

Abstract
We investigate the probability of fixation of a new mutation arising in a metapopulation that ranges over a heterogeneous selective environment. Using simulations, we test the performance of several approximations of this probability, including a new analytical approximation based on separation of the timescales of selection and migration. We extend all approximations to multideme metapopulations with arbitrary population structure. Our simulations show that no single approximation produces accurate predictions of fixation probabilities for all cases of potential interest. At the limits of low and high migration, previously published approximations are found to be highly accurate. The new separation-of-timescales approach provides the best approximations for intermediate rates of migration among habitats, provided selection is not too intense. For nonzero migration and relatively strong selection, all approximations perform poorly. However, the probability of fixation is bounded above and below by the approximations based on low and high migration limits. Surprisingly, in our simulations with symmetric migration, heterogeneous selection in a metapopulation never decreased—and sometimes substantially increased—the probability of fixation of a new allele compared to metapopulations experiencing homogeneous selection with the same mean selection intensity.
THE fate of new alleles is determined by selection, drift, migration, mutation, and the way these processes vary and interact over space and time. Heterogeneity is the hallmark of biology at all levels, including spatial variation in selection. Such spatial heterogeneity can substantially affect the course of evolution, including the probability of fixation of a new mutation.
One of the most striking results in population genetics is that a new mutation, even if it is favorable and arises in an extremely large population, has a low probability of fixation. This occurs because, when an allele is rare, it is present only in a few copies and therefore has a high probability of leaving no descendants due to the stochastic nature of reproduction. Haldane (1927, on the basis of a suggestion from Fisher 1922) showed for beneficial alleles in an ideal population that the probability of fixation of a new mutation was only ∼2s, where s is the fitness advantage of a heterozygote carrying the new mutation. Thus a new mutation with a 1% advantage has only an ∼2% chance of eventual fixation and a 98% chance of being lost. These results were generalized by Kimura (1957, 1962) to deleterious alleles, arbitrary dominance, and nonideal populations. Kimura showed that even deleterious alleles have a reasonable probability of fixation if the effective population size (Ne) is small enough, an idea that had been proposed qualitatively by Wright (1931). Kimura used a diffusion approach to solve the problem, and this has proven to be a powerful tool for subsequent refinements.
These results, as powerful as they are, apply only to closed panmictic populations and ignore the fact that most species are distributed over a spatial range, sometimes subdivided into local populations with nonrandom mating among populations. The first study to compute the probability of fixation in a spatially structured population was by Maruyama (1970), who found for the island model that the probability of fixation of an additively acting beneficial allele is also 2s, unchanged by the population structure. Nagylaki (1980, 1982) confirmed this result for all structured populations with conservative migration, that is, when migration does not change local population size. (These results also require that all demes contribute exactly equally to the next generation on a per capita basis.) Barton (1993) showed that this “invariance” result fails in two special cases that include local extinction and recolonization; in these cases the probability of fixation in a structured population was not well predicted by 2s.
Whitlock (2003) showed that Kimura's (1962) diffusion approximation could be extended to a broad range of structured populations, allowing for arbitrary dominance, arbitrary sign of selection, and complex metapopulation structure. These extensions produce good approximations to the probability of fixation with extinction and colonization, asymmetric migration (such as source-sink dynamics), stepping-stone migration (with isolation by distance), etc. For example, the probability of fixation of a new beneficial allele assuming “soft selection” (i.e., selection does not affect demography) is given by 2sNe/NTOT(1 − FST), where NTOT is the total census size of the metapopulation and FST is Wright's standardized description of the genetic variance among populations.
Importantly, all these authors assumed that the action of selection is uniform over space—that all localities and habitats share the same quantitative relationship between fitness and genotype. Many species span a diversity of habitats; therefore for many loci the pattern of selection is expected to vary over space.
Spatially variable selection adds substantial complication to the calculation of the probability of fixation of a new mutation in a structured population. Several approaches to addressing this challenge have been taken. Pollak (1966) used branching process calculations to find an expression for the probability of fixation of an allele that is on average beneficial. Nagylaki (1980) found in the limit of high migration (in other words, when there is little effect of the population's spatial structure) that the probability of fixation could be determined by substituting the arithmetic mean value of the selection coefficient into Kimura's panmictic formula (in effect, the structure of selection also does not matter). At the opposite extreme, Tachida and Iizuka (1991) found the probability of fixation in the weak migration limit, by assuming that migration was sufficiently rare that the mutation either fixes or is lost within each deme between migration events. In this limit, the probability of fixation of both deleterious and beneficial alleles is substantially higher than would be predicted by the mean selection coefficient. Recently, a more general analytical approach was proposed by Gavrilets and Gibson (2002), who derived formulas for the probability of fixation for both beneficial and deleterious alleles in a symmetric two-deme system.
All of these approaches have limitations. They assume that the new mutations act additively on fitness, an assumption that we also make in this article. The branching process results apply only to beneficial alleles (Pollak 1966). Tachida and Iizuka (1991) and Nagylaki (1980) have assumed either very weak or very strong migration, respectively. They all assume temporal homogeneity, meaning that the selection coefficients do not change locally or globally over time. Both Tachida and Iizuka (1991) and Gavrilets and Gibson (2002) assumed that there are only two demes in the system, and the latter article furthermore assumes that the two demes are equal in size with symmetric migration between them.
In this article, we review and extend results on the probability of fixation of a new mutation in a heterogeneous selective environment. We have four goals. First, we evaluate existing approximations, using a mixture of simulation and exact Markov chain calculations. Most of the methods in the literature perform well given their stated assumptions, although there are exceptions. Second, we extend these results to more complex spatial population structures, building on results found in Whitlock (2003) for uniform selection with complex population structure. We also relax the symmetry assumptions of previous models and develop new theory that allows calculation of the probability of fixation with local extinction, source-sink dynamics, stepping-stone structure, etc. Third, we seek improved approximations for the probability of fixation of a new mutation that is deleterious on average, a case for which we find previous approximations to be inaccurate. Finally, we use the best approximations to evaluate the overall effect of spatially variable selection on the probability of fixation relative to the fixation probability in a uniform environment with the same mean selection intensity, with a surprising conclusion. Spatial heterogeneity in selection increases the probability of fixation of new alleles, relative to an allele with the same mean selection coefficient.
METHODS
In this section, we describe a basic model of a spatially structured population that occupies two ecologically distinct habitats. We wish to infer the probability of global fixation of a new mutation that appears in one of the habitats. We then present several methods that can be used to assess this probability. Two of the methods, Monte Carlo simulation and finite Markov chain methods, are accurate but also highly computationally intensive. Simulations, as tested by comparison to exact Markov chain calculations, were used to test the accuracy of various approximations to the fixation probability. These approximations include numerical solution of a two-dimensional diffusion equation and three analytic approximations of the solution of this equation. One of the analytic approximations is a new approach based on separation of timescales and therefore is developed in its own subsection. We also extend all the approximate methods for computing probabilities of fixation, including those previously published, to allow unequal habitat sizes, asymmetric migration, and multiple demes within each habitat type.
Definitions:
We limit our attention here to a case where there are only two kinds of habitats, labeled 1 and 2. These habitats may differ in their selection pressures, but selection acts additively within each. We consider a locus at which an ancestral allele and a novel mutant allele are segregating. The fitnesses of the three diploid genotypes depend on two selection coefficients s1 and s2, such that the relative fitnesses of the ancestral homozygote, heterozygote, and new homozygote are given by 1:1 + si:1 + 2si in habitat i. Thus, if si > 0, then the allele is favored by selection in habitat i. If si < 0, then the new allele is deleterious in the ith habitat. The selection coefficients are assumed constant over time. In practice, we assume that the si are small (see below).
We write pi for the mutant allele frequency in habitat i, and qi = 1 − pi. The number of diploid individuals in habitat i is held constant and defined as Ni. It is convenient to keep track of the proportion of all individuals that live in habitat 1, and for this we use φ, which by definition is N1/N, where N = N1 + N2. The variance effective size of the whole system is written as Ne. Migration occurs between the two habitats, with the backward migration rate (i.e., the proportion of individuals in population i that are immigrants each generation) given by mi.
We also use averages of many of these quantities weighted by habitat frequency, which is indicated by an overbar. For example, the mean selection coefficient is given by 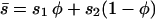 , and the mean allele frequency is given by
, and the mean allele frequency is given by 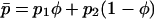 . Finally, the (unweighted) difference in allele frequency between the two habitats is given by
. Finally, the (unweighted) difference in allele frequency between the two habitats is given by 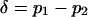 . Note that if the system consists of two demes, the population can be completely described in terms of
. Note that if the system consists of two demes, the population can be completely described in terms of 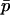 and δ because
and δ because
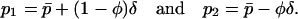 |
(1) |
Accurate methods: simulations and finite Markov chains:
The simulation program used in Whitlock (2003) was modified to allow heterogeneous selection (two different habitats with distinct selection coefficients in each). Selection can favor or disfavor an allele, and this can be different in the two different habitat types. The program also allows for a wide range of possible population demography, ranging from the simple two-deme symmetric migration case to multiple demes with local extinction and recolonization, asymmetric migration, source-sink dynamics, isolation by distance, etc.
These simulations have been extensively checked for the uniform selection case. Furthermore, the new simulations with variable selection were validated using an exact method—finite Markov chain methods (e.g., Kemeny and Snell 1976; Ewens 1979)—to compute fixation probabilities in the two-deme model (details not shown). In practice, the Markov chain calculations (implemented in the program Matlab) were computationally feasible only for demes containing <30 individuals. Extensive checks were performed to compare simulation results to the exact Markov chain results, to test the accuracy of the simulation program. In all cases, the simulations matched the Markov chain results within the appropriate confidence interval.
High- and low-migration limit approximations:
Previous authors have already determined the probability of fixation in the limits of very high or very low migration rates, and we briefly review their findings here. Nagylaki (1980) found that in the limit of high migration (m1, m2 ≫ |s1|, |s2|), the probability of fixation can be determined from the mean selection coefficient 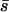 , by
, by
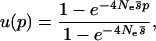 |
(2) |
where p is the initial frequency of the allele calculated over the whole metapopulation. In the high-migration limit, the correlation between the location of an allele and its selection coefficient is negligible, so any allele experiences selection in proportion to the mean selection coefficient.
In contrast, in the limit of low migration, each new allele is either fixed or lost in a habitat before migration can carry even a single copy to the other habitat. In this case the probability of fixation is
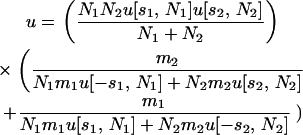 |
(3) |
(Equation 6 of Tachida and Iizuka 1991), where u[si, Ni] is the probability of fixation of a single allele in habitat i with selection coefficient si and effective size Ne,i. This u[si, Ni] is given by Kimura's (1964) formula,
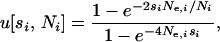 |
(4) |
where Ne,i is the local variance effective population size.
Two-deme approximations:
The probability of fixation in the two-deme system with initial frequencies p1 and p2, respectively, can be approximated for general migration rates by the solution u(p1, p2) of a diffusion equation. By definition, N1 = φN and N2 = (1 − φ)N, where N is the total population size and φ is the relative frequency of patch 1. In addition, assume that 2Nsi and 2Nmi have finite limits as N → ∞ (call these respective limits Si and Mi). Rescaling time so that 1 unit corresponds to 2N generations, letting N → ∞, and ignoring higher-order terms yields the following boundary value diffusion problem for u(p1, p2), the approximate probability of fixation in a two-deme system with initial frequencies p1 and p2,
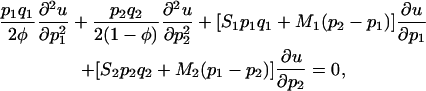 |
(5) |
with boundary conditions u(0, 0) = 0 and u(1, 1) = 1. Except for the rescaling, this is identical to Equation 8 in Tachida and Iizuka (1991). Note too that we have used the alternate parameterization N = N1 + N2 and 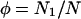 to emphasize that N1 and N2 cannot be varied separately. In addition, solutions to (5) should yield good approximations provided N is large and the selection coefficients and migration rates are all of order N−1 (Karlin and Taylor 1981). To solve this system numerically, we used finite differencing (Press et al. 1992) and sparse matrix functions of the software package Matlab (Mathworks, Natick, MA).
to emphasize that N1 and N2 cannot be varied separately. In addition, solutions to (5) should yield good approximations provided N is large and the selection coefficients and migration rates are all of order N−1 (Karlin and Taylor 1981). To solve this system numerically, we used finite differencing (Press et al. 1992) and sparse matrix functions of the software package Matlab (Mathworks, Natick, MA).
We obtained analytic approximations to u(p1, p2) by extending the singular perturbation approach described in the Appendix of Gavrilets and Gibson (2002) to Equation 5, which now accounts for asymmetric migration and unequal population sizes. If the new mutant is at a net selective advantage over both demes (i.e., if 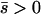 ), then the probability of fixation given frequencies p1 and p2 is approximately
), then the probability of fixation given frequencies p1 and p2 is approximately
 |
(6a) |
where α and β are positive solutions of the algebraic system
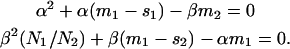 |
(6b) |
If the new mutant is net deleterious (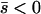 ), then the approximate solution is
), then the approximate solution is
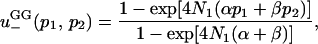 |
(7a) |
where α and β are positive solutions of
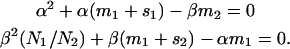 |
(7b) |
Finally, if the mutant is net neutral among the demes (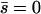 ), the approximate probability of fixation is
), the approximate probability of fixation is
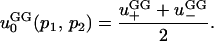 |
(8) |
Equations 5–8 reduce to expressions (7)–(11) of Gavrilets and Gibson (2002) when N1 = N2 and m1 = m2.
Separation of timescales: diffusion approximations based on the mean allele frequency:
The above approximations either were developed for migration extremes or perform poorly for alleles that are net deleterious (see two-deme results below). In this section, we seek a new diffusion approximation for intermediate migration rates (where m is of order s or more) that performs well for both net deleterious and net beneficial alleles.
With only two habitats, it is possible to rewrite the two variables used in the two-dimensional diffusion process (p1 and p2), without any loss of information, in terms of the overall mean frequency of the mutant allele, 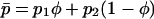 , and among-deme frequency difference,
, and among-deme frequency difference, 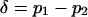 (see Equation 1). A key observation underlying our new approach is that, for many conditions, these two quantities are closely related, such that δ can be well approximated given
(see Equation 1). A key observation underlying our new approach is that, for many conditions, these two quantities are closely related, such that δ can be well approximated given 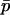 , reducing the dimensionality of the problem. This approximation should work well if population size is large (so that the metapopulation should evolve on a path close to that expected deterministically) and if δ initially changes rapidly relative to the changes in
, reducing the dimensionality of the problem. This approximation should work well if population size is large (so that the metapopulation should evolve on a path close to that expected deterministically) and if δ initially changes rapidly relative to the changes in 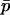 (which in practice requires that the migration rate between populations is large relative to the magnitude of selection). This latter assumption is the “separation of timescales” in the title of this section: that the difference in allele frequency between populations will approach its quasi-equilibrium quickly relative to the rate of change in the mean allele frequency. Under these circumstances, a one-dimensional diffusion based on
(which in practice requires that the migration rate between populations is large relative to the magnitude of selection). This latter assumption is the “separation of timescales” in the title of this section: that the difference in allele frequency between populations will approach its quasi-equilibrium quickly relative to the rate of change in the mean allele frequency. Under these circumstances, a one-dimensional diffusion based on 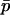 may be sufficient to predict the fixation dynamics of the two-habitat system.
may be sufficient to predict the fixation dynamics of the two-habitat system.
[Note that our sense of separation of timescales is different from that typical of the literature of the coalescent in structured populations. Our approach shares more in common with the quasi-linkage equilibrium approach (Barton and Turelli 1991).]
Kimura (1962) derived the one-dimensional diffusion equations necessary to predict the probability of fixation for alleles with arbitrary dominance, treating both beneficial and deleterious alleles and accounting for the differences between the effective size and the census size of a population. Kimura's results dealt exclusively with single panmictic populations, but Whitlock (2003) has shown that these equations can be extended to address spatially structured populations as well. The equations require that we be able to write the mean and the variance of the change in mean allele frequency per generation (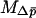 and
and 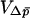 , respectively). It also requires that these quantities be time homogeneous, that is, that the mean and variance of change are functions of
, respectively). It also requires that these quantities be time homogeneous, that is, that the mean and variance of change are functions of  alone. With populations that are structured over space, this assumption is never strictly true, but Whitlock (2003) showed by simulation that violations of the strict assumptions of the diffusion were often not important (namely, when the strength of selection was not much greater than the typical immigration rate to demes) and the one-dimensional diffusion equations would work quite well with simple modifications.
alone. With populations that are structured over space, this assumption is never strictly true, but Whitlock (2003) showed by simulation that violations of the strict assumptions of the diffusion were often not important (namely, when the strength of selection was not much greater than the typical immigration rate to demes) and the one-dimensional diffusion equations would work quite well with simple modifications.
We seek to develop a one-dimensional diffusion approximation based on 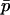 . This requires that we express, to a reasonable level of accuracy, the expected changes in allele frequency within demes as a function solely of the mean overall allele frequency:
. This requires that we express, to a reasonable level of accuracy, the expected changes in allele frequency within demes as a function solely of the mean overall allele frequency:
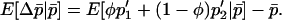 |
(9) |
Within demes, allele frequencies after selection are
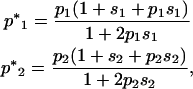 |
(10) |
where the asterisk indicates the value after selection. Subsequent migration is expected to cause allele frequencies to change to, by definition, 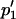 and
and 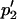 :
:
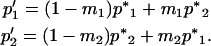 |
(11) |
Recall from (1) that the allele frequencies of the two demes, at any time, can be written as functions of the mean allele frequency 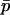 and the between-deme difference in allele frequency, δ. The difference δ can be further broken down into the deterministically expected difference in allele frequency between the demes given
and the between-deme difference in allele frequency, δ. The difference δ can be further broken down into the deterministically expected difference in allele frequency between the demes given 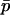 , which we call
, which we call 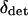 , and the chance deviation around the deterministic expectation, which we call δdrift. So
, and the chance deviation around the deterministic expectation, which we call δdrift. So 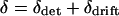 .
.
We now argue that 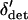 and the first conditional moments of
and the first conditional moments of 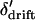 can be well approximated using functions of
can be well approximated using functions of 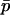 alone under certain conditions. First, note that
alone under certain conditions. First, note that
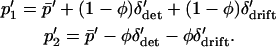 |
(12) |
Using expressions (10) expanded to second order in  , taking the expectations of p′1 and p′2 in (11) conditional on
, taking the expectations of p′1 and p′2 in (11) conditional on 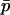 , and applying the substitutions (12) will result in terms involving
, and applying the substitutions (12) will result in terms involving 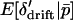 ,
, 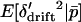 , and
, and 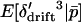 . The conditional expectation of
. The conditional expectation of 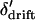 is 0, but the expectations of the squared and cubed terms are nonzero in finite populations. Whitlock (2002) showed that when selection is weak relative to migration (|si| ≤ mi), the deviations around the expected allele frequency can be approximated in terms of FST. To later allow for subdivision of the habitats into local populations, we refer to the F-statistic describing deviations in habitat allele frequency from the expected allele frequency as FHT. Following Whitlock (2002), the expected values
is 0, but the expectations of the squared and cubed terms are nonzero in finite populations. Whitlock (2002) showed that when selection is weak relative to migration (|si| ≤ mi), the deviations around the expected allele frequency can be approximated in terms of FST. To later allow for subdivision of the habitats into local populations, we refer to the F-statistic describing deviations in habitat allele frequency from the expected allele frequency as FHT. Following Whitlock (2002), the expected values 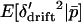 , and
, and 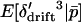 are approximately
are approximately
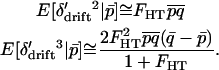 |
(13) |
These terms are calculated from the conditional expectations of (pi′ + δdrift)2 and (pi′ + δdrift)3 and by solving for the required terms.
Because 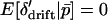 ,
, 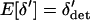 each generation. As we argue below, under some conditions δdet changes at a rate that is fast relative to the rate of change in
each generation. As we argue below, under some conditions δdet changes at a rate that is fast relative to the rate of change in 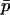 . Given this, we can find a quasi-equilibrium value for δdet, and hence δ, given a value of
. Given this, we can find a quasi-equilibrium value for δdet, and hence δ, given a value of 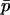 . This is the separation of timescales in the title of this section. We find the deterministic difference between populations by setting the δdrift terms identically to zero and solving
. This is the separation of timescales in the title of this section. We find the deterministic difference between populations by setting the δdrift terms identically to zero and solving
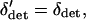 |
(14) |
where 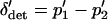 and
and 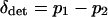 . Solving this equation and expanding in a Taylor series to order |si|2, we find an approximation for the difference in allele frequencies between demes given
. Solving this equation and expanding in a Taylor series to order |si|2, we find an approximation for the difference in allele frequencies between demes given 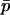 ,
,
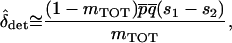 |
(15) |
where 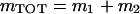 . Simulations suggest that approximation (15) for
. Simulations suggest that approximation (15) for 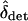 works well when the total rate of migration is greater than the maximum strength of selection,
works well when the total rate of migration is greater than the maximum strength of selection, 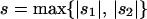 , but fails when migration rates are small relative to s. Moreover, it can be shown using arguments parallel to those in Nagylaki (1992, pp. 177–181) that if mTOT > s and migration is approximately symmetric, then δ will quickly converge to
, but fails when migration rates are small relative to s. Moreover, it can be shown using arguments parallel to those in Nagylaki (1992, pp. 177–181) that if mTOT > s and migration is approximately symmetric, then δ will quickly converge to 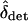 . The approximation should fail when migration rates are small relative to the maximal strength of selection. Developing more rigorous approximations of
. The approximation should fail when migration rates are small relative to the maximal strength of selection. Developing more rigorous approximations of 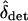 represents a fruitful avenue for future research.
represents a fruitful avenue for future research.
We can now write the expected change in 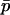 over a generation as a function of
over a generation as a function of  ,
, 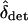 , and FHT. FHT is not much affected by
, and FHT. FHT is not much affected by 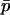 , and under the conditions given in the previous paragraph we can write
, and under the conditions given in the previous paragraph we can write 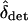 as a function of constants and
as a function of constants and 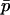 . This means that, when the strength of selection is weak compared to migration, we can write a recurrence equation for
. This means that, when the strength of selection is weak compared to migration, we can write a recurrence equation for 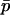 that depends only on
that depends only on 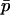 , allowing a one-dimensional diffusion approach to the probability of fixation. Expanding (9) in a series after substituting Equations 11, 12, 13, and 15, and ignoring terms of order mi2, si2, misi, or higher, we find
, allowing a one-dimensional diffusion approach to the probability of fixation. Expanding (9) in a series after substituting Equations 11, 12, 13, and 15, and ignoring terms of order mi2, si2, misi, or higher, we find
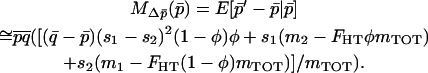 |
(16) |
The variance term 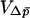 is given by
is given by
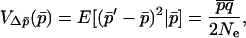 |
(17) |
where Ne is the effective size of the whole system (given by Whitlock and Barton 1997 for most cases). This term is approximately unaffected by heterogeneous selection. The major effects of heterogeneous selection appear in the mean allele frequency change term (16).
With 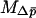 and
and  thus defined, Kimura's (1962) one-dimensional diffusion results can then be employed using
thus defined, Kimura's (1962) one-dimensional diffusion results can then be employed using 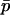 in place of p to approximate the probability of fixation. The approximate probability of fixation is
in place of p to approximate the probability of fixation. The approximate probability of fixation is
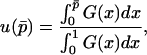 |
(18) |
where 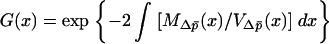 .
.
This derivation has required many assumptions, in particular about the predictability of δ from mean allele frequency, the use of neutral FST, and the applicability of the diffusion approximation. Simulations were used to test whether these various assumptions severely compromise its accuracy or precision.
Note that at no point in the derivation was any assumption made about the sign of the mean selection coefficient. Equation 18 is therefore valid for both beneficial and deleterious alleles.
Extensions to multiple demes:
The approximations above can be extended to multiple demes, by allowing each of the two selective habitats to contain more than one deme. For the separation-of-timescales approach, we again need to specify the conditional mean and variance of change in the average allele frequency, 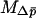 and
and 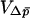 , respectively. The effective population size of such a system can be found using the equations in Whitlock and Barton (1997) for many cases, giving the necessary Ne to calculate
, respectively. The effective population size of such a system can be found using the equations in Whitlock and Barton (1997) for many cases, giving the necessary Ne to calculate 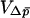 . The equation for
. The equation for 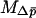 is different in the multiple-deme case than in the two-deme case. If we consider selection within each habitat to be acting independently (between migration events) from selection in the other habitat, then the response to selection over all the demes within each habitat can be described using the equations in Whitlock (2003) for homogeneous selection in a structured population. With soft selection and additive gene action, as has been assumed here, the expected change in allele frequency for each habitat is reduced by a factor (1 − FSH), where FSH is Wright's fixation index calculated for demes within a habitat. Assuming that the population structure in the two habitats is similar, this results in the effective strength of selection within each habitat being reduced by a factor (1 − FSH), such that the effective strength of selection in habitat i is
is different in the multiple-deme case than in the two-deme case. If we consider selection within each habitat to be acting independently (between migration events) from selection in the other habitat, then the response to selection over all the demes within each habitat can be described using the equations in Whitlock (2003) for homogeneous selection in a structured population. With soft selection and additive gene action, as has been assumed here, the expected change in allele frequency for each habitat is reduced by a factor (1 − FSH), where FSH is Wright's fixation index calculated for demes within a habitat. Assuming that the population structure in the two habitats is similar, this results in the effective strength of selection within each habitat being reduced by a factor (1 − FSH), such that the effective strength of selection in habitat i is 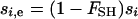 . Equation 16 can be used with this substitution for si. Note that, as in Whitlock (2003), FSH is calculated weighted by local population size.
. Equation 16 can be used with this substitution for si. Note that, as in Whitlock (2003), FSH is calculated weighted by local population size.
TWO-DEME RESULTS
Several different domains are defined by the assumptions of different approaches that determine the best approximation for solutions to a particular case, on the basis of simulation results (Table 1). These domains are delimited by whether the new allele is beneficial or deleterious on average and by how strong selection is relative to the rate of migration. We discuss the cases of beneficial and deleterious alleles separately.
TABLE 1.
Theoretical parameter domains of applicability for, and actual performance of, two-deme approximations to the probability of fixation
| Method
|
||||
|---|---|---|---|---|
| Domain | High-migration limit (Equation 2) | Low-migration limit (Equations 3 and 4) | Singular perturbation (Equations 6–8) | Separation of timescales (Equations 16–18) |
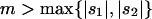 |
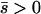 a a
|
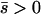 |
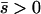 |
|
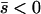 a a
|
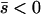 b b
|
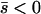 |
||
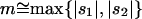 |
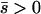 |
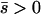 |
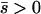 |
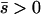 a a
|
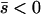 |
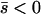 |
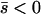 b b
|
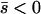 a a
|
|
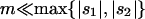 |
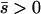 a a
|
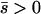 |
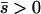 b b
|
|
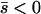 a a
|
 b b
|
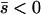 b b
|
||
Best, or most convenient, approximation within the parameter domain.
Poor performance by method within the parameter domain.
Net beneficial alleles:
When alleles are on average beneficial (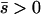 ), the relative performance of four different approximations (strong-migration approximation, weak-migration approximation, separation of timescales, and singular perturbation) varies as a function of migration rate (Figure 1). The relative power of selection and migration can be divided roughly into three regimes: the case where migration is stronger than selection, the case where the allele would be stably polymorphic in an infinite population because local selection is stronger than migration, and the limiting case of very weak migration. Different techniques work better in these three different regimes. We discuss them in turn.
), the relative performance of four different approximations (strong-migration approximation, weak-migration approximation, separation of timescales, and singular perturbation) varies as a function of migration rate (Figure 1). The relative power of selection and migration can be divided roughly into three regimes: the case where migration is stronger than selection, the case where the allele would be stably polymorphic in an infinite population because local selection is stronger than migration, and the limiting case of very weak migration. Different techniques work better in these three different regimes. We discuss them in turn.
Figure 1.—

The probability of fixation of a net beneficial allele as a function of migration rate in a two-deme model. The dots present results from 107 simulations. The bottom horizontal dashed line is the high-migration limit (Nagylaki 1980); the top dashed line is the low-migration limit due to Tachida and Iizuka (1991); the solid line shows the results from the separation-of-timescale approximation; and the dotted line shows the results of the singular perturbation approximation, which in this case is exactly equivalent to the results of Gavrilets and Gibson (2002). For these cases, the migration rate was symmetric between the two demes, and the other parameters used were N1 = N2 = 50, s1 = 0.01, and s2 = −0.005.
As Nagylaki (1980) showed, when migration is common the fate of the allele is determined by its mean selection coefficient (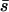 ). This seems intuitive: with large migration rates an allele moves rapidly from habitat to habitat, and its success would therefore be determined by its effects averaged over these habitats. In other words, with large migration rates, little or no association builds between habitat type and allelic type. Three methods should apply to this case (using the mean selection coefficient in standard diffusion equations, the singular perturbation approximation, and the separation-of-timescales approach derived above), and all work relatively well, but using the mean selection coefficient in the standard diffusion expression (i.e., the high-migration limit) is most straightforward.
). This seems intuitive: with large migration rates an allele moves rapidly from habitat to habitat, and its success would therefore be determined by its effects averaged over these habitats. In other words, with large migration rates, little or no association builds between habitat type and allelic type. Three methods should apply to this case (using the mean selection coefficient in standard diffusion equations, the singular perturbation approximation, and the separation-of-timescales approach derived above), and all work relatively well, but using the mean selection coefficient in the standard diffusion expression (i.e., the high-migration limit) is most straightforward.
When the strength of selection is large relative to the migration rate, a new allele is more likely to fix than would have been expected by its mean selection coefficient. This is a result of the evolved association between habitat type and allelic identity. Selection causes alleles to be more common in localities where they are favored relative to their overall frequencies (Figure 2), which means that alleles are somewhat more likely to persist locally than would be expected if alleles were distributed at random over the range of the species. With lower rates of migration, these associations build up to sufficiently high levels as to qualitatively affect the probability of fixation of new alleles. This effect can be seen on the left-hand side of Figure 1, especially where the rate of migration is lower than the minimum magnitude of selection for or against the allele.
Figure 2.—

The difference in allele frequency between habitats, as a function of the overall mean allele frequency. A positive number for this difference indicates that the deme where the allele is favored has a higher frequency of the allele. For this example, there are two equal, large demes with symmetric migration rate 0.025. s1 = 0.01 and s2 = −0.005. The graph traces the deterministic trajectory from an initial state when the allele is rare in both populations to when the allele has reached global fixation.
For migration rates about equal to either selection coefficient, the separation-of-timescales approach works better (Figure 1.) If migration rates are low enough (such that the locus would be stably polymorphic in an infinite population, with the conditions given in Karlin 1982 or Gavrilets and Gibson 2002), then the separation-of-timescales approach fails drastically. This is expected, because the assumptions of the separation-of-timescales approach require that the between-deme difference in allele frequency evolves quickly to quasi-equilibrium relative to changes in the mean allele frequency, which is not the case when migration rates are low. The singular perturbation approximation is also less accurate than the separation-of-timescales approximation when the local effective population size is not large (Figure 3).
Figure 3.—

The effects of increasing symmetric population size on the various approximations for the two-deme model. The meaning of the lines is as in Figure 1, with the dashed line indicating the high-migration limit. Migration is symmetric at m1 = m2 = 0.025, with s1 = 0.01 and s2 = −0.005.
Finally, in the limit as the migration rate approaches zero, the approximation of Tachida and Iizuka (1991) works very well. This approximation assumes that the fate of an allele within a deme is resolved between successive migration events, and so it works well only for very low migration rates, perhaps on the order of those expected between incipient species.
For all of the examples we have simulated, the relationship between probability of fixation and migration rate is monotonic, with the probability of fixation bounded between the low-migration and high-migration limit values. We hypothesize that these therefore represent likely upper and lower bounds, respectively, for intermediate migration levels.
Net deleterious alleles:
When the average effect of the new allele is deleterious (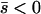 ), the allele is not expected to fix deterministically, but detrimental alleles sometimes fix due to the stochastic effects of genetic drift. Again, three domains, defined by the relative strengths of migration and selection, dictate which approximations are most accurate in determining the probability of fixation of the new allele (Figure 4 and Table 1). With strong migration (
), the allele is not expected to fix deterministically, but detrimental alleles sometimes fix due to the stochastic effects of genetic drift. Again, three domains, defined by the relative strengths of migration and selection, dictate which approximations are most accurate in determining the probability of fixation of the new allele (Figure 4 and Table 1). With strong migration (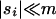 ), the process of fixation is well described by Kimura's single-population diffusion results, using the mean selection coefficient and mean allele frequency.
), the process of fixation is well described by Kimura's single-population diffusion results, using the mean selection coefficient and mean allele frequency.
Figure 4.—

The probability of fixation of net-deleterious alleles as a function of migration rate. The meanings of the lines are the same as in Figure 1. The high- and low-migration limits (dashed lines) work well in their respective domains, and the separation-of-timescales approximation (solid curve) is adequate for |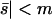 , but the singular perturbation approximation (dotted curve) is unsatisfactory. N1 = N2 = 50, s1 = 0.01, and s2 = −0.02.
, but the singular perturbation approximation (dotted curve) is unsatisfactory. N1 = N2 = 50, s1 = 0.01, and s2 = −0.02.
For migration rates of approximately the same order as the mean strength of selection, the separation-of-timescales approximation works fairly well. Like the case with net beneficial alleles, the probability of fixation is somewhat higher than would be predicted from the mean selection coefficient alone, again because the alleles are relatively more frequent in habitats in which they are favored, causing these habitats to affect allele frequency change more than the deleterious habitats. The separation-of-timescales approach fails, however, when the conditions are consistent with a stable internal equilibrium of allele frequency, i.e., when in an infinite population the allele would remain indefinitely polymorphic at a balance between migration and selection. Fixation probabilities in this range of parameter space can be approached only via numerical solutions to the full two-dimensional diffusion approximation, or by exact Markov chain calculations (feasible only for very small populations), or by simulation. Each of these is computer intensive, although the numerical solution of the diffusion can be computed quickly even on a personal computer.
The singular perturbation approximation due to Gavrilets and Gibson (2002) can give qualitatively incorrect results for alleles that are on average deleterious (Figure 4). In fact, the equations in Gavrilets and Gibson (2002) give the probability of fixation conditioned on the locality of the introduction of the new allele, and these equations yield the counterintuitive (and counterfactual) conclusion that the allele is more likely to fix if it is initially introduced into the habitat where it is selected against than it would be if introduced into the favorable habitat. This result is incorrect, as we have observed by exact Markov chain calculations, simulation, and numerical solutions of the two-dimensional diffusion equation (results not shown).
Finally, if the migration rate is very small, the allele is either fixed or lost in each population between migration events. In this case, equation 6 of Tachida and Iizuka (1991) again provides extremely accurate predictions of the probability of fixation.
Unequal habitat size:
One extension made in this article is to allow for unequal population size between the two habitats, a case treated in the past only for the limits of very large (Nagylaki 1980) or very small (Tachida and Iizuka 1991) migration. As shown in Figure 5, the separation-of-timescales approach is very successful in dealing with asymmetric migration, and the extension of the singular perturbation approximation is moderately successful. For the examples presented in Figure 5, a constant fraction of the individuals in each deme stay where they were born, and the remainder migrate to the other deme. (In other words, the forward migration rate was held constant.) Following this dispersal, the population sizes of each habitat are then regulated to their carrying capacities: 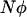 and
and 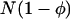 . This means that the backward migration rate into the habitat with the lower population size is higher than the migration rate into the larger habitat. In these cases, the diffusion approximation based on mean allele frequency using the separation of timescales predicts the probability of fixation very well.
. This means that the backward migration rate into the habitat with the lower population size is higher than the migration rate into the larger habitat. In these cases, the diffusion approximation based on mean allele frequency using the separation of timescales predicts the probability of fixation very well.
Figure 5.—

The probability of fixation in the two-deme model as a function of the relative population size of the two demes. The lines have the same meaning as in Figure 1, with the dashed line indicating the high-migration limit. Emigration rates are held constant for each individual at 0.025, which has the effect of increasing the backward migration rate for the smaller population relative to the larger population. Parameter values used are: N1 + N2 = 200, s1 = 0.01, and s2 = −0.005.
MULTIPLE-DEME RESULTS
With multiple demes, a much broader array of possible demographic structures becomes possible: local demes can go extinct and be recolonized; some demes can be more productive than others; populations can be geographically arranged in a wide variety of ways, etc. We used simulations to test the value of the separation-of-timescales approximation and the singular perturbation approximations as extended in the previous sections to account for multiple demes and more complex metapopulation structure. Here we consider cases in which dispersal among demes across habitat boundaries is not different from dispersal among demes within habitats.
Figure 6 shows that with local extinction and colonization, both approaches perform fairly well for net beneficial alleles. Moreover, in all cases, if the rate of migration between habitats is large relative to the magnitudes of the selection coefficients, using the mean selection coefficient in the equations of Whitlock (2003) gives accurate predictions of the probability of fixation. For metapopulations of this size, deleterious alleles have a negligibly small probability of fixation unless their net selection coefficients are small, in which case the high migration limit (using 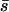 ) is appropriate.
) is appropriate.
Figure 6.—

The probability of fixation with the extinction/colonization model and multiple demes, as a function of the proportion of demes that occur in habitat 1. Lines follow the same meanings as in previous figures. Here there were 100 demes of 100 individuals each, with migration into each deme equal to 0.025. Other parameters are: s1 = 0.02, s2 = −0.01; number of individuals colonizing new demes, four; extinction rate 0.05 per generation; and the probability of common origin of the colonists, 0 (see Whitlock and McCauley 1990). The singular perturbation approximation (dotted line) and the separation-of-timescales (solid line) approximation work reasonably well, while the high-migration approximation (dashed line) quantitatively fails for these parameters.
The contributions of different demes can be very asymmetric. Figure 7 shows the probability of fixation as a function of the relative emigration rates of “source” populations and “sink” populations, where the former have high contributions to subsequent generations and the latter have lower contributions. In these examples, which correspond to similar examples in Whitlock (2003) but with variable selection, each deme has a constant size, but the source populations contribute more individuals to the migrant pool. The FST and effective population size were calculated as in Whitlock (2003), following the eigenvalue approach of Nagylaki (1980). As the contribution of the sinks declines, the effective population size of the whole metapopulation also is reduced, causing the probability of fixation to be reduced for net beneficial alleles and increased for deleterious alleles compared to a metapopulation without source-sink structure. Again, the separation-of-timescales approach is very successful in predicting the probability of fixation in this case (Figure 7).
Figure 7.—

Probability of fixation with a source-sink population in a heterogeneous environment uncorrelated with demography. The model and parameters here are the same as in Figure 3 of Whitlock (2003) except that selection in one habitat was s1 = 0.01 and in the other equally common habitat was s2 = −0.005. There were 100 demes each with 100 individuals, and the immigration rate into the sources was 0.2, while for the sinks the immigration rate was 0.25. Sink populations contributed to the migrant pool in proportion to the value on the x-axis.
DISCUSSION
Environmental heterogeneity can substantially affect the probability of fixation of new alleles. If environments are differentially productive, then the effective population size will be reduced relative to the census population size and the probabilities of fixation of new alleles are reduced for beneficial alleles and enhanced for deleterious alleles (Whitlock 2003). This is true whether selection on these alleles is heterogeneous across space or not. In addition, as we have shown by induction from simulations in this article, if the selection coefficient of a new allele varies across space, then its probability of fixation is equal to or larger than would be expected on the basis of the allele's mean selective effect over the range of values we considered in our simulations. This result holds regardless of the strength of migration or selection, although the magnitude of this “fixation boost” depends on the circumstances. We have confirmed this result only for cases with symmetric migration. The enhancement is small when migration is high and quite substantial in the limit of low migration.
Our findings, which emerge from an assortment of analytical and numerical methods, show that accurately predicting the probability of fixation of new mutations in heterogeneous environments is complicated; no single approximation works best under all circumstances (Table 1). It is useful perhaps to attempt to give a heuristic division of possible cases with an idea of which mathematical approach performs best. In the limit of low migration rates (when each deme becomes fixed for a single allele in the time between migration events), the approach developed by Tachida and Iizuka (1991) works very well for all parameter values we have tested. At the other extreme, in the limit of high migration rates, such that the migration rate is much higher than the maximum magnitude of selection across habitats, the system behaves approximately as though the alleles experienced the average selection coefficient everywhere, as suggested by Nagylaki (1980). These low- and high-migration limits seem to set the upper and lower bounds, respectively, for the probability of fixation.
For intermediate migration rates, neither of these limits is particularly accurate (nor, of course, were they claimed to be). Other approximations do better. In particular, the singular perturbation approximation of the two-dimensional PDEs developed by Gavrilets and Gibson (2002) and extended here works fairly well for alleles that on average are beneficial, but it can fail badly for alleles with a net deleterious effect. By comparison, the separation-of-timescales argument developed in this article works well for either beneficial or deleterious alleles, provided that the strength of selection is not strong relative to the migration rates. However, this approximation also fails badly for weak migration (approximately corresponding to cases with stable deterministic internal polymorphisms). This leaves a case for which there is no acceptable analytic approximation: when the new mutation is on average deleterious (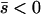 ) and migration is somewhat weaker than selection (max mi < min|si|). The probability of fixation, however, seems to be bounded above by the low-migration limit and bounded below by the high-migration limit.
) and migration is somewhat weaker than selection (max mi < min|si|). The probability of fixation, however, seems to be bounded above by the low-migration limit and bounded below by the high-migration limit.
It is not clear to what extent adaptation is limited by the probability of fixation of new mutations, because we do not know enough about mutation rates or the distribution of new mutational effects. If mutation rates are limiting the rate of evolution, then the deviations in probability of fixation caused by environmental heterogeneity could have some effect on the rate of adaptation. For sufficiently strong migration, the rate of change in mean fitness across a heterogeneous environment should be determined by the mean selective effect of new alleles across all habitats (i.e., by 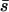 ). For weaker migration, though, alleles are more likely to fix than predicted by their mean selective effect. One interesting ramification of this is that alternate alleles at the same locus can all have elevated probabilities of fixation depending on which one is already fixed in the population. While this does not in itself bias the mean fitness at equilibrium, it would cause alleles to replace each other faster than expected by uniform selection theory. For metapopulations of any size, however, the rate of these substitutions will be very slow, because the time required for the fixation of alleles maintained by balancing spatial selection can be very long.
). For weaker migration, though, alleles are more likely to fix than predicted by their mean selective effect. One interesting ramification of this is that alternate alleles at the same locus can all have elevated probabilities of fixation depending on which one is already fixed in the population. While this does not in itself bias the mean fitness at equilibrium, it would cause alleles to replace each other faster than expected by uniform selection theory. For metapopulations of any size, however, the rate of these substitutions will be very slow, because the time required for the fixation of alleles maintained by balancing spatial selection can be very long.
In this article we have made several assumptions that limit the generality of our conclusions. Throughout we have assumed that the selective effects of these alleles do not influence the demographic properties of the demes in which they occur (i.e., soft selection). Clearly, in some cases, adaptation to a local environment will affect the productivity of the population in that habitat, and the dynamics of fixation will be changed. Moreover, we have assumed that the demographic properties of each deme are independent of its habitat type, particularly in the multiple-deme extensions in the latter part of this article. Clearly the metapopulation properties of demes can—and probably do—covary with habitat type (e.g., extinction rates may be lower in higher-quality habitats). In the simulations, we have focused on cases with symmetric migration, and we have not yet explored the case of weak, asymmetric migration. It is not yet clear whether the increase in probability of fixation would appear in these cases, although we suspect that it would. Finally, and perhaps most importantly, we have examined the effects of only one locus segregating in the population. This is clearly unrealistic in cases of multiple habitats connected by limited migration, when many selectively important alleles are likely to be segregating in the metapopulation at the same time. Linkage disequilibria between multiple loci all involved in local adaptation can result in correlated selection on each allele in the linkage group. As a result, the effective selection against “foreign” alleles in a habitat may be increased, in some cases very substantially. The effect of these limitations on our overall conclusion—that spatially variable selection enhances the fixation of new mutations—is an important open question.
Acknowledgments
The authors thank Sally Otto, Cort Griswold, and the SOWD group at the University of British Columbia (UBC) for very helpful comments on the manuscript. John Wakeley and two anonymous referees made extremely useful comments on the manuscript we initially submitted, for which they have our gratitude. We thank David Watkins for advice on the numerical analysis of the two-deme diffusion equation. Most of this research was completed while R.G. was on sabbatical at UBC; he thanks UBC and its Department of Zoology for generously hosting his stay. This research was supported by the Natural Science and Engineering Research Council (Canada) and the National Science Foundation (United States).
References
- Barton, N. H., 1993. The probability of fixation of a favoured allele in a subdivided population. Genet. Res. 62: 149–158. [Google Scholar]
- Barton, N. H., and M. Turelli, 1991. Natural and sexual selection on many loci. Genetics 127: 229–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens, W. J., 1979. Mathematical Population Genetics. Springer-Verlag, Berlin.
- Fisher, R. A., 1922. On the dominance ratio. Proc. R. Soc. Edinb. 42: 321–341. [Google Scholar]
- Gavrilets, S., and N. Gibson, 2002. Fixation probabilities in a spatially heterogeneous environment. Popul. Ecol. 44: 51–58. [Google Scholar]
- Haldane, J. B. S., 1927. A mathematical theory of natural and artificial selection. V. Selection and mutation. Proc. Camb. Philos. Soc. 23: 838–844. [Google Scholar]
- Karlin, S., 1982. Classifications of selection-migration structures and conditions for a protected polymorphism. Evol. Biol. 14: 61–204. [Google Scholar]
- Karlin, S., and H. M. Taylor, 1981. A Second Course in Stochastic Processes. Academic Press, New York.
- Kemeny, J. G., and L. Snell, 1976. Finite Markov Chains. Springer-Verlag, Berlin/Heidelberg, Germany/New York.
- Kimura, M., 1957. Some problems of stochastic processes in genetics. Ann. Math. Stat. 28: 882–901. [Google Scholar]
- Kimura, M., 1962. On the probability of fixation of mutant genes in a population. Genetics 47: 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1964. Diffusion models in population genetics. J. Appl. Probab. 1: 177–232. [Google Scholar]
- Maruyama, T., 1970. On the fixation probability of mutant genes in a subdivided population. Genet. Res. 15: 221–225. [DOI] [PubMed] [Google Scholar]
- Nagylaki, T., 1980. The strong-migration limit in geographically structured populations. J. Math. Biol. 9: 101–114. [DOI] [PubMed] [Google Scholar]
- Nagylaki, T., 1982. Geographical invariance in population genetics. J. Theor. Biol. 99: 159–172. [DOI] [PubMed] [Google Scholar]
- Nagylaki, T., 1992. Introduction to Theoretical Population Genetics. Springer-Verlag, Berlin.
- Pollak, E., 1966. On the survival of a gene in a subdivided population. J. Appl. Probab. 3: 142–155. [Google Scholar]
- Press, W. H., S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, 1992. Numerical Recipes in C: The Art of Scientific Computing. Cambridge University Press, Cambridge, UK.
- Tachida, H., and M. Iizuka, 1991. Fixation probability in spatially changing environments. Genet. Res. 58: 243–251. [DOI] [PubMed] [Google Scholar]
- Whitlock, M. C., 2002. Selection, load, and inbreeding depression in a large metapopulation. Genetics 160: 1191–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock, M. C., 2003. Fixation probability and time in a metapopulation. Genetics 164: 767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock, M. C., and N. H. Barton, 1997. The effective size of a subdivided population. Genetics 146: 427–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock, M. C., and D. E. McCauley, 1990. Some population genetic consequences of colony formation and extinction: genetic correlations within founding groups. Evolution 44: 1717–1724. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16: 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]