

Abstract
In Arabidopsis thaliana, significant efforts to determine the extent of genomic variation between phenotypically divergent accessions are under way, but virtually nothing is known about variation at the transcription level. We used microarrays to examine variation in transcript abundance among three inbred lines and two pairs of reciprocal F1 hybrids of the highly self-fertilizing species Arabidopsis. Composite additive genetic effects for gene expression were estimated from pairwise comparisons of the three accessions Columbia (Col), Landsberg erecta (Ler), and Cape Verde Islands (Cvi). For the pair Col and Ler, 27.0% of the 4876 genes exhibited additive genetic effects in their expression (α = 0.001) vs. 32.2 and 37.5% for Cvi with Ler and Col, respectively. Significant differential expression ranged from 32.45 down to 1.10 in fold change and typically differed by a factor of 1.56. Maternal or paternal transmission affected only a few genes, suggesting that the reciprocal effects observed in the two crosses analyzed were minimal. Dominance effects were estimated from the comparisons of hybrids with the corresponding midparent value. The percentage of genes showing dominance at the expression level in the F1 hybrids ranged from 6.4 to 21.1% (α = 0.001). Breakdown of these numbers of genes according to the magnitude of the dominance ratio revealed heterosis for expression for on average 9% of the genes. Further advances in the genetic analysis of gene expression variation may contribute to a better understanding of its role in affecting quantitative trait variation at the phenotypic level.
NOW that the complete genome sequence of Arabidopsis thaliana (L.) Heynh. is known, a fundamental next objective is to determine the function of its ∼30,000 genes (Arabidopsis Genome Initiative 2000) and to assess the extent of genotypic variation in phenotypically divergent accessions. Genotypic variation of accessions may initially express itself as naturally occurring variation of gene expression, thus constituting an important intermediate genetic determinant of phenotypic variation. Developing an understanding of the extent of nonadditivity in transcription (the degree and direction of dominance and epigenetic effects on gene expression) may provide us with insight into the extent of transcriptional interaction networks: a means to establish models of molecular mechanisms that help us to explain and predict complex genetic phenomena, such as heterosis.
Microarrays constitute a potentially powerful tool to study the genetic basis of natural variation in gene expression. Although not exhaustively covering the total biodiversity, microarray studies have revealed numerous differences in transcript abundance of even up to one-third of the genes, both within and between closely related species (Primig et al. 2000; Jin et al. 2001; Brem et al. 2002; Enard et al. 2002; Oleksiak et al. 2002; Cheung et al. 2003; Hsieh et al. 2003; Ranz et al. 2003; Schadt et al. 2003; Townsend et al. 2003; Fay et al. 2004; Gibson et al. 2004). For Arabidopsis, significant efforts are under way to determine the extent of genomic variation between phenotypically divergent accessions (http://walnut.usc.edu/2010). At present, the extent to which accessions differ in transcript abundance and the level at which these differences can be attributed to genetic effects are unknown.
Crossing of two inbred lines results in F1 progeny that is heterozygous for all loci at which the parental lines contain different alleles. The difference between the mean phenotype of the resulting F1 and the midparent value, often called hybrid vigor or heterosis, is a function of both dominance and additive × additive epistasis (Lynch and Walsh 1998). Ignoring epistasis, a simple explanation for heterosis is the presence of complementary sets of deleterious recessive genes in the two parental lines and the masking of their effects in the F1 heterozygotes. Hybrids of Arabidopsis and their respective parental lines may offer an attractive model system to investigate the genetic basis of heterosis with genomic tools. Here, we show that this model provides an ideal framework to estimate the proportion of genes expressed in a dominance fashion in a particular hybrid combination. However, the extent to which such changes are caused either by a large number of genes of relatively small effects or by only a single or a few major segregating factors cannot easily be estimated from data obtained from pairs of lines and their first-generation derivates.
Here, we started a genome-wide genetic analysis of variability in gene expression in Arabidopsis, in a number of accessions and their subsequent F1 hybrids using a loop design (Churchill and Oliver 2001). With such a quantitative approach, two major questions can be addressed immediately:
What fraction of genes show significant differences at the expression level between Arabidopsis accessions; i.e., are the additive genetic effects for gene expression different from zero?
Which proportion of genes is expressed in a nonadditive, i.e., dominance fashion in a particular hybrid?
MATERIALS AND METHODS
Microarray preparation:
The Arabidopsis 6K microarray used consisted of 6008 cDNA fragments, of which 5834 were from the Incyte Unigene collection (Arabidopsis Gem I; Incyte, Palo Alto, CA), and 408 positive and negative controls, of which 384 were from the Lucidea Microarray ScoreCard v1.1 (GE Healthcare Bio-Sciences, Little Chalfont, UK), which were spotted in duplicate and distant from each other; for details see http://www.microarrays.be/service.htm/ currently available arrays.
Culture conditions and sampling:
Columbia (Col), Landsberg erecta (Ler), and Cape Verde Islands (Cvi) seeds were obtained from the Nottingham Arabidopsis Stock Centre (http://nasc.nott.ac.uk/) and used as parental lines. The two pairs of reciprocal F1 hybrids (Col × Ler, Ler × Col and Ler × Cvi, Cvi × Ler) were produced by manual pollination. Crossing Col and Cvi resulted in poor F1 seed germination. The resulting reciprocal F1 hybrids could not be taken into further analysis. In total, 90 seeds of each Arabidopsis genotype were plated on agar-solidified culture medium [1× MS (Duchefa, Haarlem, The Netherlands), 0.5 g liter−1 MES, pH 6.0, 1 g liter−1 sucrose, and 0.6% plant tissue culture agar (LabM, Bury, UK)] in three 150 × 25-mm round petri dishes (type Integrid; BD Falcon, Franklin Lakes, NJ). After sowing, plates were cold stratified at 4° for 7 days and subsequently transferred to a growth chamber kept at 22° with a 16-hr photoperiod of 65 μE m−2 sec−1 photosynthetically active radiation supplied by white fluorescent tubes. Plants were grown together under the same conditions and plates were rotated randomly on a regular basis.
To measure purely genetic differences in expression profiles, we used mature leaves as biological material, with constant environment and developmental time. On day 21 after germination, the first leaf pair of 60 plants (20 plants/plate) was dissected. To avoid effects of the circadian clock to expression changes, tissues were collected between 6 and 8 hr after dawn. For the three parental lines, leaves were immediately pooled. For the F1 hybrids, leaves were pooled only after the hybrid proved to be heterogeneous at marker loci. In addition to the sampling at day 21 after germination, the whole procedure was repeated with harvesting the first leaf pair at day 24 after germination, to cover the transient period at which the first leaf pair reached maturity (De Veylder et al. 2002).
Target labeling and hybridizations:
We prepared total RNA from the sample pools using TRIzol reagent (Invitrogen, Carlsbad, CA). Total RNA (5 μg) of each sample was reverse transcribed and amplified according to a modified protocol for in vitro transcription and subsequently fluorescently labeled with Cy5 or Cy3 (GE Healthcare Bio-Sciences) (http://www.microarrays.be/service.htm/protocols). Hybridization and washing were performed in an automated hybridization station (GE Healthcare Bio-Sciences) for a 16-hr cycle (http://www.microarrays.be/service.htm/protocols). The arrays were scanned at 532 and 635 nm by a Generation III scanner (GE Healthcare Bio-Sciences) and images were analyzed with an ArrayVision analyzer (Imaging Research, Ontario, Canada). Spot intensities were measured as artifact-removed total intensities (http://www.microarrays.be/service.htm/principal_measures) without correction for background.
Experimental design:
We constructed a loop design (Figure 1), eliminating the need for a reference sample with all its drawbacks, such as inefficient use of microarrays (half of the measurements concern the reference sample, which is presumably of little or no interest) and a large cost in degrees of freedom, hence resulting in lower significances in t- or F-statistics.
Figure 1.

The loop design, consisting of nine replicated dye-swap experiments in which two independent pools of first leaf pair samples of the seven genotypes were compared in an unequal treatment replication structure, on a total of 18 cDNA microarrays. The seven genotypes hybridized on the microarrays were the three homozygous accessions Col, Ler, and Cvi and the two pairs of reciprocal F1 hybrids obtained by crossing Col and Ler and Ler and Cvi. These microarrays had duplicated sets of 6008 Arabidopsis cDNA clones (of which 4876 were used in the analysis), representing almost one-quarter of the Arabidopsis gene repertoire, and 520 control cDNAs. Each microarray is represented by an arrow and connects the two sampled genotypes hybridized to it. The samples at the tail and head of each arrow are labeled with Cy3 and Cy5, respectively. In this arrangement, each sample is labeled equally often with Cy5 and with Cy3, ensuring that expression levels are free from biases due to dye effects. The pairings of pools sampled at days 21 and 24 after germination are represented by the loop in clockwise and anticlockwise directions, respectively.
Statistical analysis of the microarray data:
For the 6008 Arabidopsis spots and a total of 24 negative control spots containing a Bacillus subtilis-specific cDNA, we first addressed within-slide normalization by plotting for each single slide a “MA plot” (Yang et al. 2002), where M = log2 (R/G) (with R representing red fluorescent dye Cy5 and G green fluorescent dye Cy3) and 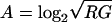 for each spot.
for each spot.
To correct for dye intensity differences, we used the robust scatter plot smoother LOWESS (Yang et al. 2002) as implemented in Genstat (Payne and Arnold 2002), to perform a local A-dependent normalization, converting M = log2(R/G) to log2{(R/G) − c(A)} = log2{R/(c(A)G)}, where c(A) is the LOWESS fit for M at A in the MA plot. The fraction of the data used for estimating the local LOWESS fit was set at 20%. On the basis of the adjusted M- and A-values for each gene, adjusted log2R and log2G signal intensities were obtained.
The data set was filtered in two steps. First, spots with a positive signal were selected as follows: we calculated median and 95th percentile of the 96 adjusted log2R and log2G signal intensities of the 24 negative control spots printed in duplicate on a single array. The 95th percentile was chosen as the signal threshold. For each gene, the adjusted log2R and log2G signal intensities were compared to the signal threshold: 1132 (18.8%) genes were below the signal threshold for more than half of the observations for each genotype and were subsequently removed from the data set. For the remaining 4876 genes, values below the 95th percentile threshold were reset to the median value of the negative control intensities. A second step of filtering involved the removal of signals showing a more than twofold difference between duplicated spots on a single array. Such signals were replaced by missing values in the further statistical analysis. This replacement involved 5% of the total of 351,072 spot measurements.
Similarly to previous studies (Jin et al. 2001; Oleksiak et al. 2002; Chhabra et al. 2003), we used the base-2 logarithm of the signals from the two channels as two separate pieces of information. Thus, the unit of analysis was an observation on an individual channel rather than first converting signals to (log-of-) the ratio of the observations on two channels. Mixed-ANOVA models were used for data analysis (Wolfinger et al. 2001). Variance components were estimated by residual maximum likelihood (REML) (Searle et al. 1992; Schabenberger and Pierce 2002), as implemented in Genstat (Payne and Arnold 2002). Following Wolfinger et al. (2001), the mixed-model analysis on the LOWESS fits to the spot measurements consisted of two steps. First, array and channel effects were removed from the expression responses by a normalization ANOVA model of the form
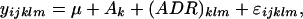 |
with i (= 1, … , 4876) indexing the selected cDNA fragments, j (= 1, … , 7) indexing the genotype, Ak representing the random array effects (k = 1, … , 18), and (ADR)klm the random replicates within array × dye combinations or channel-replicate effects with k = 1, … , 18 arrays, l = 1, … , 2 dyes, and m = 1, … , 2 replicates. This model was completed by the random error ɛijklm. Array and channel effects were chosen as random because we interpreted these effects as nuisance parameters for which the assumption of normality would be reasonable. Taking these effects as random will facilitate the analysis, because now only two variance components need to be estimated for those terms, after which the estimated effects, or best linear unbiased predictions (BLUPs) (Searle et al. 1992), can be created relatively simply.
The estimated residuals from the normalization ANOVA, rijklm, were subjected to 4876 gene-specific ANOVA models of the form
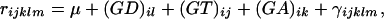 |
partitioning gene-specific variation into fixed gene-specific dye effects (GD)il, fixed gene-specific genotype (treatment) effects (GT)ij, random spot effects (GA)ik, and random error γijklm. In these gene-specific models, the spot effect accounted for the spot-to-spot variability inherent to spotted microarray data.
Although duplicate spots were not printed independently (namely after probe resampling by the printing pens), we treated them as independent replicates because they were clearly deposited remote from each other. A diagnostic check between residuals of duplicates showed no correlation or systematic bias (data not shown). Thus, we obtained 72 measurements for each of the 6008 Arabidopsis genes.
From the REML analysis as implemented in Genstat (Payne and Arnold 2002), for each gene we saved the vector of estimated genotype effects with the corresponding estimated variance-covariance matrix. These vectors of genotype effects of length 7, containing the gene-specific estimated effects for Col, Ler, Cvi, Col × Ler, Ler × Col, Ler × Cvi, and Cvi × Ler, were used to estimate composite additive, dominance, and reciprocal effects. For example, concentrating on the parents Col and Ler and their F1 Col × Ler, and assuming that the expression for Col is higher than that for Ler, we could estimate the composite additive genetic effect for gene expression, i.e., the sum of the additive genetic effects across all loci involved, by applying the contrast (1 −1 0 0 0 0 0) to the vector of estimated genotype effects. Of course, this contrast is an estimate of the composite additive genetic effect only under the rather restrictive assumption of no epistasis. Furthermore, when the additive genes are dispersed between the two parents, additive genetic variation will remain hidden. Nevertheless, this simple estimator will at least give a rough indication of additive genetic differences between parent pairs. Analogously, the contrast (−0.5 −0.5 0 1 0 0 0 0) will produce an estimate for the composite dominance effect under the assumption of absence of epistatic effects. Finally, estimates for reciprocal effects can be obtained from the contrast vectors (0 0 0 1 −1 0 0) for Col × Ler vs. Ler × Col and (0 0 0 0 0 1 −1) for Ler × Cvi vs. Cvi × Ler.
The contrast vectors used to produce the estimates for additive, dominance, and reciprocal effects can be used to produce estimates for the variances of these parameters by the standard rules for obtaining variances for linear contrasts (see, for example, Kuehl 2000). When c represents the contrast vector and V the estimated variance-covariance matrix of the estimates for the genotypic effects, c′Vc, with c′ the transpose of c, gives the variance corresponding to the contrast c.
Test statistics for contrasts between genotype effects can be constructed from the parameter estimates divided by their standard errors. These ratios were supposed to follow approximately a t-distribution with the degrees of freedom equal to the degrees of freedom for the error term in the gene-specific model. On the basis of the t-approximation to the test statistics for the contrasts, P-values were calculated that subsequently were transformed into false discovery rates, Q-values, according to Storey and Tibshirani (2003). The false discovery rate expresses the number of false positives, i.e., expression differences that in reality are null, but that are falsely declared significant, compared to the total number of significant differences. The Q-values were used to assess the significance of individual contrasts. This measure for significance seemed to be a more attractive choice for dealing with the problem of multiple testing than competing methods such as the classical Bonferroni correction (Kuehl 2000) and the false discovery rate (Benjamini and Hochberg 1995). It adaptively, i.e., on the basis of the observed P-values, combines the avoidance of large numbers of false positives with the identification of larger numbers of truly different gene expressions. For the calculation of Q-values, P-values for individual contrasts as saved from Genstat were introduced into the software QVALUE, written by Alan Dabney and John Storey (http://genomine.org/qvalue/); default parameter settings were used to estimate the proportion of features that are truly null.
The [d]/[a] ratio, with [d] the composite dominance effect and [a] the composite additive effect (Kearsey and Pooni 1996; Lynch and Walsh 1998), also referred to as potence, or hp (Griffing 1990), provides a standardized measure of the F1 hybrid expression level relative to the average of the parental levels. In the case of individual, single loci, |hp| > 1 can be explained only by an overdominance gene action. In a quantitative genetic situation, in which genotypic differences at many loci are involved, |hp| > 1 results from the dominance effects at many genes, differing in magnitude and even in sign, as well as from interactions between alleles at different loci.
To investigate the importance of the nonadditive gene expression in relation to the additive gene expression, we investigated the potence for all genes with significant dominance effects. To avoid working with ratios of effects with troublesome statistical properties, we translated the potence ratio into a difference of additive and dominance effects as follows. We constructed 99.8% confidence intervals for the differences [d] − [a] (or F1 minus P1) for positive estimates of [d] and confidence intervals for [d] + [a] (or F1 minus P2) for negative estimates of [d], under the convention that P1 is greater than P2. For the genes with a positive estimate for [d], when the confidence interval constructed for the difference [d] − [a] included zero, the hypothesis [d] − [a] = 0 was not rejected at a test level α = 0.002, and hence hp = 1. When the confidence interval did not include zero and was positive, then hp > 1, whereas hp < 1 when the confidence interval was negative. An analogous procedure was applied for the sum [d] + [a] in the case of a negative estimate for [d].
Functional classification:
An Arabidopsis Genome Initiative (AGI) code was assigned to every Arabidopsis clone spotted on the microarray, through evaluation of the most significant BLAST (Altschul et al. 1990) comparisons of the nucleic acid sequences against two databanks of Arabidopsis genomic sequences. One databank contains the cDNA sequences of all genes as defined in the Arabidopsis genome annotation (release 5.0) by The Institute of Genome Research (TIGR) (ftp://ftp.tigr.org/pub/data/a_thaliana/ath1/SEQ-UENCES/ATH1.cdna) and the other contains the genomic sequences of all genes enlarged with 500 bases of flanking sequences as defined in MatDB by the Munich Information Center for Protein Sequences (ftp://ftpmips.gsf.de/cress/arabidna/arabi_-genomicplus500_v110703.gz). On the basis of their AGI code, all 6008 Arabidopsis clones were functionally annotated according to MatDB annotations (http://mips.gsf.de/) and again to TAIR annotations (http://www.arabidopsis.org/) on the basis of TIGR's latest Arabidopsis Genome Release, TIGR 5 (http://www.tigr.org/). General functional classification of the genes is based on the corresponding gene ontology (GO) terms (http://www.geneontology.org/GO.current.annotations.html) assigned by TIGR for Arabidopsis. These data were parsed into a local database, and queries were written to extract and count the uppermost tree specifications for the GO network (biological process, cellular component, and molecular function) as set by the AmiGO arrangement (http://www.godatabase.org/cgi-bin/go.cgi). In the case of double-category assignment, the biologically most informative category was chosen or a choice was made supported by additional biological information.
An 11 × 4 contingency table of counts for functional gene categories vs. potency categories for gene action was created. Because the number of counts is smaller than five for the two classes with |hp| < 1 (but different from zero) and for the functional classes motor activity, nutrient reservoir activity, and obsolete molecular function, these two dominance ratio classes and the three functional categories were left out of the testing. Association between functional gene category and potency category was tested by a goodness-of-fit test for the 11 × 4 table. To identify cells that contributed most to the overall test statistic, contributions of individual cells to this statistic were calculated.
RESULTS
To answer to what extent genes are differentially expressed between the three accessions Col, Ler, and Cvi, we tested for differences between the accession effects by constructing t-tests on the three contrasts corresponding to the three pairwise differences of interest between the three accessions for each of the 4876 genes. Visualizations of the significance and the magnitude of the genotype effects were provided by the volcano plots for each pairwise accession contrast (Figure 2a; supplementary Figure S1, C and D, at http://www.genetics.org/supplemental/). These volcano plots contrast significance on the −log10(Q) scale against expression difference on the log2 scale. Genes toward the left and right on each plot had large expression differences and those toward the top were highly significant. Expression differences between accessions with Q-values ≤0.001 were called significant, resulting in a false discovery rate (FDR) of 0.1% among the significant features. Here and below, we used as a rule of thumb to choose that level of false discovery as a cutoff for significance that produced about one to two false discoveries in the whole set of significant differential expressions. Hence, we found 1827 (37.5%), 1569 (32.2%), and 1316 (27.0%) genes with significant additive gene expression between Col and Cvi, Ler and Cvi, and Col and Ler, respectively (Table 1), ranging from 32.45 down to 1.10 in fold change, with a median fold change value of 1.56 (Table 1). The small effects, with low variances and consequently high significance, are excluded from methods that assess significance only on the basis of a fold-change criterion. For example, 84.7% of the genes differentially expressed between Col and Ler (Q < 0.001) would be missed, if only those genes having at least a 2-fold difference in expression were selected. On the other hand, we also found 8 genes with large genotype effects (>2-fold difference) between Col and Ler that did not achieve significance at the α = 0.001 level because of their high variance (left or right of the hypothetical vertical line on a volcano plot that demarcates the 2-fold magnitude of difference, but below the horizontal line that demarcates the chosen significance threshold Q = 0.001). Figure 3 displays a number of box plots of which the first three refer to the absolute magnitude of the significant additive effects that were estimated as half the expression difference between two accessions for each of the three pairwise comparisons. The range of magnitude of the effects was almost consistent across the three pairwise comparisons. The respective gene lists are provided at http://www.psb.ugent.be/gexpr/.
Figure 2.


Volcano plots contrasting the significance [−log10(q) on the ordinate] and the magnitude of the expression difference (log2 on the abscissa) for the Col vs. Ler (a) and the Col × Ler vs. Ler × Col (b) comparison. Each cross represents one of the 4876 genes. The bottom horizontal dashed line corresponds to the FDR acceptance level of Q = 0.001 (−log10 = 3) for the accession contrast and of Q = 0.01 (−log10 = 2) for the reciprocal hybrid contrast.
TABLE 1.
Number of genes with significant pairwise genotypic or reciprocal differences between accessions (Q < 0.001) or reciprocal hybrids (Q < 0.01), respectively
| No. of genes (%)
|
Fold change
|
|||
|---|---|---|---|---|
| Pairwise comparisons | Min | Median | Max | |
| Acceptance level 0.001 | ||||
| Col vs. Ler | 1316 (27.0) | 1.15 | 1.53 | 13.96 |
| Col vs. Cvi | 1569 (32.2) | 1.10 | 1.59 | 30.04 |
| Ler vs. Cvi | 1827 (37.5) | 1.12 | 1.55 | 32.45 |
| Acceptance level 0.01 | ||||
| Col × Ler vs. Ler × Col | 313 (6.4) | 1.18 | 1.55 | 4.36 |
| Ler × Cvi vs. Cvi × Ler | 178 (3.7) | 1.16 | 1.54 | 7.49 |
The percentage (of 4876 total clones) is given in parentheses. The minimum, median, and maximum fold changes in gene expression observed among the clones with a Q-value less than the indicated acceptance level are shown.
Figure 3.

Box plots displaying the absolute magnitude of the estimated composite additive ([a]), dominance ([d]), and reciprocal effects (r) on the log2 scale. a[1], a[2], and a[3] were estimated as half the expression difference between Col and Ler, Col and Cvi, and Ler and Cvi, respectively; d[1], d[2], d[3], and d[4] were estimated from the contrasts of the hybrids Col × Ler, Ler × Col, Ler × Cvi, and Cvi × Ler, respectively, vs. the corresponding midparent value; r[1] and r[2] were estimated from the contrasts Col × Ler vs. Ler × Col and Ler × Cvi vs. Cvi × Ler, respectively.
The direct comparison of reciprocal hybrids might answer the question of whether transmission of genes through either the male or the female parent had an effect on their expression (also referred to as reciprocal effects). In the Col × Ler vs. Ler × Col and the Cvi × Ler vs. Ler × Cvi comparisons, 313 (6.4%) and 178 (3.7%) genes showed significant differences in expression (Q < 0.01), respectively (Table 1). Conspicuously, the volcano plot depicting the reciprocal hybrids (Figure 2b; supplementary Figure S1E at http://www.genetics.org/supplemental/) had a less “explosive” character than the one that shows the parental lines (Figure 2a; supplementary Figure S1, C and D, at http://www.genetics.org/supplemental/), reflecting a smaller range of fold changes and significances of gene expression differences (Table 1). The magnitude of the reciprocal effects measured in the Col × Ler vs. Ler × Col comparison was slightly higher than that in the Ler × Cvi vs. Cvi × Ler comparison (Figure 3). All these differentially expressed genes were nuclear encoded, and 38 of them were common in the two respective gene lists and were tabulated (see http://www.psb.ugent.be/gexpr/).
As we wanted to know the proportion of genes expressed in a nonadditive, i.e., dominance fashion in a particular hybrid, for each of the 4876 genes and for each of the four hybrids, we tested whether [d] ≠ 0. The number of nonadditively expressed genes with a Q-value ≤0.001 ranged from 313 (6.4%; Ler × Cvi), to 577 (11.8%; Cvi × Ler), to 645 (13.2%; Col × Ler), to 1028 (21.1%; Ler × Col). The equal distribution of the absolute magnitude of these significant composite dominance effects across the four hybrids is presented in Figure 3, in which the composite dominance effects are consistently higher in magnitude than the composite additive effects. So, by considering gene expression levels as quantitative traits, we estimated traditional quantitative genetic parameters, such as [a] and [d] for our expression data. The interpretation of the estimates depends, however, on whether the genetic effects were the outcome of a single locus or of multiple loci. In a multilocus situation, the estimate for [a] for each gene transcript represents the net balance of the na genes involved in the composite additive effect 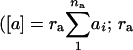 is the coefficient of gene association/dispersion), while the estimate for [d] represents the effects of the nd genes being involved in the composite dominance effect
is the coefficient of gene association/dispersion), while the estimate for [d] represents the effects of the nd genes being involved in the composite dominance effect 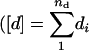 ) (Kearsey and Pooni 1996). For our type of data, however, individual gene effects could not be estimated: [a] gives very little about individual additive gene effects, whereas [d] indicates the direction of dominance on the majority of the k genes, weighted by the magnitudes of their effects.
) (Kearsey and Pooni 1996). For our type of data, however, individual gene effects could not be estimated: [a] gives very little about individual additive gene effects, whereas [d] indicates the direction of dominance on the majority of the k genes, weighted by the magnitudes of their effects.
Heterosis can be defined as [d]/[a] > 1 or < −1 (Gardner et al. 1953), where this ratio is referred to as the dominance ratio or potence, hp (Griffing 1990), providing a standardized measure for genetic nonadditivity. In the case of individual, single loci, |hp| > 1 can be explained only by overdominance. In a quantitative genetic situation in which genotypic differences at many loci are involved, |hp| = |[d]/[a]| > 1 results from the dominance effects at many genes, differing in magnitude and even in sign, as well as from interactions between alleles at different loci.
To further break down the number of nonadditively expressed genes according to their hp, we constructed 99.8% confidence intervals for the differences [d] − [a] (if d > 0) and [d] + [a] (if [d] < 0) (see materials and methods and http://www.psb.ugent.be/gexpr/). Depending on the sign of [d], the potence was classified as either negative or positive. Figure 4 reveals that among the nonadditively expressed genes in the four hybrids, genes clearly manifesting heterosis (|hp| > 1) at the expression level are most prevalent.
Figure 4.

Breakdown of the nonadditively expressed genes in each of the four hybrids according their dominance ratio hp. For the differences [d] − [a] (if [d] > 0) and [d] + [a] (if [d] < 0), 99.8% confidence intervals were constructed. Depending on the sign of [d], the dominance ratio was classified as either negative or positive.
In an attempt to examine the nature of the genes expressed significantly in a dominance way, genes were categorized according their molecular function (see materials and methods), taking into account their dominance ratio (Table 2). We tested for association between the functional classification of a gene and its dominance ratio by using a χ2-goodness-of-fit test at a level α of 0.01 (see materials and methods). A calculated χ2 = 129.19 ≥ χ(30,1%)2 provided strong evidence for an overall association between the categorical variables functional category and dominance ratio. To identify those cells that contributed most to the overall test statistic, contributions of individual cells to this statistic were calculated. We called cells in Table 2 having a contribution to the overall test statistic >4 significant and put these in italics. It is clear from Table 2 that the heterotic expression pattern of genes with a signal transducer activity shifted significantly from negative to positive. In contrast, the functional categories “catalytic activity,” “structural molecule activity,” and “translation regulatory activity” were significantly depleted for genes exhibiting hp > +1.
TABLE 2.
Functional classification of the nonadditively expressed genes based on their GO terms and their exhibited dominance ratio
| Function | hp < −1 | hp = −1 | hp = +1 | hp > +1 |
|---|---|---|---|---|
| Antioxidant activity | 19 | 9 | 4 | 19 |
| Binding (ligand) | 46 | 33 | 18 | 67 |
| Catalytic activity (enzyme activity) | 151 | 107 | 78 | 191 |
| Chaperone activity | 8 | 5 | 9 | 21 |
| Enzyme regulator activity | 10 | 6 | 5 | 9 |
| Signal transducer activity | 34 | 20 | 29 | 151 |
| Structural molecule activity | 25 | 19 | 6 | 9 |
| Transcription regulator activity | 105 | 40 | 37 | 139 |
| Translation regulator activity | 17 | 12 | 9 | 7 |
| Transporter activity | 66 | 30 | 33 | 123 |
| Molecular function unknown | 236 | 125 | 76 | 313 |
The 11 × 4 contingency table shows the counts of genes expressed significantly in a dominance way categorized according their molecular function (see materials and methods), taking into account their dominance ratio. The calculated χ2 = 129.19 ≥ χ(30,1%)2 = 50.89 provides strong evidence for an overall association between the categorical variables functional category and dominance ratio. Cells with a contribution to the χ2-statistic >4 were called significant and are in italics.
DISCUSSION
Our study provides an initial assessment of natural variation of gene expression and constitutes the first direct measurements of the magnitude of additivity, dominance, and heterosis of transcript abundance at a genome-wide level in diploid tissue of Arabidopsis. We show that a substantial number of genes are nonadditively expressed. Similar findings of nonadditive gene expression in a hybrid situation were found by Auger et al. (2005), who examined the amount of various transcripts in hybrid and inbred individuals in maize, and by Gibson et al. (2004), by assessing the degree of nonadditive gene expression in Drosophila. These results as well as ours indicate that basic genetic parameters, such as additivity and the lack thereof, can be readily observed and quantified for gene expression in a limited set of inbred strains and F1 crosses.
Previously, some microarray-based transcript studies have suggested that when different races or accessions of a species are compared, the underlying genome structure (insertions, deletions, and polymorphisms) itself may, to some extent, account for hybridization differences between otherwise comparable samples. In the course of a reanalysis of an Affymetrix-based experiment, designed to compare transcript abundance among human, chimp, and orangutan, Hsieh et al. (2003) noted biases in the directionality and significance of changes in expression, which led them to question whether the Affymetrix technology is really suitable for any comparison of genetically divergent or polymorphic species. It is obvious that these discrepancies can be circumvented by performing control hybridizations to genomic DNA prior to RNA hybridization to exclude oligonucleotide features that exhibit differences arising at the DNA (rather than at the RNA) level, such as SNPs. However, because Affymetrix arrays are still relatively expensive, for the purposes of this study and others whose aim is comparative analysis of different Arabidopsis accessions, we consider a cDNA microarray (although with less dense coverage of the genome) to be a far more low-cost alternative than a full-genome oligonucleotide array, because such genotype polymorphisms are not considered to pose an issue for hybridization to cDNA microarrays.
The degree to which gene expression is additive (α = 0.001) and therefore heritable for the three pairwise comparisons of the three accessions Col, Ler, and Cvi (27.0, 32.2, and 37.5%, respectively) is higher than that found between different strains of yeast (24%) (Brem et al. 2002), between Fundulus individuals within the same population (18%) (Oleksiak et al. 2002), and in Drosophila melanogaster (25%) (Jin et al. 2001; Gibson et al. 2004). In these latter studies, however, P-value cutoffs used might have been too stringent to achieve at least qualitatively what the Q-value directly achieves. On the other hand, the frequency of such genes might be overestimated somewhat in our study because we considered data from replicate spots on a single array as genuine replicates, but this generosity has been compensated by setting the FDR acceptance rate at the stringent α-level of 0.001. Table 1 and Figure 2a show that a minimum number of eight observations per genotype provide a statistic resolution strong enough to draw significant conclusions from genotype effects as low as 1.1-fold. This result is in good agreement with both theoretical and empirical studies in a variety of species, suggesting that six replicates of several treatments are sufficient to detect reliably differences in transcript abundance as small as 1.2-fold by using ANOVA (Wolfinger et al. 2001) or Bayesian statistical methods (Efron and Tibshirani 2002). Furthermore, the volcano plots show clearly that fold change does not follow statistical significance and because fold change does not acknowledge gene-specific error variation, it does not provide an appropriate criterion for singling out differentially expressed genes.
The few genes with significant reciprocal effects are indicative either of epigenetic phenomena, such as genomic imprinting, or of cytoplasmic effects (Kollipara et al. 2002). At this stage, the two effects cannot be distinguished. However, to determine that maternal effects are not involved in the observed expression differences, reciprocal F1 hybrids could be self-pollinated and expression phenotypes of the resulting progeny evaluated. Considerable reduction of the gene expression differences between the self-pollinated progeny of the reciprocal F1 hybrids would suggest that any maternal effect on the expression phenotypes is negligible.
In our study, dominance for gene expression (α = 0.001), estimated from the comparisons of hybrids with the corresponding midparent value, was clearly present at from 6.4 up to 21.1% of the genes. Meyer et al. (2004) have shown that alternative pollination methods (hand- vs. self-pollination) have significant effects on seed size and early seedling growth rate in Arabidopsis. Therefore, one could argue that part of the apparent dominance effects on gene expression may simply be attributable to the alternative pollination methods applied to obtain parental and F1 seeds. However, Meyer et al. (2004) also have observed that when the numbers of siliques on the self- and hand-pollinated mother plants were equal, the differences on seed weight and dry shoot mass disappeared. In the pollination procedure that we applied, the numbers of siliques on both mother plants and parental controls were not restricted and, hence, can be considered as equal. Therefore, we believe that the dominance effects on gene expression observed in the F1 hybrids of our study are mainly of genetic origin, although we cannot completely rule out a pollination effect.
Genetic interpretation of the nonadditive or dominance gene effects depends on the complexity of the underlying system. If genotype differences were due to a single locus, [d] would be strictly a measure of the locus-specific dominance effect. However, when genotypic differences at many loci are involved, the composition and magnitude of the dominance component [d] is a function of many positive and negative dominance effects. For our type of data, the difference between the two situations could not be tested.
On the basis of χ2-statistics, some functional gene categories were found to be significantly either enriched or depleted for genes with |hp| > 1. Significant enrichment for |hp| > 1 in cellular processes, such as signal transduction, may indicate that these processes are more enhanced in a heterozygous than in a homozygous genetic background. Although only a fraction of all the genes have been analyzed here at a single developmental stage, it is already tempting to speculate that the phenomenon of heterosis at the level of maintenance processes may be positively associated with signal transduction. A more elaborate and systematic recording of such molecular phenotypes (as in a diallel), combined with the morphological phenotypes and class of gene ontology, potentially gives rise to very rich data sets (compendium data) that over time can be mined for pathway information and provide the foundation for the modeling of complex genetic interactions (genetic networks).
On average, 9.4% of the transcriptome clearly manifested heterosis, which is almost twice the percentage found in Drosophila (Gibson et al. 2004). An hp > +1.0 implies that [d] > [a] or 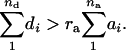 This relationship tells us that, as long as
This relationship tells us that, as long as 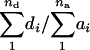 is greater than the degree of gene dispersion ra, then there will be heterosis. Therefore, very little dominance at individual genes is required to produce quite considerable heterosis at the expression level when the genes are dispersed between the two parents (Kearsey and Pooni 1996). The distinction between the degree of dominance and the degree of association ra, either or both of which can give rise to heterosis, may have very great scientific and economical importance. Unfortunately, neither of the two conditions can be estimated without analyzing (the variation in) segregating populations. Therefore, this sets the stage for further genetic analysis to search for the sequence variants that control or influence gene expression or more specifically the loci underlying the superiority of the hybrid for some gene expression phenotypes. This can possibly be done by using linkage analysis to map expression control elements (Cheung and Spielman 2002) or expression quantitative trait loci (Schadt et al. 2003), as has been done in yeast (Brem et al. 2002), mouse, man, and maize (Schadt et al. 2003). As the two crosses analyzed here correspond to the two sets of recombinant inbred lines (Lister and Dean 1993; Alonso-Blanco et al. 1998), which have been and are still frequently utilized for quantitative trait loci mapping of many morphological traits in Arabidopsis (Alonso-Blanco and Koornneef 2000), mapping the loci underlying the observed variation in gene expression between Col, Ler, and Cvi can readily be initiated.
is greater than the degree of gene dispersion ra, then there will be heterosis. Therefore, very little dominance at individual genes is required to produce quite considerable heterosis at the expression level when the genes are dispersed between the two parents (Kearsey and Pooni 1996). The distinction between the degree of dominance and the degree of association ra, either or both of which can give rise to heterosis, may have very great scientific and economical importance. Unfortunately, neither of the two conditions can be estimated without analyzing (the variation in) segregating populations. Therefore, this sets the stage for further genetic analysis to search for the sequence variants that control or influence gene expression or more specifically the loci underlying the superiority of the hybrid for some gene expression phenotypes. This can possibly be done by using linkage analysis to map expression control elements (Cheung and Spielman 2002) or expression quantitative trait loci (Schadt et al. 2003), as has been done in yeast (Brem et al. 2002), mouse, man, and maize (Schadt et al. 2003). As the two crosses analyzed here correspond to the two sets of recombinant inbred lines (Lister and Dean 1993; Alonso-Blanco et al. 1998), which have been and are still frequently utilized for quantitative trait loci mapping of many morphological traits in Arabidopsis (Alonso-Blanco and Koornneef 2000), mapping the loci underlying the observed variation in gene expression between Col, Ler, and Cvi can readily be initiated.
Acknowledgments
The authors thank Hilde Van den Daele and Debbie Rombaut for excellent technical assistance and Steven Vercruysse and Roel Sterken for providing the gene annotation and functional classification. This work was supported by grants from the Interuniversity Poles of Attraction Programme-Belgian Science Policy (P5/13), the European Union (QLK3-CT-2002-02035), and the Ministerie van de Vlaamse Gemeenschap-Landbouwkundig Onderzoek (IWT/020716).
References
- Alonso-Blanco, C., and M. Koornneef, 2000. Naturally occurring variation in Arabidopsis: an underexploited resource for plant genetics. Trends Plant Sci. 5: 22–29. [DOI] [PubMed] [Google Scholar]
- Alonso-Blanco, C., A. J. M. Peeters, M. Koornneef, C. Lister, C. Dean et al., 1998. Development of an AFLP based linkage map of Ler, Col and Cvi Arabidopsis thaliana ecotypes and construction of a Ler/Cvi recombinant inbred line population. Plant J. 14: 259–271. [DOI] [PubMed] [Google Scholar]
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers and D. J. Lipman, 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative, 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- Auger, D. L., A. D. Gray, T. S. Ream, A. Kato, E. H. Coe, Jr. et al., 2005. Nonadditive gene expression in diploid and triploid hybrids in maize. Genetics 169: 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57: 289–300. [Google Scholar]
- Brem, R. B., G. Yvert, R. Clinton and L. Kruglyak, 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755. [DOI] [PubMed] [Google Scholar]
- Cheung, V. G., and R. S. Spielman, 2002. The genetics of variation in gene expression. Nat. Genet. 32 (Suppl.): 522–525. [DOI] [PubMed] [Google Scholar]
- Cheung, V. G., L. K. Conlin, T. M. Weber, M. Arcaro, K.-Y. Jen et al., 2003. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat. Genet. 33: 422–425. [DOI] [PubMed] [Google Scholar]
- Chhabra, S. R., K. R. Shockley, S. B. Conners, K. L. Scott, R. D. Wolfinger et al., 2003. Carbohydrate-induced differential gene expression patterns in the hyperthermophilic bacterium Thermotoga maritima. J. Biol. Chem. 278: 7540–7552. [DOI] [PubMed] [Google Scholar]
- Churchill, G. A., and B. Oliver, 2001. Sex, flies and microarrays. Nat. Genet. 29: 355–356. [DOI] [PubMed] [Google Scholar]
- De Veylder, L., T. Beeckman, G. T. S. Beemster, J. De Almeida Engler, S. Ormenese et al., 2002. Control of proliferation, endoreduplication and differentiation by the Arabidopsis E2Fa/DPa transcription factor. EMBO J. 21: 1360–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron, B., and R. Tibshirani, 2002. Empirical Bayes methods and false discovery rates for microarrays. Genet. Epidemiol. 23: 70–86. [DOI] [PubMed] [Google Scholar]
- Enard, W., P. Khaitovich, J. Klose, S. Zöllner, F. Heissig et al., 2002. Intra- and interspecific variation in primate gene expression patterns. Science 296: 340–343. [DOI] [PubMed] [Google Scholar]
- Fay, J. C., H. L. McCullough, P. D. Sniegowski and M. B. Eisen, 2004. Population genetic variation in gene expression is associated with phenotypic variation in Saccharomyces cerevisiae. Genome Biol. 5: R26.1–R26.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner, C. O., P. H. Harvey, R. E. Comstock and H. F. Robinson, 1953. Dominance of genes controlling quantitative characters in maize. Agron. J. 45: 185–191. [Google Scholar]
- Gibson, G., R. Riley-Berger, L. Harshman, A. Kopp, S. Vacha et al., 2004. Extensive sex-specific nonadditivity of gene expression in Drosophila melanogaster. Genetics 167: 1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffing, B., 1990. Use of a controlled-nutrient experiment to test heterosis hypotheses. Genetics 126: 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, W.-P., T.-M. Chu, R. D. Wolfinger and G. Gibson, 2003. Mixed-model reanalysis of primate data suggests tissue and species biases in oligonucleotide-based gene expression profiles. Genetics 165: 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, W., R. M. Riley, R. D. Wolfinger, K. P. White, G. Passador-Gurgel et al., 2001. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat. Genet. 29: 389–395. [DOI] [PubMed] [Google Scholar]
- Kearsey, M. J., and H. S. Pooni, 1996. The Genetical Analysis of Quantitative Traits. Chapman & Hall, London.
- Kollipara, K. P., I. N. Saab, R. D. Wych, M. J. Lauer and G. W. Singletary, 2002. Expression profiling of reciprocal maize hybrids divergent for cold germination and desiccation tolerance. Plant Physiol. 129: 974–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehl, R. O., 2000. Design of Experiments: Statistical Principles of Research Design and Analysis, Ed. 2. Duxbury Press, Pacific Grove, CA.
- Lister, C., and C. Dean, 1993. Recombinant inbred lines for mapping RFLP and phenotypic markers in Arabidopsis thaliana. Plant J. 4: 745–750. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Meyer, R. C., O. Törjék, M. Becher and T. Altmann, 2004. Heterosis of biomass production in Arabidopsis. Establishment during early development. Plant Physiol. 134: 1813–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiak, M. F., G. A. Churchill and D. L. Crawford, 2002. Variation in gene expression within and among natural populations. Nat. Genet. 32: 261–266. [DOI] [PubMed] [Google Scholar]
- Payne, R. W., and G. M. Arnold, 2002. Genstat Release 6.1 Reference Manual—Part 3: Procedure Library PL14. VSN International, Oxford.
- Primig, M., R. M. Williams, E. A. Winzeler, G. G. Tevzadze, A. R. Conway et al., 2000. The core meiotic transcriptome in budding yeasts. Nat. Genet. 26: 415–423. [DOI] [PubMed] [Google Scholar]
- Ranz, J. M., C. I. Castillo-Davis, C. D. Meiklejohn and D. L. Hartl, 2003. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science 300: 1742–1745. [DOI] [PubMed] [Google Scholar]
- Schabenberger, O., and F. J. Pierce, 2002. Contemporary Statistical Models for the Plant and Soil Sciences. CRC Press, Boca Raton, FL.
- Schadt, E. E., S. A. Monks, T. A. Drake, A. J. Lusis, N. Che et al., 2003. Genetics of gene expression surveyed in maize, mouse and man. Nature 422: 297–302. [DOI] [PubMed] [Google Scholar]
- Searle, S. R., G. Casella and C. E. Mcculloch, 1992. Variance Components (Wiley Series in Probability and Mathematical Statistics: Applied Probability and Statistics). Wiley, New York.
- Storey, J. D., and R. Tibshirani, 2003. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 100: 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend, J. P., D. Cavalieri and D. L. Hartl, 2003. Population genetic variation in genome-wide gene expression. Mol. Biol. Evol. 20: 955–963. [DOI] [PubMed] [Google Scholar]
- Wolfinger, R. D., G. Gibson, E. D. Wolfinger, L. Bennett, H. Hamadeh et al., 2001. Assessing gene significance from cDNA microarray expression data via mixed models. J. Comput. Biol. 8: 625–637. [DOI] [PubMed] [Google Scholar]
- Yang, Y. H., S. Dudoit, P. Luu, D. M. Lin, V. Peng et al., 2002. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res. 30: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]