

Abstract
Comparison of population differentiation in neutral marker genes and in genes coding quantitative traits by means of FST and QST indexes has become commonplace practice. While the properties and estimation of FST have been the subject of much interest, little is known about the precision and possible bias in QST estimates. Using both simulated and real data, we investigated the precision and bias in QST estimates and various methods of estimating the precision. We found that precision of QST estimates for typical data sets (i.e., with <20 populations) was poor. Of the methods for estimating the precision, a simulation method, a parametric bootstrap, and the Bayesian approach returned the most precise estimates of the confidence intervals.
COMPARATIVE studies of population differentiation in marker genes and genes coding quantitative traits have become popular during recent years (reviewed in Merilä and Crnokrak 2001; McKay and Latta 2002). These studies are based on the realization (Wright 1969) that the degree of quantitative trait differentiation among populations, as measured by the QST index (Spitze 1993), is comparable to that of the FST index, estimated from neutral marker genes. The relative magnitudes of these two indexes are therefore informative about the role of natural selection and genetic drift as a cause of the observed degree of population differentiation in quantitative traits in question. In other words, if QST > FST, then the differentiation is likely to be the result of directional selection, whereas if FST ≫ QST, then genetic drift is a plausible explanation for the observed degree of differentiation. However, these interpretations are subject to a number of restrictive assumptions (e.g., Merilä and Crnokrak 2001), and other potential problems and pitfalls with these comparisons have also surfaced (e.g., Crnokrak and Merilä 2002; Hendry 2002; Morgan et al. 2005).
Two particular problems that have as yet received little attention are the precision and possible bias in the estimates of QST. There are three reasons to suspect that the quality of QST estimates may be poor. First, the components of a QST estimate are variance components, which are typically estimated from small numbers of sampling units characteristic of wild populations (i.e., relatively few populations are sampled). In general, estimates of variance components tend to have low precision, in part because they have to include uncertainty in the mean as well. This problem is particularly acute in QST studies, because the aim of many comparative studies of population differentiation is to make inferences about pairwise differences among a small number of populations (reviewed in Merilä and Crnokrak 2001). Second, QST is typically estimated using “plug-in” estimates of the variance components; i.e., point estimates of the variance components are estimated and then plugged into the equation for QST (Equation 1a or 1b below). This in itself can lead to a bias in the estimates, as the expected value of a ratio is not the same as the ratio of the expectations. Finally, the estimation of the precision of a variance is sensitive to outliers and departures from normality (e.g., Miller 1997), problems that are typical of real data.
Overall, it appears that evolutionary studies are predisposed to produce QST estimates of low precision. However, although the confidence intervals in many empirical studies suggest low precision (e.g., Koskinen et al. 2002; Palo et al. 2003), this has not yet been investigated in detail.
One problem is that there are different methods of estimating standard errors or confidence intervals for QST, and these may differ in their precision and bias. Several approaches to estimating precision have been tried, ranging from bootstrap methods (e.g., Spitze 1993; Koskinen et al. 2002) and a delta method approximation (e.g., Morgan et al. 2001), both of which are based on a maximum-likelihood approach, to a Bayesian analysis (e.g., Palo et al. 2003). The variance of QST is straightforward to estimate in a contemporary Bayesian framework, as the whole posterior distribution is estimated, so that the distributions of any variables calculated from the posterior are also correct. For estimates based on maximum likelihood, however, the variance is estimated indirectly, either by using a resampling scheme such as the jackknife or bootstrap or by using an approximation (e.g., a delta method). These methods are generally correct only asymptotically, and the amount of data needed to be close enough to the asymptotic state has to be evaluated. This is particularly a problem for QST studies where, as already pointed out, the number of populations studied is typically low.
The resampling methods have a further problem that it is not clear what level in the experimental design should be resampled. At first sight, it would appear to be sufficient use a nonparametric bootstrap over individuals (as in, for example, Spitze 1993; Koskinen et al. 2002). However, this turns out to be incorrect. The nonparametric bootstrap works by resampling over independent units (Davison and Hinkley 1997), and the observations on the individuals are correlated: for example, individuals from the same family tend to have similar phenotypes. Davison and Hinkley (1997, pp. 100–102) discuss this problem for the bootstrap, pointing out that the resampling should be over the highest level in the hierarchical structure (here the population). However, they show that this will lead to biased estimates, particularly when only a few populations are in the data set. Perhaps surprisingly, they also show that the bias is greater if the bootstrap is carried out at two levels (i.e., population and sire here). They also raise the possibility of bootstrapping the residuals from the model, but without being confident about how well it will work for any particular problem.
Statistically, the problem here is very similar to the estimation of the standard error of heritability. The statistical properties of the jackknife (over families) (Knapp et al. 1989), the delta method, and a parametric method (similar to the parametric bootstrap used below) (Hohls 1997) have been investigated, and overall both the jackknife and the parametric methods worked reasonably well, while the delta method needed a lot of data to perform well. A crucial practical difference between heritability and QST is that while QST is typically estimated with only a few populations, for heritability a larger number of sires (which perform an equivalent role in the statistic) are usually used.
Our aim here is to compare performance of different methods in estimating QST in terms of their precision and possible bias. First we examine the performance of the commonly used restricted maximum-likelihood (REML) estimator, using simulations to see the effects of the actual value of QST and the number of populations in the sample on the bias and variability of the estimated point values. Then we examine several methods for estimating the standard error and confidence limits of the estimates, using both simulated data and a real data set. Although a smaller standard error might seem better, this can mean that the error associated with a statistic is being underestimated, leading to undue confidence in the statistic. Here we concentrate on the coverage of the methods to evaluate their performance, i.e., the proportion of times that a confidence interval contains the true value.
METHODS
Point estimation:
All of the data sets used here have the same extended NCI (North Carolina I) design. Within each population, five males are taken and each is mated with two females. Five offspring from the cross are measured. The response is therefore modeled as a function of the random effects population, sire (nested within population), and dam (nested within sire and population). As the additive variance in a NCI design is four times the sire variance component (Lynch and Walsh 1998), QST can be calculated as
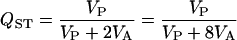 |
(1a) |
or
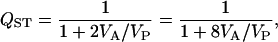 |
(1b) |
where VP is the population variance, VS is the sire variance, and VA is the additive variance (Spitze 1993). The second form is sometimes more useful in estimating the confidence limits for QST (see below).
Point estimates for QST have usually been obtained by fitting the model to the data using REML. Most of the methods used here are based on this approach as well, but as the experimental design is always balanced, the estimates are identical to those from a least-squares fit.
Precision estimation:
Several methods for estimating the precision of the QST estimates are outlined below. Three properties are worth noting for each estimator: (1) although the point estimate can be considered nonparametric (if it is viewed as a least-squares estimate), several of the methods for estimating the precision of the QST estimates rely on making parametric assumptions, in essence that the data and the variance components are normally distributed; (2) some of the precision estimates outlined below also attempt to estimate the bias due to using the simple REML estimates of the variance components as plug-in estimates for QST; and (3) some of the estimators are appropriate only for data from a balanced design. These properties are noted in the descriptions.
Delta method:
An approximate method for calculating the bias and variance of a statistic is to expand it as a Taylor series about the true value and examine the expectations of the lower-order terms. In general (e.g., Lynch and Walsh 1998, Appendix 1), if f(x, y) is a function of x and y with mean mf and variance sf2, then
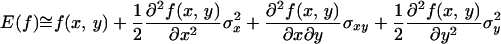 |
(2) |
and
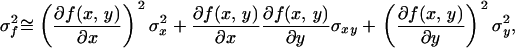 |
(3) |
where 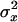 and
and 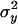 are the variances of x and y, respectively, σxy is the covariance between x and y, and f is evaluated at the true values of x and y. For QST we get
are the variances of x and y, respectively, σxy is the covariance between x and y, and f is evaluated at the true values of x and y. For QST we get
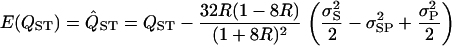 |
(4) |
and
 |
(5) |
where R = VS/VP, and the (co)variances are for the standard deviations on the log scale (Pinheiro and Bates 2000, Chap. 2). This shows that we should expect a negative bias when  (i.e., the value of QST calculated from the REML estimates of VP and VS will on average be less than the true value). The approximate confidence interval can then be calculated as
(i.e., the value of QST calculated from the REML estimates of VP and VS will on average be less than the true value). The approximate confidence interval can then be calculated as 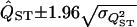 .
.
An alternative method for calculating the confidence intervals is to calculate the intervals for the difference in log variances and back-transform these to the limits for QST. This takes advantage of the better asymptotic properties of the difference in the log variances, as well as of the monotonicity of the transformation. The 95% confidence limits are calculated as
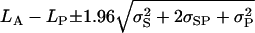 |
(6) |
and these limits are then transformed with Equation 1b.
The delta method assumes that the likelihood is dominated by its lower-order terms in the Taylor series expansion (i.e., the variances and covariances), so that the confidence limits assume that the statistics are normally distributed. Both assumptions are asymptotic, i.e., they are reasonable for a large amount of data, and the approximation will become better as the sample size increases. The method is nonparametric, as it does not make any assumption about the distribution of the statistics, and it also does not require the data to be balanced. A delta method has been used by Morgan et al. (2001) and Podolsky and Holtsford (1995), although no details of the calculations are given.
Nonparametric bootstrap I:
The nonparametric bootstrap works by resampling the data (or portions of it) with replacement and calculating the statistic on the resampled data. The variance of the resampled statistic is approximately that of the statistic itself (Davidson and Hinkley 1997). The bootstrap can be carried out in several ways for these data, and here we try several methods: resampling over (i) populations, (ii) sires, (iii) dams, (iv) individuals, and (v) populations and sires. Of these, i and v were discussed by Davidson and Hinkley (1997), and iv has been used in practice. ii and iii are included for completeness. For each level, 1000 simulations were made, and the variance components were estimated by REML, from which QST was calculated.
All different approaches for the nonparametric bootstrap also estimate the bias of the statistic. They are free from distributional assumptions, but it is unclear if they can be used for unbalanced data without modification. Bootstrapping over individuals has been used previously by Spitze (1993), Koskinen et al. (2002), and Morgan et al. (2005).
Nonparametric bootstrap II:
Here, the bootstrapping is done over residuals, extending an idea suggested by Davidson and Hinkley (1997). A slight correction to the residuals is needed as the raw residuals have excess variation, due to the estimation of the means. If we define xp as the pth population effect (i.e., the difference between the population's mean and grand mean, p = 1, … , P) and xps as the sire effect (i.e., the difference between the sire's mean and the sire's population's mean, s = 1, … , S), then we resample the xp's and xps's with replacement and calculate the bootstrapped data as
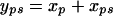 |
(7) |
and the corrected values as
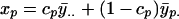 |
(8a) |
 |
(8b) |
where
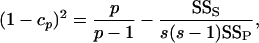 |
(9a) |
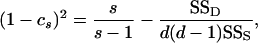 |
(9b) |
or cp or cs = 1 if (9a) or (9b), respectively, is negative, and SSP, SSS, and SSD are the population, sire, and dam sums of squares, respectively. This should retain the second-order properties of the data. This method is free from distributional assumptions and also estimates the bias in the statistic, but cannot be extended to unbalanced data without further modification. It has not been used previously in QST estimation.
Jackknife:
The jackknife, like the bootstrap, is a resampling method designed to reduce the bias in an estimate as well as estimate the variance (Miller 1974). The jackknife is carried out by removing each experimental unit in turn and calculating the focal statistic, θ−i, from this reduced data set. Pseudo-values are then calculated,
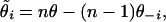 |
(10) |
where n is the number of units in the complete data set, and q is the statistic calculated for the whole data set. The mean and standard deviation of the θ̃i's then give the point estimate and standard error. These can then be assumed to approximately follow a t-distribution with n − 1 d.f., and this can be used for calculating confidence limits for θ.
Here θ = log(VS/VP) rather than QST as the statistic of interest, to improve the distributional approximation (Miller 1974), and the confidence limits are calculated on this scale and then back-transformed to QST using Equation 1b. As with the bootstrap, the jackknife can be carried out at different levels, and here, for completeness, four levels are examined: jackknifing over populations, sires, dams, and individuals. This method does not make distributional assumptions about the data or need a balanced data set and does provide an estimate of the bias. It has not, to our knowledge, been used to estimate QST.
Parametric bootstrap:
A parametric bootstrap simulates a statistic by simulating either the statistic itself or secondary statistics that are used to calculate the statistic of interest (Davidson and Hinkley 1997). For balanced data, it is known that the variance at each level is proportional to a chi-square distribution (e.g., Searle 1971). If our sums of squares are SSP, SSS, SSD, and SSE for the population, sire, dam, and residual effects, respectively, then the variance components can be estimated as
 |
(11) |
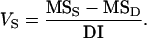 |
(12) |
The likelihood distribution can therefore be estimated by simulating SSP, SSS, and SSD from their distributions, calculating VP and VS from (10) and (11) above, and hence QST from (1). The use of the chi-square distributions relies on the assumption that the data are normally distributed so, as the name suggests, the method is parametric. For the nonbalanced data, the variance components are correlated, and no analytic results are available. Hence, the method requires that the data are balanced. It does, however, estimate the bias. This method was used by Morgan et al. (2005), who noted that it gave larger confidence intervals than the nonparametric bootstrap over individuals.
Direct simulation of data:
From the formal frequentist view of probability (which is the approach that underlies maximum-likelihood estimation), the confidence limits give the limits within which we would expect to see the statistic of interest, given that the model and maximum-likelihood estimates are correct. In principle, therefore, we can simply simulate the data, given the maximum-likelihood (or REML) estimates and the model, and for each of the simulations calculate the estimated QST. The distribution of these simulated values can then be used to calculate the confidence limits. This method is parametric, as it relies on simulating the data, and it estimates the bias. It does not require that the data be balanced and has not, to our knowledge, been used in the estimation of QST.
Bayesian analysis:
All of the previous methods use REML to estimate a point value and then estimate the confidence limits indirectly. The Bayesian approach estimates the full posterior distribution for the model and data, from which the distribution of QST can be calculated directly (Gelman et al. 2004). Prior distributions for the parameters need to be specified, and here they were designed to be as uninformative as possible. The overall mean was given a normally distributed prior with mean zero and variance of 106. The population, sire, dam, and residual standard deviations in the model were given uniform prior distributions between zero and 1000 (see supplementary material at http://www.genetics.org/supplemental and Gelman 2005 for a justification for this prior). The model was fitted by Markov chain Monte Carlo, using WinBUGS1.4 (Spiegelhalter et al. 1999). Two chains were run, and after a burn-in of 5000 iterations, the next 10,000 iterations were taken from each chain. Convergence was assessed using the Brooks-Gelman-Rubin statistic (Brooks and Gelman 1998).
This method is parametric and is applicable to unbalanced data. No bias is defined for the Bayesian method, as the whole distribution is obtained, not just a point estimate. It has previously been used in QST estimation (Palo et al. 2003; Cano et al. 2004). These analyses used a fuller model, in which information about the additive variance in the dam and individual levels was also used. This was not done here, to keep the models identical, so that comparisons are made only across estimation methods.
Performance of methods:
For all of the analyses, the data have a similar structure, based on that of the real data set. There are several populations (four unless otherwise stated). Within each population, there were five sires. Each sire was crossed with two dams, and five individuals were measured per dam, giving a total of 50 observations per population. This is therefore a NCI design (Lynch and Walsh 1998), with dams nested within sires. This gives a data set with a balanced design, which makes the estimation easier, and means that a larger number of methods for estimating the standard error of QST are available. The response variable is assumed to be normally distributed.
Simulated data:
The bias and variation in the point estimates of QST were examined by simulating data with known parameter values and comparing the known and estimated parameters. The properties of the variance estimates were also examined with simulated data. All of the simulated data had an overall mean of zero and both dam and residual variances were set to 0.2. The population and sire variances were set so that they summed to 1, and this variance was partitioned into the two components to give the QST desired. Random effects and the response were all modeled as being normally distributed. This means that the assumptions needed for the parametric estimates above are automatically fulfilled.
Effect of QST:
The effects of different values of QST were examined for values of QST between 0.1 and 0.9. For each value of QST, 1000 replicates of the data with the structure outlined above (with four populations) were simulated. QST was estimated by REML, using the point estimates of the population and sire variances. The variation in the point estimates reflects the underlying sampling variation. The estimated bias is the difference between the mean of the estimated values and the true value.
Effect of number of populations:
The effects of the number of populations on the bias and variation in the point estimates were examined by creating simulated data as above, with values of QST of 0.5 and 0.9, and the number of populations was varied between 5 and 35. As above, for each combination of QST and number of populations, 1000 simulated data sets were created and QST was estimated by REML.
Coverage:
Coverage is defined as the proportion of times the true value of a parameter is contained within the estimated confidence limits. Clearly, if the estimated confidence interval is correct then for a 95% confidence interval this should be 95%. The coverage properties of the different confidence limit estimators were examined by using the estimators to estimate the confidence limits for simulated data. Data sets were created with the design and parameters as outlined above, with either 4 or 10 populations and with QST = 0.5 or 0.8. For each combination of number of populations and value of QST, 400 replicate data sets were created. For each estimator, the proportion of simulations where the true value was contained within the 95% confidence interval was recorded.
Empirical data:
The empirical data come from an experiment described by Palo et al. (2003), which was designed to study adaptation in the common frog, Rana temporaria. The response variable is weight at metamorphosis, measured to the nearest milligram. While the original data contained different food and temperature treatments, here only the low-food and cold-temperature treatments were used to simplify the analyses. To create a balanced data set as described above, one of the populations (population U; Laugen et al. 2003) was removed, and then further observations were removed at random until the balanced data set had been created. In two crosses, this left the data set one individual short, so for these an extra individual with a weight equal to the mean effect of that cross was added. In addition, the analysis here treats the data as if they came from an NCI design (i.e., it ignores the information that females might have been mated to several males).
As has already been noted, some of the estimation methods make distributional assumptions about the data—in particular, that the residuals and variance components are normally distributed, with equal variances. For the empirical data, the assumption that the residuals and random effects are all normally distributed seems reasonable (Figure 1), and there do not seem to be any large outliers (Figures 1 and 2). The assumption of homogeneity of variances across units does not seem to be severely violated, although there is some evidence that population 2 has less variation than the others (Figure 2).
Figure 1.—

Normal probability plots for estimated effects of (a) population, (b) sires, (c) dams, and (d) individuals. If normality is a reasonable assumption, then the points should lie along the straight lines.
Figure 2.—

Plots of estimated effect sizes: (a) sires plotted against population, (b) dams plotted against population, (c) individuals plotted against population, (d) dams plotted against sire, (e) individuals plotted against sire, and (f) individuals, plotted against dam.
RESULTS
Simulated data:
Effect of QST:
The simulations show that there is some bias in the REML estimates of QST (Figures 3 and 4). However, unless the actual value of QST is large, the bias is small and can probably be neglected. But for large values of QST there is an appreciable downward bias. For example, when QST = 0.8, the bias is −0.05, while for QST = 0.9 this is already −0.10 (Figure 3). The other point of note is that the variance in the estimates is large for all values of QST. In particular, for intermediate values of QST (between ∼0.4 and 0.7), virtually all possible values of QST lie within the 95% confidence limits.
Figure 3.—

Estimated bias and precision of QST, estimated by REML, for simulated data sets with different values of QST. The boxes show the interquartile range (i.e., from the 25% to the 75% quantile), the horizontal lines in the boxes show the mean, and the whiskers show the 95% confidence intervals. The diagonal line is a 1:1 correspondence between actual and estimated QST.
Figure 4.—

Estimated bias and precision of QST, estimated by REML, for simulated data sets with different numbers of populations. The boxes show the interquartile range (i.e., from the 25% to the 75% quantile), the lines in the boxes show the mean, and the whiskers show the 95% confidence intervals. The solid lines are a 1:1 correspondence between actual and estimated QST. (a) QST = 0.9; (b) QST = 0.5.
Effect of number of populations:
The effects of using different numbers of populations are shown in Figure 4. When QST = 0.9, the bias decreases as the number of populations increases, although it is not eliminated. For QST = 0.5, the bias is much less. Naturally, the variation in QST also decreases with the number of populations in the study, with most of the improvement occurring up to 20 populations for both values of QST examined (Figure 4).
Coverage:
When the coverage of the different methods for estimating the precision of QST is examined, we see that many of the methods perform poorly (Figure 5). The delta and nonparametric bootstrap methods are almost uniformly bad (with the strange exception of the bootstrap over dams when QST = 0.8; we know of no reason why this method should work). The jackknife over populations works well when QST = 0.5, but it fails for QST = 0.8. The parametric bootstrap, simulation method, and Bayesian method all give coverages that are near the actual 95%, even if they do not always fall within the allowable range.
Figure 5.—

The percentage of times that the nominal 95% interval misses the true QST for different methods of confidence interval estimation. The shaded area shows the 95% confidence region if the actual coverage is correct.
Empirical data:
The point estimate for the QST in the data as obtained with REML estimation was 0.82. We would expect this to be biased downward, as shown in the delta method calculations and the analysis of the simulated data (Figure 3). This bias is captured in the point estimates for the delta method, the bootstrap over the residuals, and the Bayesian method. The estimated standard errors and confidence intervals from different approaches are shown in Figure 6. The jackknife estimates tend to give the highest estimated standard error. The standard delta method fails badly: it gives an upper limit of 1.37, somewhat larger than the maximum possible value of 1. The bootstrap over the sires gives the smallest estimated standard error and confidence interval, but the method performs poorly in terms of coverage, suggesting that the small confidence interval is due to poor estimation of the standard error, leading to undue confidence in the parameter estimate: the coverage suggests that the true value can be outside the confidence limits too frequently.
Figure 6.—

Point estimates, 95% confidence limits, and standard errors (Std Error) for QST from different estimation methods for a real data set.
DISCUSSION
The results of this study show that the precision of the QST estimates—irrespective of the estimation method used—is very low, especially when the number of study populations is low. Furthermore, there is an appreciable downward bias in QST estimates when the actual QST is high. However, even more alarming is the poor performance of several of the methods for estimating the confidence limits of QST, although the parametric bootstrap, the simulation method, and the Bayesian approach all give reasonable results. We discuss each of these findings in turn.
The bias is appreciable only at high values of QST (> ∼0.7). This suggests that it is of little practical concern: generally when QST is high enough for the bias to be a problem, the conclusions of the study will be that it too high to be explained by genetic drift anyway (exceptions would occur if FST were also very high).
While the bias in QST estimates is of concern only for highly differentiated populations and traits, the low precision of the estimates is of more of concern as it occurs whenever the number of populations is low. This is irrespective of the actual degree of differentiation between populations. Unfortunately, studies of quantitative trait differentiation usually use only a small number of populations. For instance, the average number of populations used in comparative studies of marker gene and quantitative trait differentiation listed in the review by Merilä and Crnokrak (2001) was about seven. The results of the present study suggest that upward of 20 populations would be needed to get reasonably precise estimates of QST. Of course, the precision will also depend on the number of sires and dams in the study, but given that most studies face severe logistic constraints in terms of size of experiments, the poor precision of estimates is likely to be more a rule than an exception. A clear recommendation is that any experiments intended to estimate QST should be carefully designed, preferably using a power analysis to optimize effort to get as good estimates as possible.
Another major factor influencing the precision of QST estimates was the chosen estimation method. Hence, the choice of method for estimation of the standard error of QST matters. A major practical concern is that most studies that have estimated the precision of QST have either used the delta method or bootstrapped over individuals, methods that were found to give very misleading results, underestimating the variance (Figure 5). The methods that performed best, giving coverages near to the nominal 95%, were all parametric: the parametric bootstrap, the simulation method, and the Bayesian approach. Of these, the parametric bootstrap works only with balanced data sets, and most real data sets will not be sufficiently accommodating, although for slightly unbalanced data using multiple imputation to “fill in” the missing values might be possible (e.g., Little and Rubin 2002). For most problems, however, this leaves either the simulation method or the Bayesian approach.
The simulation method is not used in statistics, perhaps because it is inefficient computationally (there are normally better ways of estimating confidence limits from one data set than by creating 1000 and fitting the model to all of those). However, it appears to work reasonably well here, and its implementation should not be too difficult in general.
The alternative for unbalanced data is to use the Bayesian approach. In principle this means that coverage concerns do not apply, as the posterior is a formally correct summary of our knowledge after the data have been analyzed. If course, this relies on the prior distributions being good summaries of our prior knowledge. In practice there may not be substantive knowledge to develop the priors from. Because of this, and because comparability across studies is often desirable, it is helpful to have prior distributions that lead to good frequentist properties, such as those properties investigated here (Bayarri and Berger 2004). Several possible priors were examined (see supplementary material at http://www.genetics.org/supplemental/) and none were found to have optimal coverage, although several gave similar results to those here.
One unfortunate feature of the results here is that the nonparametric methods all perform poorly. Clearly, if the parametric assumptions are reasonable, then this is not a problem. However, the assumptions underlying the parametric methods do need to be checked (Waldmann et al. 2005), as was done here. If the assumptions are not correct then remedial action may be needed. For example, the effects of outliers can be checked by comparing analyses with and without them. Heterogeneity of additive variance is more difficult to deal with statistically, but the challenge is as much one for biology as for statistics: finding ways of characterizing divergence in populations where the level of genetic variation within populations has also diverged.
The main difference between the results here and those from studies looking at the estimation of heritability is that here the jackknife performs badly. This may be because of the difference in sample size (Knapp et al. 1989 used a minimum of 20 families) or because of the more complex structure of the experiment simulated here. In general, as the performance of all of the approaches employed here will improve with increasing sample sizes, and more sires are used in the calculation of heritability than populations are used to calculate QST, the problems should not be as severe as in the case of estimation of QST. However, when the sample sizes are small, as for many studies dealing with wild populations, the problems may materialize. Hence, caution should be exercised when trying to estimate heritabilities and their standard errors from small amounts of data. Conversely, the results of Knapp et al. (1989) suggest that jackknife standard errors for QST for data taken from at least 20 populations will probably be reasonably accurate.
In conclusion, the results of this study provide a cautionary note about the poor precision in QST estimates obtained with different estimation methods. Recognition of these problems is an important first step toward developing more accurate and precise approaches for estimation of the degree of population differentiation in quantitative traits, and while methods based on parametric assumptions can provide solutions, there is still no general solution to problems caused by these assumptions not being valid.
Acknowledgments
We thank J. M. Cano Arias for comments on the manuscript. Our research was supported through grants from the Academy of Finland to R.B.O. (project no. 205371) and J.M.
APPENDIX: DERIVATION OF DELTA METHOD EQUATIONS
If we write the log of the standard deviation for the population and sire levels as lP and lS, respectively, then 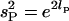 , and
, and 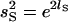 . We also define
. We also define 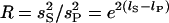 . Then
. Then
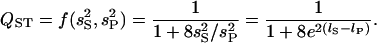 |
(A1) |
By taking a Taylor series expansion around the actual value of QST, the approximate bias (E(QST) − QST) and variance can be estimated,
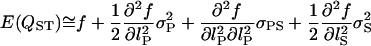 |
(A2) |
and
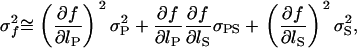 |
(A3) |
where f is evaluated at the true estimates of sP and sS. After some calculation, we get
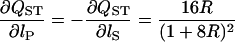 |
(A4) |
so that
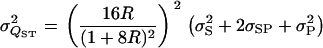 |
(A5) |
Some more algebra shows us that  , giving
, giving
 |
(A6) |
The bias is therefore
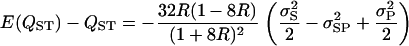 |
(A7) |
References
- Bayarri, M. J., and J. O. Berger, 2004. The interplay of Bayesian and frequentist analysis. Stat. Sci. 19: 58–80. [Google Scholar]
- Brooks, S. P., and A. Gelman, 1998. Alternative methods for monitoring convergence of iterative simulations. J. Comput. Graph. Stat. 7: 434–455. [Google Scholar]
- Cano, J. M., A. Laurila, J. Palo and J. Merilä, 2004. Population differentiation in G matrix structure in response to natural selection in Rana temporaria. Evolution 58: 2013–2020. [DOI] [PubMed] [Google Scholar]
- Crnokrak, P., and J. Merilä, 2002. Genetic population divergence: markers and traits. Trends Ecol. Evol. 17: 501. [Google Scholar]
- Davison, A. C., and D. V. Hinkley, 1997. Bootstrap Methods and Their Applications. Cambridge University Press, Cambridge, UK.
- Gelman, A. J., 2005. Prior distributions for variance parameters in hierarchical models. Bayesian Anal. (in press).
- Gelman, A. J., J. B. Carlin, H. S. Stern and D. B. Rubin, 2004. Bayesian Data Analysis, Ed. 2. Chapman & Hall, London.
- Hendry, A. P., 2002. QST>=< FST? Trends Ecol. Evol. 17: 502. [Google Scholar]
- Hohls, T., 1997. Reliability of confidence interval estimators under various nested design parental sample sizes. Biomet. J. 40: 85–98. [Google Scholar]
- Knapp, S. J., W. C. Bridges and M.-H. Yang, 1989. Nonparametric confidence interval estimators for heritability and expected selection response. Genetics 121: 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskinen, M. O., T. O. Haugen and C. R. Primmer, 2002. Contemporary Fisherian life-history evolution in small salmonid populations. Nature 419: 826–830. [DOI] [PubMed] [Google Scholar]
- Little, R. J. A., and D. B. Rubin, 2002. Statistical Analysis with Missing Data, Ed. 2. John Wiley & Sons, New York.
- Laugen, T. A., A. Laurila, K. Räsänen and J. Merilä, 2003. Latitudinal countergradient variation in the common frog (Rana temporaria) developmental rates—evidence for local adaptation. J. Evol. Biol. 16: 996–1005. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- McKay, J. M., and R. G. Latta, 2002. Adaptive population divergence: markers, QTL and traits. Trends Ecol. Evol. 17: 285–291. [Google Scholar]
- Merilä, J., and P. Crnokrak, 2001. Comparison of genetic differentiation at marker loci and quantitative traits. J. Evol. Biol. 14: 892–903. [Google Scholar]
- Miller, R. G., 1974. The jackknife—a review. Biometrika 61: 1–15. [Google Scholar]
- Miller, R. G., 1997. Beyond ANOVA: Basics of Applied Statistics. Chapman & Hall, London.
- Morgan, K. K., J. Hicks, K. Spitze, L. Latta, M. E. Pfrender et al., 2001. Patterns of genetic architecture for life-history traits and molecular markers in a subdivided species. Evolution 55: 1753–1761. [DOI] [PubMed] [Google Scholar]
- Morgan, T. J., M. A. Evans, T. Garland, Jr., J. G. Swallow and P. A. Carter, 2005. Molecular and quantitative genetic divergence among populations of house mice with known evolutionary histories. Heredity 94: 518–525. [DOI] [PubMed] [Google Scholar]
- Palo, J. U., R. B. O'Hara, A. T. Laugen, A. Laurila, C. R. Primmer et al., 2003. Latitudinal divergence of common frog (Rana temporaria) life history traits by natural selection: evidence from a comparison of molecular and quantitative genetic data. Mol. Ecol. 12: 1963–1978. [DOI] [PubMed] [Google Scholar]
- Pinheiro, J., and D. M. Bates, 2000. Mixed Effects Models in S and S-PLUS. Springer-Verlag, New York.
- Podolsky, R. H., and T. P. Holtsford, 1995. Population structure of morphological traits in Clarkia dudleyana. I. Comparison of FST between allozymes and morphological traits. Genetics 140: 733–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle, S. R., 1971. Linear Models. John Wiley & Sons, New York.
- Speigelhalter, D. J., A. Thomas and N. G. Best, 1999. WinBUGS Version 1.2 User Manual. MRC Biostatistics Unit, Cambridge.
- Spitze, K., 1993. Population structure in Daphnia obtusa: quantitative genetic and allozymic variation. Genetics 135: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann, P., M. R. García-Gil and M. J. Sillanpää, 2005. Comparing Bayesian estimates of genetic differentiation of molecular markers and quantitative traits: an application to Pinus sylvestris. Heredity 94: 623–629. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1969. Evolution and the Genetics of Populations, Vol. 2: The Theory of Gene Frequencies. University of Chicago Press, Chicago.