of Guelph, Guelph, ON, Canada.
- Ilahi, H., and H. N. Kadarmideen, 2004. Bayesian segregation analysis of milk flow in Swiss dairy cattle using Gibbs sampling. Genet. Sel. Evol. 36: 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janss, L. L. G., 1998. MAGGIC: a package of subroutines for genetic analyses with Gibbs sampling, pp. 459–460 in Proceedings of the 6th World Congress on Genetics Applied to Livestock Production, vol. 27. Organizing Committee on 6th WCGALP, Armidale, Australia.
- Janss, L. L. G., and N. M. Bolder, 2000. Heritabilities of and genetic relationships between salmonella resistance traits in broilers. J. Anim. Sci. 78: 2287–2291. [DOI] [PubMed] [Google Scholar]
- Janss, L. L. G., R. Thompson and J. A. M. van Arendonk, 1995. Application of Gibbs sampling for inference in a mixed major gene-polygenic inheritance model in animal populations. Theor. Appl. Genet. 91: 1137–1147. [DOI] [PubMed] [Google Scholar]
- Janss, L. L. G., J. A. M. van Arendonk and E. W. Brascamp, 1997. Bayesian statistical analyses for presence of single major genes affecting meat quality traits in crossed pig population. Genetics 145: 395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadarmideen, H. N., and J. C. M. Dekkers, 2001. Generalized marker regression and interval QTL mapping methods for binary traits in half-sib family designs. J. Anim. Breed. Genet. 118: 297–309. [Google Scholar]
- Kadarmideen, H. N., L. L. G. Janss and J. C. M. Dekkers, 2000. a Power of QTL mapping for polygenic binary traits using generalized and regression interval mapping in multi-family half-sib designs. Genet. Res. 76: 305–317. [DOI] [PubMed] [Google Scholar]
- Kadarmideen, H. N., R. Thompson and G. Simm, 2000. b Linear and threshold model genetic parameters for disease, fertility and milk production in dairy cattle. Anim. Sci. 71: 411–419. [Google Scholar]
- Kadarmideen, H. N., R. Rekaya and D. Gianola, 2001. Genetic parameters for clinical mastitis in Holstein-Friesians: a Bayesian analysis. Anim. Sci. 73: 229–240. [Google Scholar]
- Kadarmideen, H. N., D. Schwörer, H. Ilahi, M. Malek and A. Hofer, 2004. Genetics of osteochondral disease and its relationship with meat quality and quantity, growth and feed conversion traits in pigs. J. Anim. Sci. 82: 3118–3127. [DOI] [PubMed] [Google Scholar]
- Lee, G. J., A. L. Archibald, G. B. Garth, A. S. Law, D. Nicholson et al., 2003. Detection of quantitative trait loci for locomotion and osteochondrosis-related traits in Large White × Meishan pigs. Anim. Sci. 76: 155–165. [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Morton, N. E., and C. J. MacLean, 1974. Analysis of family resemblance. III. Complex segregation of quantitative traits. Am. J. Hum. Genet. 26: 489–503. [PMC free article] [PubMed] [Google Scholar]
- Neàs, A., M. Dvoák and J. Zatloukal, 1999. Incidence of osteochondrosis in dogs and its late diagnosis. Acta Vet. Brno 68: 131–139. [Google Scholar]
- Rebai, A., 1997. Comparison of methods for regression interval mapping in QTL analysis with non-normal traits. Genet. Res. 69: 69–74. [Google Scholar]
- Scott, D. W., 1992. Multivariate Density Estimation: Theory, Practice, and Visualization. John Wiley & Sons, New York.
- Seidler, A. U., V. Bolm-Audor, H. Heiskel, N. Henkel, B. Roth-Küver et al., 2001. The role of cumulative physical work load in lumbar spine disease: risk factors for lumbar osteochondrosis and spondylosis associated with chronic complaints. Occup. Environ. Med. 58: 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaller, G., L. Dempfle and I. Hoeschele, 1996. Maximum-likelihood analysis of rare binary traits under different modes of inheritance. Genetics 143: 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trostle, S. S., R. G. Nicoll, L. J. Forrest and M. D. Markel, 1997. Clinical and radiographic findings, treatment, and outcome in cattle with osteochondrosis: 29 cases (1986–1996). J. Am. Vet. Med. Assoc. 211: 1566. [PubMed] [Google Scholar]
- Wang, C. S., J. J. Rutledge and D. Gianola, 1994. Bayesian analysis of mixed linear models via Gibbs sampling with an application to litter size in Iberian pigs. Genet. Sel. Evol. 26: 91–115. [Google Scholar]
- Wright, S., 1934. An analysis of variability in number of digits in an inbred strain of guinea pigs. Genetics 19: 506–536. [DOI] [PMC free article] [PubMed] [Google Scholar]