

Abstract
There is currently considerable interest in genetic analysis of quantitative traits such as blood pressure and body mass index. Despite the fact that these traits change throughout life they are commonly analyzed only at a single time point. The genetic basis of such traits can be better understood by collecting and effectively analyzing longitudinal data. Analyses of these data are complicated by the need to incorporate information from complex pedigree structures and genetic markers. We propose conducting longitudinal quantitative trait locus (QTL) analyses on such data sets by using a flexible random regression estimation technique. The relationship between genetic effects at different ages is efficiently modeled using covariance functions (CFs). Using simulated data we show that the change in genetic effects over time can be well characterized using CFs and that including parameters to model the change in effect with age can provide substantial increases in power to detect QTL compared with repeated measure or univariate techniques. The asymptotic distributions of the methods used are investigated and methods for overcoming the practical difficulties in fitting CFs are discussed. The CF-based techniques should allow efficient multivariate analyses of many data sets in human and natural population genetics.
QUANTITATIVE traits such as cholesterol levels in humans, milk yield in dairy cows, and fruit size in tomatoes are known to change over time; they are inherently longitudinal in nature. A major aim of genetics is to better understand the composition of such traits. With the advent of inexpensive molecular marker technology a wide variety of quantitative trait locus (QTL) mapping techniques have been developed to allow the dissection of quantitative traits in outbred populations (e.g., Haseman and Elston 1972; Goldgar 1990; Amos 1994; Hoeschele et al. 1997; Almasy and Blangero 1998; George et al. 2000). While these allow the extraction of information from univariate data (one trait measure per individual), techniques for QTL mapping when there are multiple trait measures are less well developed.
Existing univariate techniques can be readily applied to data measured at different stages of life but such approaches fail to capture the correlations between the components underlying traits such as cholesterol. At the other extreme, analyses are readily performed if we are prepared to assume that there is no change in the genetic composition of the trait over life [i.e., that the measures made are simply repeated realizations of exactly the same trait (Lynch and Walsh 1998)]. Neither of these approaches is satisfactory for many traits. A further alternative involves treating the individual trait measures (taken at different times) as distinct trait measures and modeling the covariance between the different traits in a multivariate analysis (e.g., Eaves et al. 1996). Such techniques, however, are difficult to apply in practice, may involve too many parameters in the model, and do not take the time element into account.
Ideally longitudinal traits would be modeled allowing for the fact that the multiple measures are ordered in time. To address this, Kirkpatrick et al. (1990) introduced covariance functions (CFs) to describe the relationship between different ages; CFs are simply continuous functions (often polynomials) that specify the covariance between two given ages. By fitting CFs with fewer parameters (e.g., a low-degree polynomial) than required to specify the full set of covariances between the different ages present in the data, the covariance structure of the data can be parsimoniously described. Using maximum-likelihood (ML)-based extensions of the Kirkpatrick et al. (1990) study, polygenic CF-based analyses of data from structured populations have been reported in recent years (Meyer 1998; Pletcher and Geyer 1999; Jaffrezic and Pletcher 2000).
In this study we extend the covariance function approach, previously applied only to polygenic effects (Meyer 1998; Kirkpatrick et al. 1990), to allow QTL mapping in a longitudinal framework. We show how the CF-based technique can be derived by extending the previously developed univariate and (unstructured covariance) multivariate approaches. Simulations are performed to investigate the properties of the different approaches available. Comparisons are made between the powers of the univariate, repeated measures, full multivariate (with unstructured covariances), and CF-based techniques.
MATERIALS AND METHODS
Theory:
Univariate model:
A method for single-trait QTL mapping, building on the theory of ML estimation of (polygenic) variance components (VC) (Lange et al. 1976; Hopper and Mathews 1982), was initially proposed by Goldgar (1990). Since then various extensions have been described in (Amos 1994; Almasy and Blangero 1998). For the univariate model we give only basic notation; for more details see Almasy and Blangero (1998).
The univariate VC model is based on the covariance between individuals i and j (with phenotypes yi, yj). This can be written in terms of the coefficient of coancestry, Θij (Lynch and Walsh 1998), and Rij, the fraction of genes shared identical-by-descent (IBD) at the QTL,
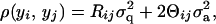 |
where 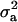 is the polygenic variance and
is the polygenic variance and 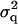 is the variance attributable to the QTL. Assuming that Rij can be estimated from marker data, the method can be applied to general pedigrees (Almasy and Blangero 1998). Assembling the Θij and Rij into matrices A and R (i.e., [A]ij = 2Θij and [R]ij = Rij), the covariance matrix can then be written as
is the variance attributable to the QTL. Assuming that Rij can be estimated from marker data, the method can be applied to general pedigrees (Almasy and Blangero 1998). Assembling the Θij and Rij into matrices A and R (i.e., [A]ij = 2Θij and [R]ij = Rij), the covariance matrix can then be written as
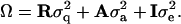 |
(1) |
Parameter estimation is performed by assuming multivariate normality of the phenotypes and applying likelihood-based methods.
Multivariate model:
The univariate variance component approach can be extended to deal with multiple-trait measures. We write the data as
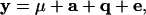 |
(2) |
where y = (y11, … , y1w, y21, … , y2w, … , yn1, … , ynw)T, μ = (μ1, … , μw, … , μ1, … , μw)T is the vector of fixed effects, a = (a11, … , a1w, a21, … , a2w, … , an1, … , anw)T is the vector of additive genetic effects, q = (q11, … , q1w, q21, … , q2w, … , qn1, … , qnw)T is the vector of QTL effects, and e = (e11, … , e1w, e21, … , e2w, … , en1, … , enw)T is the vector of environmental effects for traits 1 to w. The phenotypic data are written with traits ordered within individuals. Let N = nw, where n is the number of individuals. Modifications for cases in which data are missing are also possible (e.g., Mrode 1996).
For many traits there will be a correlation between the different trait measures within an individual. We can rewrite Equation 1, accounting for the covariances between relatives and between multiple-trait values as
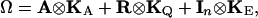 |
(3) |
where KA is a w × w matrix of additive genetic covariances between traits, KQ is a w × w matrix of additive QTL covariances between traits, and KE is a w × w matrix of environmental covariances between traits. ⊗ denotes the direct product of two matrices. We refer to this as the full multivariate model. When there are more than a few traits, estimation of the w(w + 1)/2 parameters in each of KA, KQ, and KE will become increasingly difficult and methods that model the data more parsimoniously will be required.
Repeatability model:
A special case of the full multivariate model where there are multiple measurements of the same trait is often called the repeatability model. This model assumes that the polygenic and QTL correlations across multiple measures are 1 and that their variances do not change over time. In this case the computational demands are considerably lower because a single parameter can be used to model the effect of the QTL and polygenic genetic effects. Since there may be environmental effects that are not constant over time there are two effects fitted alongside the QTL and polygenic effects. The first of these, commonly called the permanent environmental effect, models environmental effects that are present in all of an individual's trait measures. The variance associated with this permanent environmental term is labeled 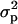 . The second effect models the additional environmental effects that are not constant over time; this is the temporary environmental term, with associated variance term denoted
. The second effect models the additional environmental effects that are not constant over time; this is the temporary environmental term, with associated variance term denoted 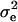 . This second term also serves as an error term for effects not modeled by the other random effects.
. This second term also serves as an error term for effects not modeled by the other random effects.
Phrasing the repeatability model in terms of the full multivariate model, the covariance matrices, KA and KQ, modeling the relationship between the different trait measures in Equation 3, are now 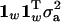 and
and 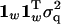 , respectively. The matrix of environmental effects KE is split into two under the repeatability model, with separate terms for the permanent and temporary environmental terms. The overall covariance matrix is hence
, respectively. The matrix of environmental effects KE is split into two under the repeatability model, with separate terms for the permanent and temporary environmental terms. The overall covariance matrix is hence
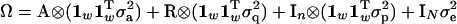 |
(4) |
with only four parameters to estimate.
Longitudinal analysis:
Although the repeatability model assumption may be a tenable one for some traits that have multiple measures over time, in most cases it will not be reasonable. Many longitudinal traits are likely to change in composition over the life of the individual and are the main focus here. For longitudinal traits it is desirable to explicitly model the relationship between age and the genetic and environmental components of the trait. To achieve this, a multivariate analysis is performed in which the unstructured covariance structure from the full multivariate model is replaced by one that utilizes the natural ordering in time of the trait measurements. Kirkpatrick et al. (1990) suggest a method suitable for “function-valued” (varying with time) traits. Although in practice the trait may be observed only at a finite number of time points [i.e., w, giving w(w + 1)/2 distinct (co)variances in a w × w covariance matrix, G], it is useful to consider a continuous function, linking the different covariance values. This continuous function, referred to as CF, is denoted 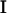 . For ages t0 and t1 the CF is
. For ages t0 and t1 the CF is
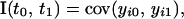 |
where yi0 and yi1 denote the trait values at times t0 and t1. Separate CFs are fitted for the QTL effect, the polygenic effect, and the permanent environmental effect, with the effects assumed to be independent of each other. The overall phenotypic CF is given by summing the component CFs.
To estimate CFs from the available data, polynomials of age can be used. While a degree w − 1 polynomial will fit the w-trait data exactly by fitting a curve through all the points, in reality a smoother curve that ignores stochastic variation (around the true curve) is required. In practice, orthogonal polynomials are used because they behave well numerically. Legendre orthogonal polynomials are used here. Such polynomials are defined on (−1, 1) and hence the age values of interest are scaled to have maximum value 1 and minimum value −1. An expression for the CF of interest, 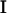 , can be written in terms of the polynomials chosen, φi(x), and a matrix of coefficients, C,
, can be written in terms of the polynomials chosen, φi(x), and a matrix of coefficients, C,
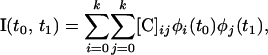 |
(5) |
where k is the degree of the polynomial chosen and t0 and t1 are the scaled ages.
Kirkpatrick et al. (1990) propose a method whereby one can estimate the matrix of coefficients, C, using least squares. Unfortunately this approach proves difficult to apply in practice and we consider instead likelihood-based analysis.
Estimation of the coefficient matrix in a general pedigree using random regression:
A random regression (RR) model is one that includes a polynomial for both fixed and random effects (Jamrozik et al. 1997; Meyer 1998; Jaffrezic and Pletcher 2000). RR is useful because the covariance between polynomials of age in a RR can be related to the covariance function coefficients of interest. The random regression model that allows this is
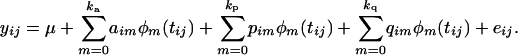 |
(6) |
ka, kp, and kq denote the degree of the polynomial for the additive genetic, permanent environmental, and QTL random effects, respectively. tij is the time at which the measure yij is taken. Each individual has wi measures, and it is possible that wi ≠ w for some individuals. The covariance structure of such a model is
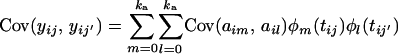 |
(7) |
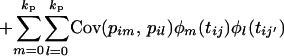 |
(8) |
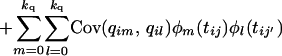 |
(9) |
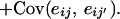 |
(10) |
Each of the covariance terms (7), (8), and (9) can now be seen to be of the same form as Equation 5. If these covariance terms can be estimated in a random regression, the covariance matrix for each of the additive genetic, permanent environment, and QTL effects is then given by the equation G = ΦCΦT, where [Φ]ij = φj(ti) (numbering the matrix indexes 0 to k).
To fit the RR model the full multivariate model is reparameterized. In this reparameterization the set of trait measures is replaced with a degree k polynomial for each effect of interest (permanent environment, polygenic, or QTL). The full multivariate model is then fitted with these polynomial coefficients regarded as correlated traits (Meyer 1998). To do this, we begin by writing Equation 6 in matrix notation,
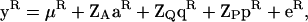 |
where 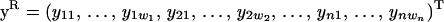 are the phenotypes,
are the phenotypes, 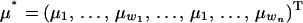 is the vector of fixed effects,
is the vector of fixed effects, 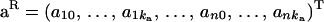 is the (ka + 1) × n vector of polygenic random regression coefficients,
is the (ka + 1) × n vector of polygenic random regression coefficients, 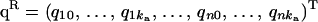 is the (kq + 1) × n vector of QTL random regression coefficients,
is the (kq + 1) × n vector of QTL random regression coefficients, 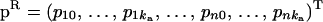 is the (kp + 1) × n vector of permanent environmental random regression coefficients, and eR is the
is the (kp + 1) × n vector of permanent environmental random regression coefficients, and eR is the 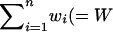 , say) vector of temporary environmental terms (note that this is w × n if all n individuals are measured for all traits, i.e., if wi = w for all i). ZA is a W × n(ka + 1) matrix of orthogonal polynomial coefficients. ZQ and ZP are defined similarly, with ka replaced by kq or kp. The covariance terms for the vector aR are given in Equation 7 and, assuming the systematic age effects have been removed by the fixed effects, can be written as
, say) vector of temporary environmental terms (note that this is w × n if all n individuals are measured for all traits, i.e., if wi = w for all i). ZA is a W × n(ka + 1) matrix of orthogonal polynomial coefficients. ZQ and ZP are defined similarly, with ka replaced by kq or kp. The covariance terms for the vector aR are given in Equation 7 and, assuming the systematic age effects have been removed by the fixed effects, can be written as 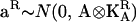 , where
, where 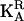 is the (ka + 1) × (ka + 1) matrix of CF coefficients (named C above) for the polygenic effects. In a similar fashion
is the (ka + 1) × (ka + 1) matrix of CF coefficients (named C above) for the polygenic effects. In a similar fashion 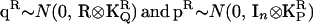 . Written as a full variance-covariance matrix,
. Written as a full variance-covariance matrix,
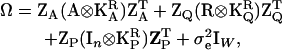 |
where 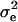 is the temporary environmental variance term. Estimation is performed by assuming multivariate normality of the phenotypes and applying likelihood-based methods. κ now has (ka + 1)(ka + 2)/2 entries for the polygenic effect and equivalent terms for the QTL and permanent environmental cases. Note that when the number of time points minus 1 equals the degree of the polynomial fitted for a RR, the number of parameters is the same for RR as for the full multivariate case; this means a separate error term (
is the temporary environmental variance term. Estimation is performed by assuming multivariate normality of the phenotypes and applying likelihood-based methods. κ now has (ka + 1)(ka + 2)/2 entries for the polygenic effect and equivalent terms for the QTL and permanent environmental cases. Note that when the number of time points minus 1 equals the degree of the polynomial fitted for a RR, the number of parameters is the same for RR as for the full multivariate case; this means a separate error term (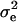 ) is no longer required.
) is no longer required.
Simulation:
To assess the properties of the models described for longitudinal data analysis, computer simulations were performed. The main interest was in QTL detection and characterization in samples of sizes realistically attainable in human and natural population genetic studies. In all simulations 150 four-sib nuclear families (900 individuals) were simulated. All individuals were given phenotypes at five evenly spaced time points. To mimic a dense marker map all individuals were typed for a highly polymorphic (20-allele) marker, completely linked to the simulated QTL. All phenotype values were generated as the sum of a permanent environmental effect, a temporary environmental effect, and a QTL effect. All effects were drawn as random effects from normal distributions with appropriate variances.
Models of QTL effect:
Three models of QTL effect over time were considered. These increase in complexity from model A to model C. First, the QTL was modeled under a repeatability model with the same QTL effect (same variance) across the five time points (simulation model A). One thousand replicates in which the QTL variance was 0.2, the permanent environment variance was 0.5, and the temporary environmental variance was 0.5 were considered (summarized in Table 1).
TABLE 1.
Parameters used in simulations A–C
| Simulation | 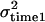 |
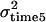 |
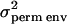 |
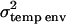 |
QTL correlation | Change in QTL variance |
|---|---|---|---|---|---|---|
| A | 0.2 | 0.2 | 0.5 | 0.5 | 1 | No change |
| B1 | 0.2 | 0.33 | 0.5 | 0.5 | 1 | Linear |
| B2 | 0.2 | 0.4 | 0.5 | 0.5 | 1 | Linear |
| B3 | 0.2 | 0.33 | 0.75 | 0.25 | 1 | Linear |
| C1 | 0.2 | 0.4 | 0.5 | 0.5 | See matrix in text | Linear |
| C2 | 0.2 | 0.4 | 0.5 | 0.5 | See matrix in text | Logarithmic |
Perm env, permanent environment; temp env, temporary environment.
Second, the QTL was modeled to increase its effect linearly over time but the QTL effects were constrained to be completely correlated over time (simulation model B). That is, there is a change in QTL variance but a “flat” correlation structure or equivalently, a repeatability model with heterogeneity of variance. Three sets of variance values, B1, B2, and B3, were considered (see Table 1). Two hundred replicates were used.
Third, the QTL was modeled to change in effect (change in variance) over time and to have QTL correlations of <1 between time points (simulation model C). The correlation structure is hence “sloping.” The specified QTL correlation matrix was
 |
This is perhaps a more realistic model of the change in genetic (QTL) effect over time than one that constrains the correlation to remain at 1. Since this represents a deviation from the assumptions of the repeatability model, any model that allows the correlations to be <1 (such as a first- or higher-degree RR) will give a better fit than the repeatability model, even when the genetic variance does not change over time. Simulation C was repeated twice (denoted C1 and C2), once with a linear increase in QTL variance and once with a logarithmic increase (see Figure 3, triangles); the parameters used are in Table 1. Two hundred replicates were generated. Note that the simulated covariance function was not generated from a polynomial. Although the true shape of the covariance function will not be known in practice, it is highly unlikely to look exactly like that generated from a polynomial.
Figure 3.

Sample results, Simulation C2.
Analysis methods applied:
Univariate, repeatability (Re), RR, and full multivariate models were used to analyze the data simulated under the simulation models described above. Note that although no polygenic effects were simulated a single term for a polygenic effect was fitted in all analysis methods. Re and first-degree RRs are applied to data from simulation model A. This allowed an empirical evaluation of the adequacy of the asymptotic approximation (to 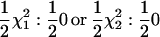 , see below) for the case where a first-degree RR is compared with the Re method. The agreement between the asymptotic distribution and the calculated statistics (see below for details of statistics used and hypothesis testing) was assessed graphically.
, see below) for the case where a first-degree RR is compared with the Re method. The agreement between the asymptotic distribution and the calculated statistics (see below for details of statistics used and hypothesis testing) was assessed graphically.
The data from simulation model B were used to evaluate the performance of the univariate, Re, and RR methods. The tests of interest with these data are the test for the significance of the slope term (power to detect change in QTL variance) and the test for the overall significance of the QTL effect (power to detect QTL). For the test of the slope term a first-degree RR is compared with a Re model. In the case of the test for overall QTL effect, a first-degree RR was evaluated alongside a Re model and univariate models (see below for details of hypothesis testing). A further test of significance of the QTL effect could be obtained by fitting a full multivariate model [i.e., 15 (co)variances] or higher-degree polynomial RRs to the data and comparing this with the univariate, Re, and first-degree RR models. Fitting these models to the data generated under simulation model B (QTL correlations equal to 1), however, proved impossible in practice. The estimation of large numbers of parameters is very difficult when the traits of interest are highly correlated. Estimation was more readily achieved with data from simulation model C where the correlation between the traits was reduced.
Finally, the Re, RR (with degrees from one to four), and full multivariate models were used to examine the data simulated under simulation model C. The full multivariate model fits five variances for the five different ages in the data and attempts to estimate separately all 10 covariances between the effects at different ages. This model should give identical likelihoods to those of the saturated fourth-degree RR model. Both fit the same number of parameters for the QTL effect (15 in all). The lower-degree RRs use polynomials to smooth the covariance function, reducing the number of parameters in the model. Under simulation model C, methods that do not model the covariance between the trait values at different ages (such as Re analysis) were expected to perform poorly and the main comparisons were between RR and full multivariate analyses.
The required likelihood maximizations were done in ASREML (Gilmour et al. 2002), with IBD estimation done in SOLAR (Almasy and Blangero 1998). One practical problem we overcame was the incorporation of IBD information into the analysis. ASREML requires the inverse of the IBD matrix as input but this matrix can be singular. To circumvent this problem we added a small value (0.0001) to the diagonal entries of each IBD matrix to render it nonsingular. This ad hoc approach has been shown to give results that are indistinguishable from those obtained from SOLAR (which does not require the inverse of the IBD matrix) in simplified univariate cases (Macgregor 2003) and was hence used in all the simulations described here. Scripts to allow application of the methods described are available on request.
Hypothesis testing:
Hypothesis testing was done by calculating P-values on the basis of asymptotic results. Hypothesis tests for the univariate model have well-known properties (Self and Liang 1987; Almasy and Blangero 1998), namely that the 2 ln likelihood-ratio (LR) test statistic for QTL vs. no QTL is distributed as 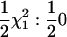 under the null hypothesis of no QTL effects. For the univariate tests of multiple time points the maximum 2 ln(LR) test statistic from the five time points and from the mean of the five trait values was used (statistic Suni); also computed was a Bonferroni-corrected version, Suni(b). Since the Re model has one variance parameter to estimate for the QTL, the 2 ln(LR) test statistic is asymptotically
under the null hypothesis of no QTL effects. For the univariate tests of multiple time points the maximum 2 ln(LR) test statistic from the five time points and from the mean of the five trait values was used (statistic Suni); also computed was a Bonferroni-corrected version, Suni(b). Since the Re model has one variance parameter to estimate for the QTL, the 2 ln(LR) test statistic is asymptotically 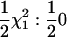 (statistic Srep).
(statistic Srep).
For the tests of RR models, the asymptotic distributions of the 2 ln(LR) statistic will also be mixtures of χ2-distributions. The simplest RR-based test is for the significance of the first-degree RR compared with that of the Re model (equal to a RR model with only the intercept fitted). This model can be tested in two ways. First, the significance of the linear term (qi1) in the RR can be tested with the covariance between the linear and the constant term (qi0) constrained to be zero. In this case twice the log-likelihood difference [2 ln(LR), statistic SΔrep1 (statistic for deviations from repeatability model), see Table 2] between the RR and the Re model is expected to be distributed as 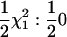 . This follows because under the null hypothesis the additional variance term is on the boundary of the parameter space. Second, if both the variance and the covariance terms are fitted in the RR (subject to the constraint that the coefficient matrix remains positive definite), the 2 ln(LR) test statistic (SΔrep2) for the RR vs. the Re model is a 50:50 mixture of
. This follows because under the null hypothesis the additional variance term is on the boundary of the parameter space. Second, if both the variance and the covariance terms are fitted in the RR (subject to the constraint that the coefficient matrix remains positive definite), the 2 ln(LR) test statistic (SΔrep2) for the RR vs. the Re model is a 50:50 mixture of 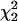 and a point mass at zero (
and a point mass at zero (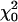 ). Note that this test statistic (with the covariance unconstrained) is not a mixture of
). Note that this test statistic (with the covariance unconstrained) is not a mixture of 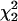 and
and 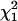 (as suggested in Stram and Lee 1994 and Meyer 1998). This is because when the variance term associated with the qi1 term is zero the covariance between the qi0 and qi1 terms must also be zero, resulting in a point mass at zero (
(as suggested in Stram and Lee 1994 and Meyer 1998). This is because when the variance term associated with the qi1 term is zero the covariance between the qi0 and qi1 terms must also be zero, resulting in a point mass at zero (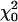 ) not at
) not at 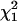 .
.
TABLE 2.
Summary of statistics, simulations A and B
| QTL RR coefficients in
|
Asymptotic distribution of 2 ln(L1/L0)
|
|||
|---|---|---|---|---|
| Test statistic | L0 | L1 | Notes | |
| SΔrep1 | qi0 | qi0, qi1 | 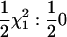 |
Deviation from Re model test |
| SΔrep2 | qi0 | qi0, qi1, cov(qi0, qi1) | 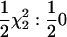 |
Deviation from Re model test |
| Srep | None | qi0 | 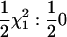 |
Power to detect QTL test |
| Suni | NA | NA | 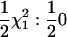 |
Power to detect QTL univariate test |
| SRR1 | None | qi0, qi1, cov(qi0, qi1) | 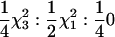 |
Power to detect QTL test |
Tests of higher-degree RR terms can be constructed analogously to those for the linear RR terms. For the test of the full-degree k + 1 RR (i.e., all elements of the CF estimated) vs. the degree k model (statistic Sk), 2 ln(LR) was compared with a 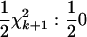 distribution. An alternative test uses the degree k + 1 RR with the correlations between the (k + 1)th diagonal term of the CF and the first k RR coefficients constrained to zero (analogous to SΔrep1 above, with the correlations between the first k coefficients left unconstrained). The 2 ln(LR) statistic comparing this constrained fit to the degree k RR [statistic Sk(c)] has a
distribution. An alternative test uses the degree k + 1 RR with the correlations between the (k + 1)th diagonal term of the CF and the first k RR coefficients constrained to zero (analogous to SΔrep1 above, with the correlations between the first k coefficients left unconstrained). The 2 ln(LR) statistic comparing this constrained fit to the degree k RR [statistic Sk(c)] has a 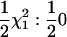 distribution. The coefficient matrix as a whole was constrained to be positive definite. In simulation C the best-fitting model was selected by increasing the degree of the RR until the addition terms were found to not significantly increase the likelihood. The higher-degree RR was deemed significantly better if the P-value for the higher-degree model was <0.01.
distribution. The coefficient matrix as a whole was constrained to be positive definite. In simulation C the best-fitting model was selected by increasing the degree of the RR until the addition terms were found to not significantly increase the likelihood. The higher-degree RR was deemed significantly better if the P-value for the higher-degree model was <0.01.
Also of interest are tests of the overall significance of the QTL terms in a RR model. The main test of interest here is the first-degree RR-based test of QTL (with constant and slope terms, together with their covariance) vs. no QTL [all three (co)variances set to zero]. The 2 ln(LR) statistic for this test (statistic SRR1) is assumed to be distributed asymptotically 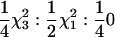 . This follows because there are two variance terms and these are on the boundary of the parameter space under the null. When performing the likelihood-ratio test, one-quarter of the time both of the variances are estimated to be positive (and their covariance can be nonzero), one-half of the time one of the variances is at zero [together with the covariance, cov(qi0, qi1), from Equation 9], and one-quarter of the time all three (co)variances are at zero. In simulation B the power of SRR1, Srep, Suni, and Suni(b) to detect the simulated QTL was assessed at three significance levels: 0.001, 0.0001 [asymptotically equivalent to a univariate base 10 logarithm of odds (LOD) of 3], and 0.00001. For reference, the statistics calculated in simulations A and B are given in Table 2.
. This follows because there are two variance terms and these are on the boundary of the parameter space under the null. When performing the likelihood-ratio test, one-quarter of the time both of the variances are estimated to be positive (and their covariance can be nonzero), one-half of the time one of the variances is at zero [together with the covariance, cov(qi0, qi1), from Equation 9], and one-quarter of the time all three (co)variances are at zero. In simulation B the power of SRR1, Srep, Suni, and Suni(b) to detect the simulated QTL was assessed at three significance levels: 0.001, 0.0001 [asymptotically equivalent to a univariate base 10 logarithm of odds (LOD) of 3], and 0.00001. For reference, the statistics calculated in simulations A and B are given in Table 2.
The RR-fitting procedure models the random deviations from a fixed curve for each regression coefficient. To ensure valid LR tests comparing different polynomial degrees for the RRs this same set of fixed effects (i.e., f0 + f1tj + f2tj2 + f3tj3 + f4tj4) was used for all fitted models. If the fixed effects are changed with the degree of the RR, the LR test is not valid. For the simulated data, no systematic change over time was simulated so while a constant (f0) and age-dependent (fi, i = 1, … , 4) fixed curve terms were fitted in the RR analyses, they were expected to yield estimates that are close to 0. For real data, suitable fixed effects (e.g., a polynomial of age with degree equal to or greater than the highest-degree random term) should be fitted to the data to ensure that the random regression coefficients model deviations from the population trajectory.
RESULTS
Simulation A:
The agreement between the expected asymptotic and simulation-based empirical distributions when fitting the RR model to data simulated to fit the repeatability model (no change in variance over time and correlation between effects at different ages equal to one) was excellent. The two statistics of interest, SΔrep1 and SΔrep2, are expected to follow 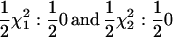 distributions, respectively. They are shown in Figure 1. For comparison the
distributions, respectively. They are shown in Figure 1. For comparison the 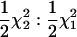 distribution is shown; this shows that neither SΔrep1 nor SΔrep2 converges to this mixture (as suggested in Stram and Lee 1994 and Meyer 1998). Note that although the covariance is not constrained to 0 in SΔrep2 the overall coefficient matrix (C) is constrained to be positive definite.
distribution is shown; this shows that neither SΔrep1 nor SΔrep2 converges to this mixture (as suggested in Stram and Lee 1994 and Meyer 1998). Note that although the covariance is not constrained to 0 in SΔrep2 the overall coefficient matrix (C) is constrained to be positive definite.
Figure 1.

Simulation A results. Fit of the statistics SΔrep1 and SΔrep2 (for data simulated under the repeatability model) to the expected asymptotic null distributions is shown.
Simulation B:
Deviations from repeatability model:
The power in each case is given in Table 3. SΔrep2 was more powerful than SΔrep1 at detecting deviations from the repeatability model. Reducing the relative amount of temporary environment (ratio of permanent to environmental variance was 75:25 instead of 50:50) resulted in the change in genetic variance over time being easier to detect.
TABLE 3.
Simulation B: power to reject the repeatability model in favor of first-degree RR
| SΔrep1 (%) | SΔrep2 (%) | |
|---|---|---|
| B1 | 5 | 41 |
| B2 | 12 | 76 |
| B3 | 16 | 75 |
Significance level set to 0.01.
Power to detect QTL: RR, Re, and univariate models:
The power to detect a simulated QTL was determined using three statistics, Suni, Srep, and SRR1. The power (proportion of 200 replicates, expressed as a percentage) at different significance levels for variance sets B1, B2, and B3 is given in Tables 4, 5, and 6, respectively.
Looking at the results from Srep in Tables 4 and 6 we see that much of the power in the repeatability analysis lies in the reduction in temporary environmental noise as a result of averaging over a number of measures; when the temporary environmental effects are small the repeatability analysis has little power to detect QTL. In contrast, the model allowing for a change in QTL effect over time (SRR1) gains power when the temporary environmental noise is reduced. This is because the change in genetic variance over time can be more readily detected, increasing the power to detect the QTL when a parameter modeling the change in QTL effect over time is fitted. Note also that a modest increase in the genetic variance at age five (compare the results when QTL variance is 0.33 with when it is 0.4, see Tables 4 and 5) has a relatively large effect upon the power when SRR1 is used; the power to detect the QTL with a LOD of 3 (asymptotic significance level 10−4) rises from 43 to 78%.
TABLE 4.
Simulation B1: power to detect QTL
| Significance level
|
|||
|---|---|---|---|
| Statistic | 10−3 | 10−4 | 10−5 |
| Srep (Re QTL vs. no QTL) | 54 | 30 | 17 |
| Suni (univariate QTL vs. no QTL) | 61 | 33 | 18 |
| Suni(b) (Bonferroni-corrected Suni) | 38 | 21 | 9 |
| SRR1 (linear RR QTL vs. no QTL) | 64 | 43 | 31 |
Data are simulated under simulation model B with QTL variance 0.2 (age 1) to 0.33 (age 5) (0.5 permanent environmental variance, 0.5 temporary environmental variance).
TABLE 6.
Simulation B3: power to detect QTL
| Significance level
|
|||
|---|---|---|---|
| Statistic | 10−3 | 10−4 | 10−5 |
| Srep (Re QTL vs. no QTL) | 30 | 13 | 5 |
| Suni (univariate QTL vs. no QTL) | 39 | 18 | 6 |
| Suni(b) (Bonferroni-corrected Suni) | 24 | 8 | 4 |
| SRR1 (linear RR QTL vs. no QTL) | 75 | 64 | 46 |
Data are simulated under simulation model B with QTL variance 0.2 (age 1) to 0.33 (age 5) (0.75 permanent environmental variance, 0.25 temporary environmental variance).
TABLE 5.
Simulation B2: power to detect QTL
| Significance level
|
|||
|---|---|---|---|
| Statistic | 10−3 | 10−4 | 10−5 |
| Srep (Re QTL vs. no QTL) | 67 | 41 | 21 |
| Suni (univariate QTL vs. no QTL) | 75 | 47 | 24 |
| Suni(b) (Bonferroni-corrected Suni) | 53 | 28 | 13 |
| SRR1 (linear RR QTL vs. no QTL) | 85 | 78 | 65 |
Data are simulated under simulation model B with QTL variance 0.2 (age 1) to 0.4 (age 5) (0.5 permanent environmental variance, 0.5 temporary environmental variance).
Since the five trait values at the five time points have correlation >0 but <1, the uncorrected Suni is anticonservative while the Suni(b) is too conservative. Assuming that the true power value at the specified significance levels can be obtained by taking a power estimate between Suni and Suni(b) we see that the repeatability and univariate methods have similar power.
Simulation C1:
The procedure outlined above was used to determine the best-fitting model for the data. Seventy-nine percent of replicates rejected, at the 1% significance level, the no-QTL model when the Re model was fitted. However, in all cases (200 replicates) the Re model was rejected in favor of the first-degree RR model (All P-values <10−5 for SΔrep1 and SΔrep2). This was unsurprising since the data were simulated so that the QTL variance changed over time and the genetic (QTL) correlations were <1. Sixty-four percent of replicates rejected the linear RR in favor of the quadratic RR when Sk was used to compare the two models. When Sk(c) was used only 23% of replicates provided evidence for the quadratic model. Using Sk(c) for the test for a cubic RR compared with the quadratic fit (for replicates where the quadratic coefficient was significant) resulted in none of the replicates indicating that the cubic fit was better. Assessing the higher-degree models (unconstrained cubic model and quartic model) proved difficult computationally, with many replicates failing to converge to a likelihood maximum. In the cubic case, roughly one-third of replicates failed to converge (using a maximum of 100 iterations in ASREML) when the unconstrained cubic model (i.e., Sk was calculated) was fitted. Taking the likelihoods as calculated (i.e., one-third of them are underestimates of the true likelihood maximum, biasing the test statistic for the significance of the cubic term downward), 7% of replicates rejected the quadratic model in favor of the cubic model. Only in 35% of cases could the full multivariate model be maximized. These results are summarized in Table 7.
TABLE 7.
Simulation C1 [QTL variance 0.2 (age 1) to 0.4 (age 5)]: best-fitting model (%)
| Model | Sk | Sk(c) |
|---|---|---|
| Repeatability | 0 | 0 |
| Linear RR | 36 | 77 |
| Quadratic RR | 57 | 23 |
| Cubic RR | 7a | 0 |
One-third of replicates failed to converge so this may be an underestimate.
For a few of the replicates all models could be maximized and a graphic representation of the results of one replicate is given in Figure 2. The variance terms from the QTL RR are expressed as a proportion of the total variance (i.e., QTL heritability). For comparison, the univariate and repeatability model results are superimposed on the same graph. This shows that the repeatability model is a poor fit to the simulated model and that the univariate results, while following the simulated model to some degree, are rather noisy. All of the polynomial-based RRs follow the simulated model well; the first-degree model offers an excellent fit with only two extra parameters fitted compared with the repeatability model. The fourth-degree polynomial follows the univariate results more closely but in this case such variations from the simulated model are simply random variation.
Figure 2.

Sample results, Simulation C1.
It is instructive to compare the results from simulations B and C. In simulation B2, when 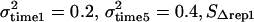 rejected the repeatability model in 12% of cases (significance level 1%). In comparison, when the data were simulated in simulation C1 with the same parameters apart from a change in the correlation structure, 100% of replicates rejected the repeatability model (significance level 1%, although in fact all rejected it at significance level 0.001%). The univariate results for simulation C1 were similar (data not shown) to those obtained for the repeatability analysis (which is equivalent to an “average across all measures” univariate analysis when there is regular age spacing) and were hence substantially less powerful than those obtained from the RR model.
rejected the repeatability model in 12% of cases (significance level 1%). In comparison, when the data were simulated in simulation C1 with the same parameters apart from a change in the correlation structure, 100% of replicates rejected the repeatability model (significance level 1%, although in fact all rejected it at significance level 0.001%). The univariate results for simulation C1 were similar (data not shown) to those obtained for the repeatability analysis (which is equivalent to an “average across all measures” univariate analysis when there is regular age spacing) and were hence substantially less powerful than those obtained from the RR model.
Simulation C2:
In simulation C2 71% of replicates rejected (significance level 1%) the no-QTL model when the Re model was fitted. In all cases (200 replicates) the Re model was rejected in favor of the first-degree RR model (all P-values <10−6 for SΔrep1 and SΔrep2). Eighty-four percent of replicates rejected the linear RR in favor of the quadratic RR when Sk was used to compare the two models. When the Sk(c) was used 89% of replicates provided evidence for the quadratic model. Note that although the likelihood ratio of Sk(c) is lower than that of Sk, since the null distributions differ Sk(c) can sometimes give smaller P-values. Using Sk(c) for the test for a cubic RR compared with the quadratic fit resulted in 4% of the replicates indicating that the cubic fit was better. This may be a slight underestimate as 5% of the replicates failed to converge to a likelihood maximum. Using the unconstrained cubic model in the test resulted in 17% of replicates rejecting the quadratic model although almost a third failed to converge fully. The quartic and full multivariate models could not be reliably fitted to these data. These results are summarized in Table 8. Although simulation C2 showed that polynomial-based CFs worked well with the simulated logarithmic increase in QTL variance with age, nonmonotonic changes in QTL variance with age were not considered here (e.g., an increase in genetic effect at earlier ages, followed by a decline in later life).
TABLE 8.
Simulation C2 [QTL variance 0.2 (age 1) to 0.4 (age 5)]: best-fitting model (%)
Almost one-third of replicates failed to converge so this may be an underestimate.
Five percent of replicates failed to converge so this may be a slight underestimate.
The results of one replicate are given in Figure 3. The values specified in the simulation model are superimposed on the graph. As expected, when the change in QTL variance is nonlinear the second- and higher-degree RRs have more utility than the first-degree model. Nonetheless, even the first-degree RR is substantially better than the repeatability model. Once again the univariate results are rather noisy; univariate methods do not utilize the natural ordering in time of the genetic effects with adjacent measures often yielding very different estimates of QTL heritability.
DISCUSSION
This article has described methods suitable for QTL analysis in complex pedigrees of data sets with longitudinal trait measures. The multivariate techniques required to effectively analyze such data are more involved than those for single-trait measures. This, together with the relative paucity of suitable data, goes some way toward explaining the lack of research in this area. Longitudinal traits are often not well described by single, cross-sectional, phenotypic measures but, as has been described, the conceptually simple full multivariate model requires the estimation of large numbers of parameters when there are more than a few time points. Since the data sets commonly available for genetic studies in human or natural populations are small, the full multivariate approach has somewhat limited application. Some longitudinal traits will be relatively highly correlated across multiple measures of the same trait compared with nonlongitudinal multivariate measures (e.g., multivariate analysis of height and weight, say). The simulations performed here showed that when traits are highly correlated the estimation of large numbers of parameters is difficult. The covariance function-based approach may have considerably more utility than the full multivariate model as it can reduce the number of parameters in the model. Fitting a polynomial with degree plus one equal to the number of age points in the data is equivalent to a full multivariate model. Fitting lower-degree polynomials smoothes the estimated covariance function, removing individual deviations that are likely to be due to stochastic variation.
The covariance function approach will be particularly useful when the data are measured at a large number of ages, perhaps with irregular gaps between measures; this is because the approach fits a polynomial through the set of ages available for each individual. Furthermore, individuals measured only for a few ages can still contribute to the analysis by providing information on the coefficients of the lower-degree polynomials (information available on constant and linear terms when there are two age measures and so on). Once the covariance function has been estimated for a given data set, predictions of future observations can be made using best linear unbiased prediction (e.g., Lynch and Walsh 1998; Mrode 1996). For example, predictions could be made about the trait value of an individual at age 60 given their measurements until age 40 or about the trait value at a particular age for children given the measures taken on their parents.
In simulation B it was shown that when there was a moderate increase in QTL variance over time fitting a first degree RR increased the power to detect the QTL. This increase in power came solely from the RR modeling the change in QTL variance. The increased efficiency of the RR in modeling any decreases in the genetic correlation between trait measures <1 was ignored by simulating data with no decline in genetic correlation with time. The increase in power was particularly large when the ratio of permanent to temporary environment was high (i.e., when most of the environmental “noise” affects all of an individual's trait measures).
At Genetic Analysis Workshop 13 (GAW13) (Almasy et al. 2003) reference is made to genes that change their variance over time as slope genes (Gauderman et al. 2003; Gee et al. 2003; Rao et al. 2003; Yang et al. 2003). The data generated under simulation model B allow a direct test for these slope genes. QTL effects, however, may not be completely correlated across ages and a more realistic simulation model (i.e., simulation model C) will allow the correlations between QTL effects at different ages to decrease.
It is not possible to know what form real-life genetic CFs take. It was assumed in simulation C that the decline in correlation followed a steady decrease with increasing time separation (i.e., we intentionally did not simulate data under the polynomial-based analysis method used for the CF-based analyses; polynomials do not generate correlation structures of the “banded” type used in simulation C). The correlation was assumed to remain relatively high over the range of ages of interest. This seems likely to be true for QTL effects (whose constituent element is one or more closely linked genes) but may be less likely to hold for polygenic effects (whose constituent elements are more heterogeneous and will change over life). The shape of possible CFs for polygenic effects has been considered by Jaffrezic and Pletcher (2000); the models considered ranged from one in which the correlation structure remained high across ages to another in which the correlation became negative at widely separated ages. They concluded that polynomial-based CFs were most effective when the correlation remained high over widely separated ages (Jaffrezic and Pletcher 2000).
The model selection (choice of polynomial fit) in simulation C was based on differences in likelihood. To ensure parsimony and robustness, we advise utilization of low-degree polynomials. Although high-degree polynomials may provide a good fit to the data, if the method is to be used for QTL detection then the benefits of improved fit may be outweighed by the increase in the degrees of freedom. It is also important to note that with most realistic sample sizes it is not possible to fit high-degree polynomials and the problem of model selection may be of little practical consequence. For genome scanning we recommend application of the Re model (zero-degree RR), the first-degree RR model, and, where data permit (large sample size), the second-degree RR model. The testing of multiple polynomial fits increases the computational load and also the number of tests done. Note that the requirement for multiple tests in QTL analysis is not new here; many QTL mapping methods for line crosses apply multiple models at each location across the genome (e.g., additive model and additive plus dominance model). If computational time is a limiting factor, scanning could be performed with moderate intermarker spacing with first-degree polynomials with regions achieving nominal significance (P < 0.05) followed up by spacing at 1-cM intervals with second-degree polynomials (or a higher degree if sufficient data are available). When we applied the RR methodology to a real data set [the Framingham Heart Study data set from GAW13 (Almasy et al. 2003)] we were not able to fit a second-degree RR (first-degree RRs were fine) to every position across the chromosomes we examined (Macgregor et al. 2003). For simplicity we used two-generation families (parents and offspring had phenotypes and genotypes) with a single linked marker in our simulations. An illustration of the application of the RR methodology to three-generation extended families and genome scan data are given in our GAW13 article (Macgregor et al. 2003). Note that since the IBD computation and phenotype modeling steps are separate in our analysis the practical application of our method is identical for single-marker or multiple-marker (multipoint) data.
Although fitting a model that estimates the full set of (co)variances in the data [there are w(w + 1)/2 to estimate when there are w trait measures] can capture the change in QTL variance over time, such methods are inefficient in most cases and are difficult to apply in practice. One of the primary aims of this article was to investigate how much information can be extracted from longitudinal data in realistic scenarios. The work here and other work on human data sets (de Andrade et al. 2002; de Andrade and Olswold 2003) indicate that approaches that do not simplify the covariance structure are unworkable in practice (the relatively small data sets do not support the estimation of large numbers of parameters). In an application of the full multivariate model to trivariate human genetic data (de Andrade et al. 2002; de Andrade and Olswold 2003) the six parameters (three variances, three covariances) could not be estimated simultaneously for all of the random effects. When the situation was approximated by three bivariate analyses, parameter estimation was possible. Given that the trivariate data in these articles (de Andrade et al. 2002; de Andrade and Olswold 2003) support the estimation of only three parameters it would probably be better to fit a first-degree RR to the full set of three traits than to fit three separate full multivariate analyses to three different subsets of the data.
In the simulations presented some simplifying assumptions were used. No fixed effects were simulated and the permanent environmental effects were assumed to be constant over time. No polygenic effects were simulated (but note that a single polygenic effect was estimated in all analysis methods). These simplifications were used to allow meaningful comparison between the methods used. The inclusion of time varying polygenic and fixed effects would increase the observed difference between the RR and the other analysis methods; one of our primary aims was assessing the effects of modeling just the QTL effect more efficiently. The simulations also ignore one of the benefits of the RR procedure (compared with full multivariate and repeatability analyses), namely the ability of the RR method to analyze data with phenotypes measured at different ages in different individuals. In the simulations all individuals were assumed to be measured at all five ages. In reality human data sets will often feature individuals measured at a large variety of different ages; a full multivariate analysis will usually require individuals at proximal ages to be grouped together, discarding information. Twenty alleles were simulated at the linked marker to approximate the situation where there is abundant marker information (through the use of either a 5-cM microsatellite marker map or the increasingly common dense SNP marker maps). The resultant polymorphism information content will generally be in the range 0.9–1.
To allow comparison between the different methods, asymptotic results were utilized. In the case where the first-degree RR was compared with the Re model, the asymptotic result was validated by simulation (Figure 1). For the test of first-degree RR QTL vs. no QTL the asymptotic result used (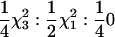 ) is the same as that used in a bivariate VC analysis by Amos et al. (2001); we note that there is disagreement in the literature on this matter, with the described asymptotic distribution from Wang (2003) appearing to disagree with the distribution described by Amos et al. (2001). We performed simulations under a no-QTL model (with constant polygenic heritability of 0.20) and found results consistent with those described in Amos et al. (2001) (data not shown). We also note that it is important to simulate either a polygenic or a QTL effect (or both) to evaluate the null distribution; in the univariate case the null distribution when neither effect is simulated appears to be
) is the same as that used in a bivariate VC analysis by Amos et al. (2001); we note that there is disagreement in the literature on this matter, with the described asymptotic distribution from Wang (2003) appearing to disagree with the distribution described by Amos et al. (2001). We performed simulations under a no-QTL model (with constant polygenic heritability of 0.20) and found results consistent with those described in Amos et al. (2001) (data not shown). We also note that it is important to simulate either a polygenic or a QTL effect (or both) to evaluate the null distribution; in the univariate case the null distribution when neither effect is simulated appears to be 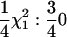 (cf. the usual
(cf. the usual 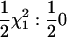 in the univariate QTL test) when a QTL plus polygenic model is compared with a polygenic model. If the distribution of the traits is not multivariate normal the asymptotic distribution may not be appropriate (incorrect type I error). In such cases gene-dropping simulations can be used to evaluate an empirical null distribution for the test of the overall significance of a QTL. Such simulations use the data structure from the actual data and generate replicates with a marker unlinked to the QTL. Although this approach requires a relatively large number of replicates for small P-values this procedure needs to be done only once per genome scan and can typically be completed in <1 day with current computing power. Instead of simply comparing the observed test statistic with the relevant percentile of the empirical distribution, the number of replicates required can be reduced either by fitting a parametric distribution to the empirical distribution (Dudbridge and Koeleman 2004) or by performing a regression of the empirical distribution on the expected asymptotic distribution. This latter approach allows the rescaling of the results so that the asymptotic distribution is valid and is implemented in the program SOLAR for univariate analysis (Almasy and Blangero 1998).
in the univariate QTL test) when a QTL plus polygenic model is compared with a polygenic model. If the distribution of the traits is not multivariate normal the asymptotic distribution may not be appropriate (incorrect type I error). In such cases gene-dropping simulations can be used to evaluate an empirical null distribution for the test of the overall significance of a QTL. Such simulations use the data structure from the actual data and generate replicates with a marker unlinked to the QTL. Although this approach requires a relatively large number of replicates for small P-values this procedure needs to be done only once per genome scan and can typically be completed in <1 day with current computing power. Instead of simply comparing the observed test statistic with the relevant percentile of the empirical distribution, the number of replicates required can be reduced either by fitting a parametric distribution to the empirical distribution (Dudbridge and Koeleman 2004) or by performing a regression of the empirical distribution on the expected asymptotic distribution. This latter approach allows the rescaling of the results so that the asymptotic distribution is valid and is implemented in the program SOLAR for univariate analysis (Almasy and Blangero 1998).
Genomewide significance can be dealt with when using the methods we describe here. For human data the P-value required for genomewide significance is generally assumed to be in the range 0.0001–0.00005 (Lander and Kruglyak 1995). Suggestive significance requires P-values <0.002. Since a number of models may be fitted per genomic location we recommend using slightly more stringent thresholds than those given in Lander and Kruglyak (1995); thresholds twofold lower than the Lander and Kruglyak levels would seem reasonable (given that no more than a few models will be utilized and they are unlikely to be uncorrelated). The P-value obtained from either the asymptotic results or gene-dropping simulations (see above) can be compared with the threshold values and significance (suggestive, genomewide) declared accordingly. For other species the relevant thresholds can be determined by taking into account the species' genome length.
One disadvantage of the RR techniques is that the method specifies the correlation structure and change in variance (over time) together. A low-degree polynomial may be adequate to model the change in variance over time but inadequate for approximating the covariance structure or vice versa. Alternative models that fit separate functions for the change in variance and the change in correlation or covariance have been proposed for polygenic effects (character process models, e.g., Pletcher and Geyer 1999; Diego et al. 2003).
Alternative techniques for longitudinal QTL mapping in structured populations have been developed recently (Ma et al. 2002; Wu et al. 2004a). Such mapping techniques assume that the QTL is a fixed effect with a specified number of alleles and this allows for sophisticated modeling of the change in QTL effect over time (Ma et al. 2002; Wu et al. 2004a). The application of such fixed-effect models has led to a greater understanding of the genetic architecture of growth in organisms such as mice and forest trees (Wu et al. 2004a,b). In contrast to this, the methodology described here is based upon random-effects modeling, where we assume that the distribution of QTL effects is normal. This allows us to circumvent the estimation of QTL allele frequencies and facilitates ready application of our method to arbitrary pedigrees. Given the matrix of QTL-specific IBD probabilities [estimated from the marker data (Almasy and Blangero 1998; George et al. 2000)] our method can be applied without modification to a wide variety of family structures. Further discussion of the differences between fixed- and random-effect QTL models is given in George et al. (2000).
A number of other methods for allowing analyses of multivariate data have been proposed. In most cases these are for distinct multiple traits (height, weight, etc.) rather than for longitudinal ones (height at age 20, at age 30, …). The simplest approach involves performing separate univariate analyses for each trait. This approach does not take advantage of the potential power gains inherent in the multivariate structure of the data. Furthermore, it is unclear how to keep the significance level at the desired level when there are multiple tests. A Bonferroni correction can be readily applied but this is almost certain to be overly conservative. The simulations performed here showed that univariate methods offered similar power to repeated measures methods and were often hence substantially less powerful than RR-based approaches. The next simplest alternative is to transform the multiple-trait values into a single summary or composite measure, thus allowing a single univariate analysis method to be used. This composite measure can be constructed such that the calculated “factor score” maximizes some parameter of interest, such as the heritability (Boomsma and Dolan 1998). Furthermore, a multivariate segregation analysis has been proposed for pedigree data (Blangero and Konigsberg 1991) and this may allow the construction of a composite measure that is particularly suitable for mapping the major gene affecting a trait. However, even in this second case where there may be more power to detect a particular QTL or locus, neither method is likely to give an optimal composite measure for other QTL or loci (Eaves et al. 1996).
A number of authors have considered extensions of the sib-pair regression methods to multivariate data (Amos et al. 1990; Allison et al. 1998; de Andrade and Olswold 2003; Huang and Jiang 2003; Mirea et al. 2003). Such methods offer advantages over the VC-based multivariate approaches in terms of computational ease but, in addition to their unsuitability for extended families, they have been shown to offer less power than VC-based approaches (for bivariate data see Amos et al. 2001).
Multivariate linkage analysis related to that described in materials and methods has been described for sib-pair data (Eaves et al. 1996) and livestock and experimental cross data (Jiang and Zeng 1995; Knott and Haley 2000; Sorensen et al. 2003) and applied to developmental dyslexia data in sib pairs (Marlow et al. 2003). The method used by Marlow et al. (2003) fitted the polygenic effect as in Equation 3 but the covariance structure of the random effect for the QTL was constrained such that correlation between any two trait measures was equal to one. This means that there are only k parameters to estimate when there are k traits [compared with k(k + 1)/2 with an unstructured QTL covariance matrix]. While this is unlikely to be true for all but the most strongly related traits, this model may allow parameter estimation in cases in which there are limited amounts of data.
In this study we have extended the RR approach to allow QTL analysis of longitudinal data. The methods described appropriately take into account the ordering in trait values over time. Computer simulations have shown that the RR-based approach offers considerable increases in power compared with univariate and repeatability-based techniques. It should be possible to take advantage of this extra power by fitting first-degree random regressions to most realistic human/natural population data sets.
Acknowledgments
We thank Robin Thompson for providing advice on the program ASREML. We thank the anonymous referees for helpful comments on a previous version of this manuscript. We gratefully acknowledge financial support from the following organizations: Akzo-Nobel (Organon), the Biotechnology and Biological Sciences Research Council, the Royal Society, and the Higher Education Funding Council for Wales.
References
- Allison, D. B., B. Thiel, P. St. Jean, R. C. Elston, M. C. Infante et al., 1998. Multiple phenotype modeling in gene-mapping studies of quantitative traits: power advantages. Am. J. Hum. Genet. 63: 1190–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasy, L., and J. Blangero, 1998. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 62: 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasy, L., L. A. Cupples, E. W. Daw, D. Levy, D. Thomas et al., 2003. Genetic analysis workshop 13: introduction to workshop summaries. Genet. Epidemiol. 25: S1–S4. [DOI] [PubMed] [Google Scholar]
- Amos, C. I., 1994. Robust variance-components approach for assessing genetic linkage in pedigrees. Am. J. Hum. Genet. 54: 535–543. [PMC free article] [PubMed] [Google Scholar]
- Amos, C. I., R. C. Elston, G. E. Bonney, B. J. B. Keats and G. S. Berenson, 1990. A multivariate method for detecting genetic-linkage, with application to a pedigree with an adverse lipoprotein phenotype. Am. J. Hum. Genet. 47: 247–254. [PMC free article] [PubMed] [Google Scholar]
- Amos, C. I., M. de Andrade and D. K. Zhu, 2001. Comparison of multivariate tests for genetic linkage. Hum. Hered. 51: 133–144. [DOI] [PubMed] [Google Scholar]
- Blangero, J., and L. W. Konigsberg, 1991. Multivariate segregation analysis using the mixed model. Genet. Epidemiol. 8: 299–316. [DOI] [PubMed] [Google Scholar]
- Boomsma, D. I., and C. V. Dolan, 1998. A comparison of power to detect a QTL in sib-pair data using multivariate phenotypes, mean phenotypes, and factor scores. Behav. Genet. 28: 329–340. [DOI] [PubMed] [Google Scholar]
- de Andrade, M., and C. Olswold, 2003. Comparison of longitudinal variance components and regression based approaches for linkage detection on chromosome 17 for systolic blood pressure. BMC Genet. 4: S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Andrade, M., R. Gueguen, S. Visvikis, C. Sass, G. Siest et al., 2002. Extension of variance components approach to incorporate temporal trends and longitudinal pedigree data analysis. Genet. Epidemiol. 22: 221–232. [DOI] [PubMed] [Google Scholar]
- Diego, V. P., L. Almasy, T. D. Dyer, J. M. P. Soler and J. Blangero, 2003. Strategy and model building in the fourth dimension: a null model for genotype (x) age interaction as a gaussian stationary stochastic process. BMC Genet. 4: S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudbridge, F., and B. P. C. Koeleman, 2004. Efficient computation of significance levels for multiple associations in large studies of correlated data, including genomewide association studies. Am. J. Hum. Genet. 75: 424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaves, L. J., M. C. Neale and H. Maes, 1996. Multivariate multipoint linkage analysis of quantitative trait loci. Behav. Genet. 26: 519–525. [DOI] [PubMed] [Google Scholar]
- Gauderman, W. J., S. Macgregor, L. Briollais, K. Scurrah, M. Tobin et al., 2003. Longitudinal data analysis in pedigree studies. Genet. Epidemiol. 25: S18–S28. [DOI] [PubMed] [Google Scholar]
- Gee, C., J. L. Morrison, D. C. Thomas and W. J. Gauderman, 2003. Segregation and linkage analysis for longitudinal measurements of a quantitative trait. BMC Genet. 4: S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George, A. W., P. M. Visscher and C. S. Haley, 2000. Mapping quantitative trait loci in complex pedigrees: a two-step variance component approach. Genetics 156: 2081–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour, A. R., B. J. Gogel, B. R. Cullis, S. J. Welham and R. Thompson, 2002. ASREML User Guide Release 1.0. VSN International, Hemel Hempstead, UK.
- Goldgar, D. E., 1990. Multipoint analysis of human quantitative genetic-variation. Am. J. Hum. Genet. 47: 957–967. [PMC free article] [PubMed] [Google Scholar]
- Haseman, J. K., and R. C. Elston, 1972. The investigation of linkage between a quantitative trait and a marker locus. Behav. Genet. 1: 11–19. [DOI] [PubMed] [Google Scholar]
- Hoeschele, I., P. Uimari, F. E. Grignola, Q. Zhang and K. M. Gage, 1997. Advances in statistical methods to map quantitative trait loci in outbred populations. Genetics 147: 1445–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper, J. L., and J. D. Mathews, 1982. Extensions to multivariate normal-models for pedigree analysis. Ann. Hum. Genet. 46: 373–383. [DOI] [PubMed] [Google Scholar]
- Huang, J. A., and Y. M. Jiang, 2003. Genetic linkage analysis of a dichotomous trait incorporating a tightly linked quantitative trait in affected sib pairs. Am. J. Hum. Genet. 72: 949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrezic, F., and S. D. Pletcher, 2000. Statistical models for estimating the genetic basis of repeated measures and other function-valued traits. Genetics 156: 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamrozik, J., L. R. Schaeffer and J. C. M. Dekkers, 1997. Genetic evaluation of dairy cattle using test day yields and random regression model. J. Dairy Sci. 80: 1217–1226. [DOI] [PubMed] [Google Scholar]
- Jiang, C. J., and Z. B. Zeng, 1995. Multiple-trait analysis of genetic-mapping for quantitative trait loci. Genetics 140: 1111–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick, M., D. Lofsvold and M. Bulmer, 1990. Analysis of the inheritance, selection and evolution of growth trajectories. Genetics 124: 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott, S. A., and C. S. Haley, 2000. Multitrait least squares for quantitative trait loci detection. Genetics 156: 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander, E., and L. Kruglyak, 1995. Genetic dissection of complex traits—guidelines for interpreting and reporting linkage results. Nat. Genet. 11: 241–247. [DOI] [PubMed] [Google Scholar]
- Lange, K., J. Westlake and M. A. Spence, 1976. Extensions to pedigree analysis. III. Variance components by the scoring method. Ann. Hum. Genet. 39: 485–491. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Ma, C. X., G. Casella and R. L. Wu, 2002. Functional mapping of quantitative trait loci underlying the character process: a theoretical framework. Genetics 161: 1751–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macgregor, S., 2003. Genetic linkage analysis in complex pedigrees. Ph.D. Thesis, University of Edinburgh, Edinburgh.
- Macgregor, S., S. A. Knott, I. White and P. M. Visscher, 2003. Longitudinal variance-components analysis of the Framingham heart study data. BMC Genet. 4: S22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow, A. J., S. E. Fisher, C. Francks, I. L. MacPhie, S. S. Cherny et al., 2003. Use of multivariate linkage analysis for dissection of a complex cognitive trait. Am. J. Hum. Genet. 72: 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, K., 1998. Estimating covariance functions for longitudinal data using a random regression model. Genet. Sel. Evol. 30: 221–240. [Google Scholar]
- Mirea, L., S. B. Bull and J. Stafford, 2003. Comparison of haseman-elston regression analyses using single, summary, and longitudinal measures of systolic blood pressure. BMC Genet. 4: S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrode, R. A., 1996. Linear Models for the Prediction of Animal Breeding Values. CAB International, Wallingford, UK.
- Pletcher, S. D., and C. J. Geyer, 1999. The genetic analysis of age-dependent traits: modeling the character process. Genetics 153: 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, S. Q., L. Li, X. Li, K. L. Moser, Z. Guo et al., 2003. Genetic linkage analysis of longitudinal hypertension phenotypes using three summary measures. BMC Genet. 4: S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self, S. G., and K. Y. Liang, 1987. Asymptotic properties of maximum-likelihood estimators and likelihood ratio tests under nonstandard conditions. J. Am. Stat. Assoc. 82: 605–610. [Google Scholar]
- Sorensen, P., M. S. Lund, B. Guldbrandtsen, J. Jensen and D. Sorensen, 2003. A comparison of bivariate and univariate qtl mapping in livestock populations. Genet. Sel. Evol. 35: 605–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stram, D. O., and J. W. Lee, 1994. Variance-components testing in the longitudinal mixed effects model. Biometrics 50: 1171–1177 [erratum: Biometrics 51: 1196 (1995)]. [PubMed] [Google Scholar]
- Wang, K., 2003. Mapping quantitative trait loci using multiple phenotypes in general pedigrees. Hum. Hered. 55: 1–15. [DOI] [PubMed] [Google Scholar]
- Wu, R. L., C. X. Ma, M. Lin and G. Casella, 2004. a A general framework for analyzing the genetic architecture of developmental characteristics. Genetics 166: 1541–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, R. L., Z. H. Wang, W. Zhao and J. M. Cheverud, 2004. b A mechanistic model for genetic machinery of ontogenetic growth. Genetics 168: 2383–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Q., I. Chazaro, J. Cui, C. Y. Guo, S. Demissie et al., 2003. Genetic analyses of longitudinal phenotype data: a comparison of univariate methods and a multivariate approach. BMC Genet. 4: S29. [DOI] [PMC free article] [PubMed] [Google Scholar]