

Abstract
Granzyme A (GzmA) induces caspase-independent cell death with morphological features of apoptosis. Here, we show that GzmA at nanomolar concentrations cleaves Ku70, a key double-strand break repair (DSBR) protein, in target cells. Ku70 is cut after Arg301, disrupting Ku complex binding to DNA. Cleaving Ku70 facilitates GzmA-mediated cell death, as silencing Ku70 by RNA interference increases DNA damage and cell death by GzmB cluster-deficient cytotoxic T lymphocytes or by GzmA and perforin, whereas Ku70 overexpression has the opposite effect. Ku70 has two known antiapoptotic effects—facilitating DSBR and sequestering bax to prevent its translocation to mitochondria. However, GzmA triggers single-stranded, not double-stranded, DNA damage, and GzmA-induced cell death does not involve bax. Therefore, Ku70 has other antiapoptotic functions in GzmA-induced cell death, which are blocked when GzmA proteolyses Ku70.
Keywords: granzyme A, Ku70, DNA repair, cytotoxic T lymphocyte
Introduction
Cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells are important for immune defence against viruses and tumours. These cells release proapoptotic mediators from cytotoxic granules to trigger target cell apoptosis (Lieberman, 2003). Granzyme A (GzmA) and B (GzmB), the most abundantly expressed granule serine proteases in activated CTLs, independently induce cell death when delivered into the target cell by perforin (PFN). GzmB activates a ubiquitous apoptotic cascade induced by cleaving caspases and key caspase pathway substrates (Lord et al, 2003). GzmA activates caspase-independent cell death with morphological features of apoptosis (Beresford et al, 1999; Shresta et al, 1999; Lieberman & Fan, 2003).
The caspase-independent GzmA pathway is characterized by single-stranded DNA damage, apoptotic morphology, mitochondrial dysfunction and loss of cell membrane integrity (Lieberman & Fan, 2003; Pardo et al, 2004; Martinvalet et al, 2005). GzmA does not activate the caspases or cleave important caspase pathway substrates. Cells that overexpress bcl-2 remain sensitive to GzmA (Beresford et al, 1999). In the mitochondrion, GzmA triggers an increase in reactive oxygen species and loss of transmembrane potential, but does not cause release of proapoptotic molecules, such as cytochrome c (Pardo et al, 2004; Martinvalet et al, 2005). GzmA targets a 270–440 kDa endoplasmic reticulum-associated complex (the SET complex), which contains three GzmA substrates—the nucleosome assembly protein SET, the DNA-binding protein HMG-2 and the base excision repair (BER) enzyme Ape1 (Beresford et al, 2001; Fan et al, 2003a; Lieberman & Fan, 2003). NM23-H1, an endo-nuclease in the SET complex, is activated to make single-stranded DNA nicks after GzmA cleaves its inhibitor, the SET protein. In the same way that GzmA does not target the same substrates as GzmB and the caspases, GzmB and the caspases do not cut most known GzmA substrates (Lieberman & Fan, 2003). Therefore, GzmA activates a parallel, independent cell death pathway.
Although GzmA is a tryptase, it is a highly selective protease. If nuclear lysates are incubated with GzmA overnight and analysed by gel electrophoresis, virtually all of the protein bands remain unchanged (Pasternack et al, 1991). Although a recent proteomics study suggests that GzmA might cleave several hundred intra-cellular substrates (Bredemeyer et al, 2004), only eight substrates (SET, Ape1, HMG-2, lamins A and B and histones H1, H2B and H3) are verified as physiological substrates within cells (Lieberman & Fan, 2003). GzmA substrates do not share a linear peptide recognition sequence. Structural studies suggest that specificity is instead determined by an extended binding domain (Bell et al, 2003; Hink-Schauer et al, 2003).
Apoptotic stimuli induce cellular repair at the same time they activate cell death. The relative balance between repair and death responses determines the ultimate fate of the cell. GzmA cleaves and inactivates Ape1, the rate-limiting BER enzyme, which interferes with the target cell's ability to repair GzmA-induced DNA damage (Fan et al, 2003b). Because of the importance of DNA repair in balancing survival and apoptosis, we investigated whether GzmA might target other DNA repair pathways. Although GzmA does not induce double-strand DNA breaks (DSBs), it cleaves and inactivates Ku70, a key double-strand break repair (DSBR) protein. The Ku complex, made up of Ku70 and Ku80, binds to DSBs early in DNA repair by non-homologous end joining to form a protein ring that supports broken DNA ends (Walker et al, 2001; Downs & Jackson, 2004). Ku bound to DSBs recruits the catalytic subunit DNA-PKcs to form the holoenzyme DNA-PK. DNA-PK then recruits XRCC4 and DNA ligase IV to rejoin the broken ends. Ku70, Ku80, DNA-PK, XRCC4 and DNA ligase IV are all crucial for DSBR; absence of any of these leads to genomic instability and increased sensitivity to DNA damage. During caspase- or GzmB-mediated apoptosis, repair of DSBs produced by the caspase-activated DNase is blocked by DNA-PKcs cleavage (Andrade et al, 1998). Ku70 also suppresses the proapototic function of bax by inhibiting bax translocation to mitochondria (Sawada et al, 2003). Here, we show that Ku70 inactivation by GzmA is important for GzmA-induced cell death, as cells overexpressing Ku70 are less susceptible, whereas cells with silenced Ku70 show enhanced GzmA-induced DNA damage and cell death.
Results
GzmA cleaves Ku70 in isolated nuclei
To determine whether Ku70 or Ku80 might be GzmA targets, GzmA-treated HeLa nuclei were analysed by Ku70 and Ku80 immunoblot. GzmA cleaves Ku70 in a dose- and time-dependent manner to a 35 kDa product, detected using a carboxy-terminal Ku70 antibody (Fig 1A). Cleavage is observed at GzmA concentrations as low as 450 nM. Neither inactive S-AGzmA (produced by mutating the active site Ser184 to Ala) nor GzmB cuts Ku70. Homologous Ku80 is not cleaved by GzmA or GzmB.
Figure 1.
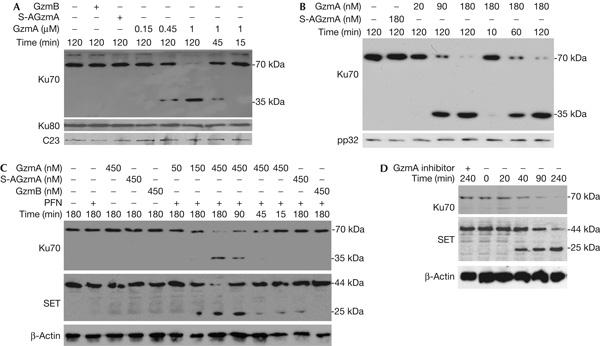
Ku70 is a granzyme A substrate. (A) Granzyme A (GzmA) cleaves Ku70, but not Ku80, in isolated nuclei. HeLa nuclei were incubated with indicated concentrations of GzmA or 1 μM S-AGzmA or GzmB at 37°C for indicated durations and analysed by immunoblot for Ku70, Ku80 or C23, a loading control. GzmA cuts Ku70 to generate a 35 kDa carboxy-terminal fragment, but Ku80 is not cleaved. Neither enzymatically inactive S-AGzmA nor GzmB cuts Ku70 or Ku80. (B) GzmA cuts recombinant Ku70. GzmA or inactive S-AGzmA was added to 0.5 μM Ku70 at 37°C for indicated durations and analysed by Ku70 immunoblot. The 35 kDa cleavage product is seen again. (C) GzmA and perforin (PFN) treatment of K562 degrades Ku70 and SET with similar kinetics. GzmA, but not GzmB or S-AGzmA, degrades Ku70 in cells. The 35 kDa cleavage fragment is also shown. (D) Ku70, like SET, is degraded in 40 min of cytotoxic T lymphocyte (CTL) attack. Ku70 and SET were not degraded when CTLs were pretreated with the GzmA inhibitor diisocoumarin. β-Actin is a loading control.
GzmA cuts Ku70 after Arg301
To test whether Ku70 is a direct substrate and identify the cleavage site, recombinant Ku70 was treated with GzmA (Fig 1B). Recombinant Ku70 was also cleaved to a 35 kDa C-terminal fragment. The cleavage site was identified by amino-terminal sequencing as Arg301 in the sequence KTKTR301TPNT. This is consistent with GzmA being a tryptase. Ku70 contains three domains—an α/β globular domain, DNA-binding β-barrel and an α-helical arm (supplementary Fig 1 online). GzmA cleaves in the β-bridge at the middle of the β-barrel domain. As DNA is threaded through the β-bridge in the Ku–DNA complex, GzmA cleavage has the potential to disrupt Ku binding to DNA.
When the cleavage site Arg was mutated to Ala, mutant Ku70 was also cleaved with comparable kinetics to produce a similar size fragment (supplementary Fig 2 online). This suggests that GzmA may cut the mutant protein at another nearby basic site, perhaps Lys297 or Lys299. We also found efficient cleavage at an alternate site when we mutated the SET cleavage site (D.Z. and J.L., unpublished). These findings support the hypothesis, based on GzmA structure and substrate specificity (Bell et al, 2003; Hink-Schauer et al, 2003), that GzmA binds to substrates by an extended exosite, perhaps with greater specificity than enzymes that recognize a short linear sequence.
Ku70 is degraded in PFN- and GzmA-treated cells
To verify that Ku70 cleavage is physiologically relevant, we treated K562 cells with GzmA and PFN (Fig 1C). With 450 nM GzmA, Ku70 is substantially reduced in 45 min. With less GzmA (150 nM), Ku70 was reduced after 3 h. The 35 kDa C-terminal fragment is again detected. The GzmA concentration needed to cleave Ku70 in vivo is comparable to that required to cleave SET (Fig 1C) and to induce cell death and DNA damage (Beresford et al, 1999). Therefore, Ku70 is a direct in vivo substrate. Ku70 cleavage occurs independently of mitochondrial damage, as it is not affected by treating target cells with a reactive oxygen species scavenger (supplementary Fig 3 online).
Ku70 is degraded after CTL attack
To verify that Ku70 is cleaved during cell-mediated lysis, we analysed cell lysates after CTL target cell incubation (Fig 1D). In 40 min of CTL attack, Ku70 is reduced, and after 90 min, it is barely detectable. The kinetics of Ku70 cleavage is similar to that of SET. However, no Ku70 degradation product is seen. The cleavage product is probably labile. The cleavage is specific—when CTLs are preincubated with the serine protease inhibitor diisocoumarin, Ku70 is not degraded. Moreover, Ku70 is unchanged in target cells treated with the NK cell line YT that expresses GzmB, but not GzmA (supplementary Fig 4 online).
Ku70 and Ku80 bind to S-AGzmA in the presence of DNA
S-AGzmA added to HeLa cell lysates immunoprecipitates with Ku70 or Ku80, demonstrating that GzmA can associate with Ku proteins in cells (Fig 2A). Recombinant glutathione S-transferase (GST)–Ku70 and GST–Ku80, but not control GST protein, also bind to S-AGzmA by GST pulldown, but only in the presence of DNA (Fig 2B). Although DNA is required for binding, binding persists when the bead-bound samples are treated with DNase I, perhaps because complexed DNA is protected from digestion. Therefore, S-AGzmA binding to Ku proteins is DNA dependent.
Figure 2.
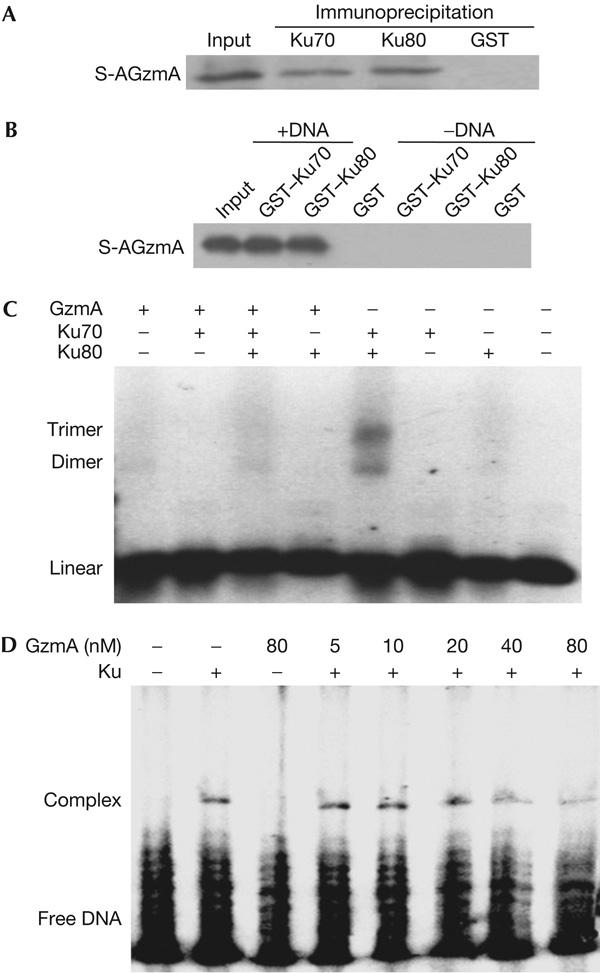
Granzyme A binds to Ku70 and Ku80 and disrupts Ku binding to DNA. (A) Ku70 and Ku80 in cell lysates bind to inactive S-AGzmA (GzmA, granzyme A). Precleared HeLa cell lysates were incubated with the indicated antibodies, and S-AGzmA and immune complexes were captured on protein G beads. GzmA binding was assayed using His-tag antibody. (B) Recombinant Ku70–GST (glutathione S-transferase) and Ku80–GST bind to S-AGzmA in vitro only in the presence of DNA. GST or Ku70–GST and Ku80–GST fusion proteins adsorbed on glutathione beads were incubated with S-AgzmA, with or without DNA. Even though the beads were treated with DNase I before extraction, S-AGzmA binding was detected with His-tag antibody only if DNA was present. (C) GzmA pretreatment blocks Ku complex association with DNA. Recombinant Ku70 and/or Ku80 that were either mock treated or preincubated with GzmA were incubated with a 600 bp DNA fragment and analysed for gel retardation by ethidium bromide staining. (D) Ku70 and Ku80 were pretreated with GzmA and then incubated with 32P-labelled oligonucleotide and analysed by electrophoretic mobility shift assay. GzmA disrupts Ku binding to DNA at nanomolar concentrations.
GzmA disrupts Ku binding to DNA
Destabilizing the interface between the α/β- and β-barrel domains within Ku70 or Ku80 releases Ku heterodimer from DNA (Jones et al, 2001; Walker et al, 2001). As GzmA cleavage disrupts the Ku70 β-bridge, GzmA would be predicted to disrupt Ku binding to DNA. Both Ku70 and Ku80 are required for DNA binding in vitro, as indicated by the retarded mobility of the test DNA fragment (Fig 2C). GzmA inhibits Ku binding to DNA. Moreover, at nanomolar concentrations, GzmA disrupts binding between Ku and DNA, as shown by electrophoretic mobility shift assay (Fig 2D).
GzmA disrupts the Ku complex in cells
To investigate whether GzmA disrupts the Ku complex within cells, an antibody that specifically recognizes the complex was used to stain K562 cells treated with GzmA and PFN. Ku signal by flow cytometry decreases in a GzmA dose-dependent manner (supplementary Fig 5 online). GzmA- and PFN-treated HeLa cells, imaged by confocal microscopy, also show reduced Ku, but undiminished Ku80, in 15 min, and further reductions occur over 2 h (Fig 3). Characteristic apoptotic changes of chromatin condensation and nuclear fragmentation, identified by propidium iodide (PI) staining, are not seen until ∼90 min. Residual Ku complex is concentrated in a few condensed foci. In cells exposed to either GzmA or PFN, nuclei do not become apoptotic and Ku staining is unchanged. These results confirm our finding that Ku70, but not Ku80, is a GzmA substrate. Furthermore, GzmA-induced Ku70 degradation and Ku complex disruption occur with kinetics similar to that of previously reported GzmA substrates (Lieberman & Fan, 2003).
Figure 3.
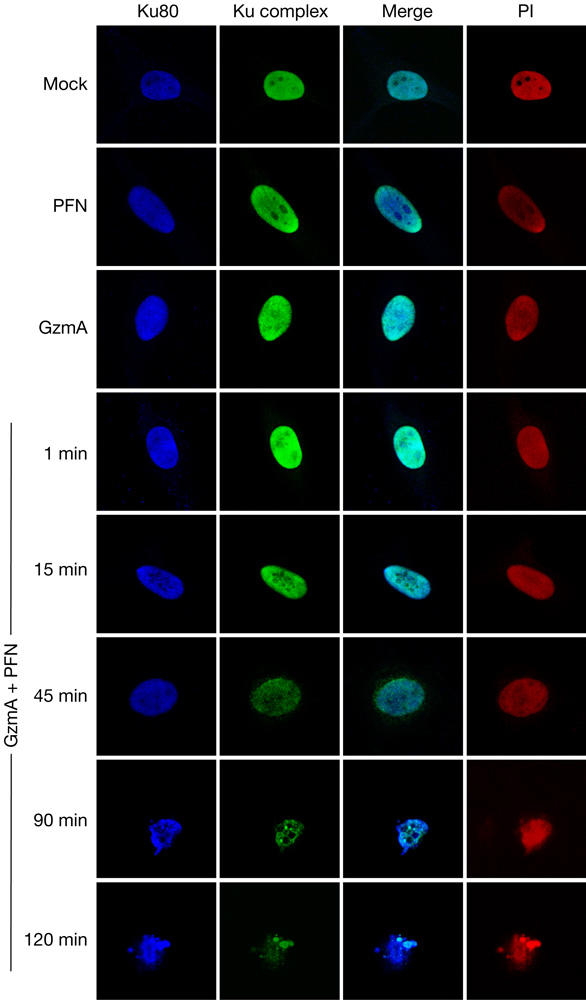
Granzyme A (GzmA) disrupts the Ku complex in vivo. Ku complex staining declines in 15 min of adding GzmA and perforin (PFN) to HeLa cells, but Ku80 staining remains bright. DNA is stained with propidium iodide (PI). Control cells treated for 2 h with GzmA or PFN show no change in Ku complex staining. Apoptotic nuclear changes are readily apparent in 90 min after GzmA and PFN treatment.
Ku70 expression influences DNA damage and death
Although GzmA is not known to cause DSBs, we suggested that Ku70 inactivation by GzmA contributes to enhancing DNA damage in cells subjected to CTL attack. To test this, we altered Ku70 expression by either silencing Ku70 using RNA interference or overexpressing Ku70. We designed four short interfering RNAs (siRNAs) to target Ku70. siRNA 1 and 2 each blocked Ku70 expression in HeLa cells 3 days later by ∼60%, and siRNA 3 and 4 blocked expression by ∼90% (Fig 4A). A mixture of all four nearly completely blocked Ku70 expression and was used in subsequent experiments. In the complementary experiment, Ku70 was overexpressed in HeLa cells transfected with pcDNA6/V5His-Ku70, but not in cells transfected with vector pcDNA6/V5His (Fig 4A). DNA fragmentation was assayed by TdT-mediated dUTP nick end labelling (TUNEL) 2 h after treating transduced HeLa cells with GzmA and PFN. Supplementary Fig 6 online shows a representative experiment and Fig 4B depicts the mean results of three independent experiments. Cells with silenced or overexpressed Ku70 had no change in background TUNEL staining, nor did cells treated with GzmA or PFN alone. However, after GzmA and PFN treatment, 38–39% of the untransfected or control cells transfected with green fluorescent protein (GFP) siRNA or pcDNA were TUNEL+. In cells overexpressing Ku70, DNA damage was reduced almost by half to 22% TUNEL+ cells (P<0.05 compared with vector control). In contrast, TUNEL+ cells increased to 57% of Ku70-silenced cells (P<0.04 compared with GFP siRNA control). These results indicate that if GzmA did not cleave Ku70, Ku70 would contribute to repairing GzmA-induced DNA damage.
Figure 4.
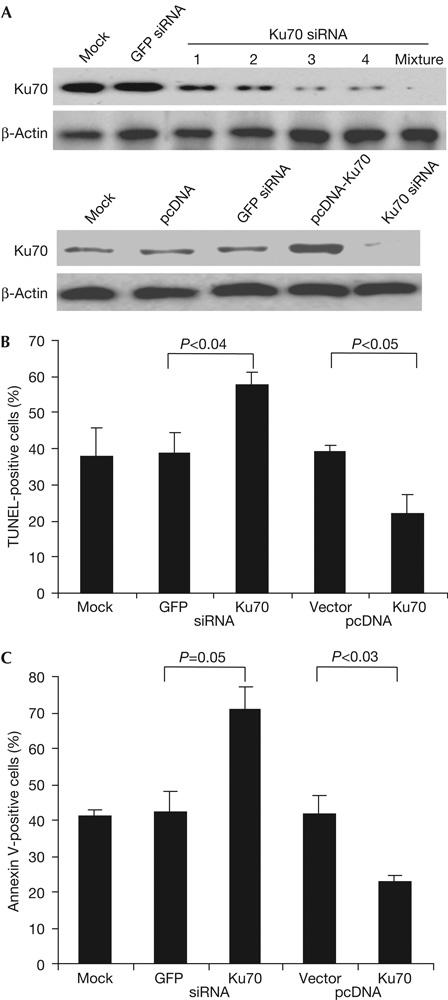
Silencing Ku70 enhances granzyme A-induced DNA fragmentation and cell death, whereas overexpressing Ku70 decreases it. (A) HeLa cells treated with Ku70 short interfering RNA (siRNA), but not with green fluorescent protein (GFP) siRNA, have reduced Ku70 protein. A mixture of all four siRNAs, used in subsequent experiments, almost completely blocks Ku70 expression. Cells transfected with pcDNA6/V5His-Ku70 overexpress Ku70. Lysates were probed 3 days after transfection. (B) Ku70 protects against granzyme A (GzmA)-induced DNA damage. The mean±s.d. of three experiments is depicted. GzmA and perforin (PFN) cause more DNA damage in HeLa cells with silenced Ku70 and less damage in cells overexpressing Ku70. TUNEL, TdT-mediated dUTP nick end labelling. (C) Apoptosis was detected by annexin V and propidium iodide staining 1 h after adding GzmA and PFN. The mean±s.d. for three experiments is depicted.
As Ku70 affected DNA damage, Ku70 could also influence GzmA-mediated cell death. Cells overexpressing or with silenced Ku70 were assayed by annexin V and PI staining after GzmA and PFN treatment. A representative experiment is shown in supplementary Fig 7 online and the means of three independent experiments are shown in Fig 4C. Transfection of siRNAs or the expression plasmids did not significantly change the background, nor did treatment with GzmA or PFN alone. At 1 h after GzmA and PFN treatment, 41±2% of mock-transfected cells became annexin V+. Silencing Ku70 increased annexin V+ cells in response to GzmA and PFN to 71±6% (P=0.05 compared with GFP siRNA) and overexpressing Ku70 decreased it to 23±1% (P<0.03 compared with vector control). Therefore, Ku70 protects cells from GzmA-induced death. The protective effect of Ku70 does not seem to act by inhibiting GzmA cleavage of other substrates; for example, SET degradation is unchanged by silencing Ku70 (supplementary Fig 8 online).
Ku70 protects cells from cell-mediated death
We next wanted to verify whether Ku70 is important during T-cell lysis. Human lymphokine-activated killer (LAK) cells were used to kill concanavalin A-sensitized HeLa cells with increased or silenced Ku70 (Fig 5A). Ku70 overexpression inhibited, but silencing Ku70 enhanced, cell-mediated death measured by 51Cr release. Whereas 39–44% of control cells were lysed, cell death was reduced in 3 independent experiments by 40% in cells that overexpressed Ku70 (24±4% cell death, P<0.02) and was increased by 74% in cells with silenced Ku70 (66±1%, P<0.05).
Figure 5.
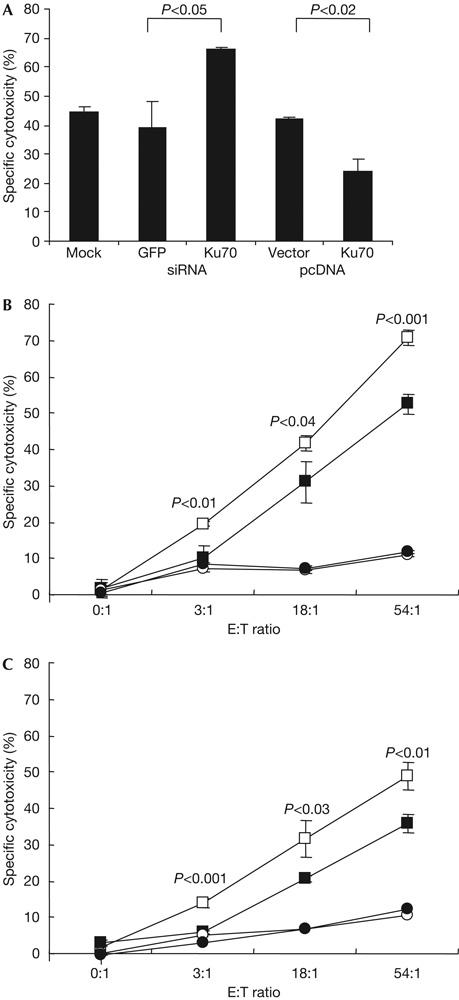
Ku70 expression protects against cytotoxic T-lymphocyte-induced cell death. (A) Ku70 expression reduces target cell susceptibility to human lymphokine-activated killer attack. Transfected HeLa cells were attacked using an effector:target (E:T) ratio of 50:1. (B,C) Ku70−/− mouse embryo fibroblasts (MEFs; open symbols) are more susceptible than Ku70+/+ MEFs (filled symbols) to cytolysis (4 h 51Cr release) by gp33-specific cytotoxic T lymphocytes (CTLs) from T-cell receptor transgenic mice expressing all granzymes (P14 mice; B or P14 × GzmB−/− mice; C). Targets were loaded with antigenic peptide (squares) or mock treated with medium (circles) before incubating with gp33-specific CTLs. P-values show significant differences at each E:T ratio.
However, LAK cells express both GzmA and GzmB. GzmB causes DSBs that might be repaired using Ku70. We therefore used CTLs from T-cell receptor (TCR) transgenic mice that express all granzymes or are genetically deficient in GzmB cluster gene expression (Pham et al, 1996) to kill mouse embryo fibroblast (MEF) targets from wild-type or Ku70−/− mice (Sawada et al, 2003) to determine whether Ku70 protects against GzmA during CTL-mediated lysis. CTLs were from P14 mice (bearing a TCR that recognizes an LCMV gp33 peptide; Pircher et al, 1989) backcrossed with wild-type (Fig 5B) or GzmB−/− (Fig 5C) mice (Pham et al, 1996); target cells were paired Ku70+/+ and Ku70−/− MEFs preincubated with no peptide or gp33 peptide. As for human targets with silenced Ku70, mouse Ku70−/− cells were more sensitive to mouse CTLs. There was significantly more cell death of Ku70−/− than Ku70+/+ target MEFs when attacked by either GzmB+/+ or GzmB−/− CTLs. Therefore, cleaving Ku70 prevents it from participating in a repair response that enables some targeted cells to escape GzmA-mediated cytolysis.
Discussion
Ku70 is a second DNA repair protein cleaved during the novel caspase-independent GzmA-mediated apoptotic pathway, joining the BER enzyme Ape1 on the short list of physiologically relevant, validated GzmA substrates. Ku70 cleavage is specific, as the Ku70 partner and homologue, Ku80, is not a target. Cleavage after Arg301 within the Ku70 β-bridge disconnects two important domains, disrupts the Ku complex and abolishes Ku–DNA complex formation. The importance of Ku70 disruption was verified by showing that silencing Ku70 increases GzmA-mediated DNA damage and cell death, whereas Ku70 overexpression reduces GzmA-induced DNA damage and death. Human cells with silenced Ku70 or mouse Ku70−/− cells are more susceptible to killer cells expressing both GzmA and GzmB or only GzmA.
Ku70 was first identified as an autoantigen in patients with polymyositis–scleroderma overlap syndrome (Mimori et al, 1981). Many autoantigens recognize molecules cleaved during apoptosis (Rosen & Casciola-Rosen, 1999). This study adds Ku70 to several other autoantigens (histones, HMG-2; Lieberman & Fan, 2003) that are substrates of GzmA, but not of GzmB or the caspases.
Ku70 targeting by GzmA was unexpected. Ku70 is a key component of the DNA-PK complex that recruits DNA ligase IV and XRCC4 for DSBR (Downs & Jackson, 2004). Although Ku70 is not a caspase substrate, DSBR during caspase- and GzmB-mediated apoptosis is foiled by DNA-PKcs cleavage (Andrade et al, 1998). Although DSBs are a hallmark of caspase-mediated apoptosis, GzmA-mediated DNA damage is single stranded. However, GzmB, present in many of the same killer cells as GzmA, activates DSBs by CAD. Therefore, destruction of Ku70, although possibly redundant in the setting of cleavage of DNA-PKcs, would be expected to inhibit repair of GzmB-induced DNA damage.
Ku70 has another antiapoptotic effect. It binds to bax and prevents it from translocating to mitochondria to release pro-apoptotic molecules, such as cytochrome c (Sawada et al, 2003). Although GzmA triggers mitochondrial damage, it does not cause proapoptotic molecule release from mitochondria (Martinvalet et al, 2005). This suggests that GzmA does not activate bax. In fact, cell death triggered by GzmA and PFN is indistinguishable in matched bax+/+ and bax−/− MEFs (not shown). However, GzmA cleavage of Ku70 would facilitate GzmB-mediated mitochondrial damage, which, at least in part, involves bax (Heibein et al, 2000; Thomas et al, 2001).
Although Ku70 protects against GzmA-induced DNA damage and cell death, the GzmA pathway does not trigger either of the two apoptotic phenomena (DSBs and bax-mediated mitochondrial damage) against which Ku70 might provide protection. This suggests that Ku70 has other unappreciated antiapoptotic functions. The most obvious function would be repair of single-stranded DNA damage. Ku can bind to single-stranded DNA in single-to-double-strand transitions, as occurs in regions of noncomplementarity, to activate DNA-PK (Morozov et al, 1994) and may also bind in a sequence-specific manner to some single-stranded DNA sequences (Torrance et al, 1998). Although structural models suggest that Ku binds to DNA as a sliding ring around a DSB (Walker et al, 2001), nearby DNA nicks on both strands might create DSBs that are not blunt-ended, through which the Ku ring could slide. In addition, Ku might be involved in regulating transcription for repair. Several studies have suggested that Ku70 directly affects transcription, independently of DNA-PK (Downs & Jackson, 2004). However, identifying how Ku70 protects against GzmA-mediated death requires further study.
Methods
Antibodies and reagents. Mouse monoclonal antibody (RJ1) to pp32 and rabbit antiserum to an N-terminal peptide of SET were previously described (Beresford et al, 2001). The following primary commercial antibodies were used: rabbit polyclonal antisera to NM23-H1 and His tag (Santa Cruz, Santa Cruz, CA, USA), ICAD (Abcam, Cambridge, MA, USA) and GST (Clontech, Mountain View, CA, USA); mouse monoclonal antibodies to β-actin (Sigma-Aldrich, St Louis, MO, USA) and Ku complex (NeoMarkers, Fremont, CA, USA); and goat polyclonal antibodies to C23 (F-18), Ku70 (C-19) and Ku80 (C-20) (Santa Cruz). Secondary antibodies were Alexa 488-conjugated donkey anti-mouse IgG (Molecular Probes, Eugene, OR, USA), Cy5-conjugated donkey anti-goat IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), phycoerythrin-conjugated donkey anti-mouse IgG (BD, San Jose, CA, USA), horseradish peroxidase (HRP)-conjugated donkey anti-goat IgG (Santa Cruz), HRP-conjugated donkey anti-rabbit and anti-mouse IgG (Amersham, Piscataway, NJ, USA). Reactive oxygen species scavenger Tiron was purchased from Sigma and used as described (Martinvalet et al, 2005).
Cell lines. HeLa and K562 cells (American Type Culture Collection, Manassas, VA, USA) were grown in DMEM supplemented with 10% fetal calf serum, 2 mM glutamine, 2 mM HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin and 50 μM β-mercaptoetha′nol. Ku70+/+ and Ku70−/− MEF cell lines, a kind gift from S. Matsuyama (Sawada et al, 2003), were maintained in the same medium. The YT cell line was grown in RPMI 1640 supplemented as above.
Recombinant proteins. Recombinant GzmA, S-AGzmA (enzymatically inactive GzmA produced by mutating the active site Ser184 to Ala) and GzmB were produced and purified as previously described (Beresford et al, 1997; Xia et al, 1998). Briefly, His-tagged GzmA expressed in Escherichia coli was purified by sequential nickel column, enterokinase treatment and cation exchange chromatography. S-AGzmA was also expressed and purified as for GzmA, with the omission of the enterokinase step. His-tagged GzmB expressed by baculovirus in SF2 cells was purified by nickel chromatography. Ku70 and Ku80 were expressed in E. coli as GST fusion proteins from GST–Ku70 and GST–Ku80 plasmids, kind gifts from S.P. Jackson (Cambridge University, Cambridge, England), and purified using glutathione-Sepharose (Pharmacia, Piscataway, NJ, USA) as described (Gell & Jackson, 1999) and GST was removed by thrombin digestion and HiTrap Benzamidine FF (Pharmacia, Piscataway, NJ, USA) chromatography following the manufacturer's protocol.
Cleavage of Ku in isolated nuclei. Nuclei were isolated from HeLa cells by NP-40 lysis (25 mM KCl, 20 mM Tris–HCl (pH 7.5), 5 mM MgCl2 and 0.2% NP-40) and washed twice with lysis buffer and once with lysis buffer without NP-40. Nuclei (1 × 107/ml) were incubated with the indicated amounts of granzymes at 37 °C for indicated durations. The reaction was stopped by adding 5 × SDS loading buffer containing protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Boiled samples were analysed by SDS–polyacrylamide gel electrophoresis and immunoblot.
For additional methods, see the supplementary information online.
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/7400622-s1.pdf).
Supplementary Material
Supplementary Material
Acknowledgments
We thank Z. Xu and D. He for technical support, P. McCaffrey for editorial assistance, A. Laouar, S.-K. Lee and Y. Feng for experimental assistance and S. Matsuyama, J.W. Shay and S.P. Jackson for generously providing reagents. This work was supported by National Institutes of Health (NIH) AI45587 (J.L.), Leukemia and Lymphoma Society (D.C.) and NIH T32 HL066987 (D.K.).
References
- Andrade F, Roy S, Nicholson D, Thornberry N, Rosen A, Casciola-Rosen L (1998) Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity 8: 451–460 [DOI] [PubMed] [Google Scholar]
- Bell JK, Goetz DH, Mahrus S, Harris JL, Fletterick RJ, Craik CS (2003) The oligomeric structure of human granzyme A is a determinant of its extended substrate specificity. Nat Struct Biol 10: 527–534 [DOI] [PubMed] [Google Scholar]
- Beresford PJ, Kam CM, Powers JC, Lieberman J (1997) Recombinant human granzyme A binds to two putative HLA-associated proteins and cleaves one of them. Proc Natl Acad Sci USA 94: 9285–9290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beresford PJ, Xia Z, Greenberg AH, Lieberman J (1999) Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity 10: 585–594 [DOI] [PubMed] [Google Scholar]
- Beresford PJ, Zhang D, Oh DY, Fan Z, Greer EL, Russo ML, Jaju M, Lieberman J (2001) Granzyme A activates an endoplasmic reticulum-associated caspase-independent nuclease to induce single-stranded DNA nicks. J Biol Chem 276: 43285–43293 [DOI] [PubMed] [Google Scholar]
- Bredemeyer AJ, Lewis RM, Malone JP, Davis AE, Gross J, Townsend RR, Ley TJ (2004) A proteomic approach for the discovery of protease substrates. Proc Natl Acad Sci USA 101: 11785–11790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs JA, Jackson SP (2004) A means to a DNA end: the many roles of Ku. Nat Rev Mol Cell Biol 5: 367–378 [DOI] [PubMed] [Google Scholar]
- Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J (2003a) Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 112: 659–672 [DOI] [PubMed] [Google Scholar]
- Fan Z, Beresford PJ, Zhang D, Xu Z, Novina CD, Yoshida A, Pommier Y, Lieberman J (2003b) Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat Immunol 4: 145–153 [DOI] [PubMed] [Google Scholar]
- Gell D, Jackson SP (1999) Mapping of protein–protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Res 27: 3494–3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heibein JA, Goping IS, Barry M, Pinkoski MJ, Shore GC, Green DR, Bleackley RC (2000) Granzyme B-mediated cytochrome c release is regulated by the bcl-2 family members bid and Bax. J Exp Med 192: 1391–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hink-Schauer C, Estebanez-Perpina E, Kurschus FC, Bode W, Jenne DE (2003) Crystal structure of the apoptosis-inducing human granzyme A dimer. Nat Struct Biol 10: 535–540 [DOI] [PubMed] [Google Scholar]
- Jones JM, Gellert M, Yang W (2001) A Ku bridge over broken DNA. Structure (Camb) 9: 881–884 [DOI] [PubMed] [Google Scholar]
- Lieberman J (2003) Cell death and immunity: the ABCs of granule-mediated cytotoxicity: new weapons in the arsenal. Nat Rev Immunol 3: 361–370 [DOI] [PubMed] [Google Scholar]
- Lieberman J, Fan Z (2003) Nuclear war: the granzyme A-bomb. Curr Opin Immunol 15: 553–559 [DOI] [PubMed] [Google Scholar]
- Lord SJ, Rajotte RV, Korbutt GS, Bleackley RC (2003) Granzyme B: a natural born killer. Immunol Rev 193: 31–38 [DOI] [PubMed] [Google Scholar]
- Martinvalet D, Zhu P, Lieberman J (2005) Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22: 355–370 [DOI] [PubMed] [Google Scholar]
- Mimori T, Akizuki M, Yamagata H, Inada S, Yoshida S, Homma M (1981) Characterization of a high molecular weight acidic nuclear protein recognized by autoantibodies in sera from patients with polymyositis–scleroderma overlap. J Clin Invest 68: 611–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov VE, Falzon M, Anderson CW, Kuff EL (1994) DNA-dependent protein kinase is activated by nicks and larger single-stranded gaps. J Biol Chem 269: 16684–16688 [PubMed] [Google Scholar]
- Pardo J, Bosque A, Brehm R, Wallich R, Naval J, Mullbacher A, Anel A, Simon MM (2004) Apoptotic pathways are selectively activated by granzyme A and/or granzyme B in CTL-mediated target cell lysis. J Cell Biol 167: 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternack MS, Bleier KJ, McInerney TN (1991) Granzyme A binding to target cell proteins. Granzyme A binds to and cleaves nucleolin in vitro. J Biol Chem 266: 14703–14708 [PubMed] [Google Scholar]
- Pham CT, MacIvor DM, Hug BA, Heusel JW, Ley TJ (1996) Long-range disruption of gene expression by a selectable marker cassette. Proc Natl Acad Sci USA 93: 13090–13095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM (1989) Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature 342: 559–561 [DOI] [PubMed] [Google Scholar]
- Rosen A, Casciola-Rosen L (1999) Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ 6: 6–12 [DOI] [PubMed] [Google Scholar]
- Sawada M, Sun W, Hayes P, Leskov K, Boothman DA, Matsuyama S (2003) Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol 5: 320–329 [DOI] [PubMed] [Google Scholar]
- Shresta S, Graubert TA, Thomas DA, Raptis SZ, Ley TJ (1999) Granzyme A initiates an alternative pathway for granule-mediated apoptosis. Immunity 10: 595–605 [DOI] [PubMed] [Google Scholar]
- Thomas DA, Scorrano L, Putcha GV, Korsmeyer SJ, Ley TJ (2001) Granzyme B can cause mitochondrial depolarization and cell death in the absence of BID, BAX, and BAK. Proc Natl Acad Sci USA 98: 14985–14990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrance H, Giffin W, Rodda DJ, Pope L, Hache RJ (1998) Sequence-specific binding of Ku autoantigen to single-stranded DNA. J Biol Chem 273: 20810–20819 [DOI] [PubMed] [Google Scholar]
- Walker JR, Corpina RA, Goldberg J (2001) Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412: 607–614 [DOI] [PubMed] [Google Scholar]
- Xia Z, Kam CM, Huang C, Powers JC, Mandle RJ, Stevens RL, Lieberman J (1998) Expression and purification of enzymatically active recombinant granzyme B in a baculovirus system. Biochem Biophys Res Commun 243: 384–389 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material