

Abstract
We show that structural type A and B bacterial ribonuclease P (RNase P) RNAs can fully replace each other in vivo despite the many reported differences in their biogenesis, biochemical/biophysical properties and enzyme function in vitro. Our findings suggest that many of the reported idiosyncrasies of type A and B enzymes either do not reflect the in vivo situation or are not crucial for RNase P function in vivo, at least under standard growth conditions. The discrimination of mature tRNA by RNase P, so far thought to prevent product inhibition of the enzyme in the presence of a large cellular excess of mature tRNA relative to the precursor form, is apparently not crucial for RNase P function in vivo.
Keywords: RNase P, in vivo complementation studies, Escherichia coli, Bacillus subtilis
Introduction
Ribonuclease P (RNase P) is an essential ribonucleoprotein enzyme responsible for the 5′-end maturation of transfer RNAs (Schön, 1999). Bacterial RNase P enzymes recognize the stacked acceptor stem/T-arm module of precursor tRNA (ptRNA) substrates (Harris & Christian, 2003, and references therein), and their RNA subunits have been shown to be catalytically active in the absence of the protein subunit (Guerrier-Takada et al, 1983). In bacteria, the RNA subunit (about 380 nt) forms a specific complex with a small basic protein of about 13 kDa, encoded by the rnpA gene. RNase P RNAs (P RNAs, encoded by the rnpB gene) from bacteria are subdivided into two distinct structural groups, termed type A (for ‘ancestral') and type B (for ‘Bacillus'; Hall & Brown, 2001). Escherichia coli and Bacillus subtilis have been the principal model systems for type A and B RNase P RNAs, respectively. Although subunits of E. coli and B. subtilis RNase P enzymes have been shown to be interchangeable in vitro (Guerrier-Takada et al, 1983), more recent studies have indicated that the differences between the type A and B RNase P RNA architectures are associated with numerous differences in their biogenesis, biochemical/biophysical properties and enzyme function in vitro. There is evidence for autolytic processing of B. subtilis P RNA 5′ and 3′ ends after association with its protein subunit (Loria & Pan, 2000), whereas the E. coli P RNA precursor is processed by RNase E at its 3′ end and by an as yet unidentified ribonuclease at the 5′ end (Lundberg & Altman, 1995). B. subtilis, but not E. coli, RNase P was found to form a specific dimer consisting of two RNA and two protein subunits (Fang et al, 2001). Dimer formation is favoured in the absence of substrate, or in the presence of a tandem tRNA substrate, but disfavoured in the presence of a monomeric ptRNA (Barrera et al, 2002). The dimeric form of B. subtilis RNase P was further observed to bind to 30S ribosomal subunits, and this association has been proposed to modulate the enzyme's activity on potential, but as yet unidentified non-tRNA substrates (Barrera & Pan, 2004). The B. subtilis RNase P holoenzyme binds to ptRNA with a much higher affinity than mature tRNA (mtRNA), but this discrimination against mtRNA seems substantially attenuated in E. coli RNase P (Tallsjö & Kirsebom, 1993; Kurz et al, 1998). Indeed, a recent study reports a 1,600-fold preference of B. subtilis RNase P for ptRNA versus mtRNA, whereas this factor is reduced to 3 in the case of the E. coli holoenzyme (Buck et al, 2005). Finally, E. coli and B. subtilis P RNAs have distinct metal ion requirements (Warnecke et al, 1999).
In this report, we present the results of in vivo complementation experiments in RNase P mutant strains of E. coli and B. subtilis. The type B RNase P RNA of B. subtilis can functionally replace the type A RNA from E. coli in vivo and vice versa. Even a single copy of the E. coli rnpB gene inserted into the B. subtilis chromosome can fully rescue the growth defect caused by repression of endogenous B. subtilis rnpB gene expression.
Results
In vivo complementation studies
The substantial differences between E. coli and B. subtilis RNase P outlined above raise the question as to whether the two RNAs can replace each other in vivo. An earlier study reported the successful complementation of an E. coli rnpB mutant strain by B. subtilis rnpB, but not by rnpB genes from the related bacteria B. brevis and B. megaterium (Waugh & Pace, 1990). Here, we describe cross-species complementation analyses in both E. coli and B. subtilis. In the E. coli mutant strain DW2/pDW160, the chromosomal rnpB gene is replaced with a chloramphenicol resistance cassette and a complementing rnpB gene is provided on a plasmid (pDW160) with a temperature-sensitive origin of replication (Waugh & Pace, 1990). Suppression of the conditionally lethal phenotype at 43°C is achieved by expression of functional P RNA from a second compatible plasmid. For complementation in B. subtilis, we constructed a conditionally lethal mutant strain (SSB318), the endogenous rnpB expression of which is dependent on isopropyl-β-D-thiogalactoside (IPTG).
Complementation in E. coli
E. coli DW2 cells were transformed with the multicopy plasmid pSP64 encoding the E. coli or B. subtilis rnpB gene (pSP64-EcrnpB or pSP64-BsrnpB; Table 1). For growth curve monitoring, corresponding transformants were cured of plasmid pDW160 (loss of kanamycin resistance), and loss of the E. coli rnpB gene was verified by colony PCR for DW2 cells harbouring pSP64-BsrnpB. Consistent with the aforementioned study (Waugh & Pace, 1990), the B. subtilis rnpB gene rescued growth of the DW2 mutant strain at the non-permissive temperature of 43°C (Table 1). Remarkably and for reasons as yet unknown, bacteria expressing B. subtilis rnpB formed larger colonies than cells transformed with the plasmid containing the homologous E. coli rnpB gene at 43°C. This was also reflected in the faster growth rate of the cured strain in liquid cultures at 37°C (64 versus 75 min doubling time in Luria-Bertani medium for DW2 cells transformed with pSP64-BsrnpB and pSP64-EcrnpB, respectively). Thus, B. subtilis P RNA can provide full RNase P function in E. coli, despite a tenfold lower affinity of this RNA for the E. coli RNase P protein (Day-Storms et al, 2004). It is possible that higher levels of expression of B. subtilis rnpB from the multicopy complementation plasmid compensate for the lower protein–RNA affinity.
Table 1.
Complementation of the Escherichia coli RNase P mutant strain by Bacillus subtilis rnpB
| |
DW2/pDW160 |
DW2 doubling time (min)* |
|
|---|---|---|---|
| RNase P RNA source | 30°C | 43°C | 37°C |
| pSP64-EcrnpB |
+++ |
+++‡ |
75±7 |
| pSP64-BsrnpB |
+++ |
+++§ |
64±4 |
| pSP64 | +++ | — | |
| Growth of mutant strains transformed with E. coli or B. subtilis rnpB was analysed on LB plates at the permissive (30°C) and non-permissive (43°C) temperature. RNase P, ribonuclease P; +++, good complementation; —, no colonies. | |||
| *Measured after elimination of pDW160. | |||
| ‡Colonies at 43°C were smaller than those at 30°C; for growth at 43°C, the phenotype of cells transformed with E. coli rnpB was set as the standard. | |||
| §Colonies at 43°C were larger than those for cells transformed with E. coli rnpB, set as the standard. | |||
Activity of hybrid holoenzymes from E. coli
E. coli DW2 bacteria expressing E. coli or B. subtilis rnpB from the complementation plasmid were cured of pDW160 and grown at 30°C for partial purification of RNase P holoenzymes. Holoenzymes were tested under standard assay conditions (10 mM Mg2+, 100 mM NH4+) for processing of ptRNAGly, as well as ptRNAG74 and ptRNAG75 variants (Fig 1), to assess cleavage efficiency and processing site selection following loss of the CCA interaction. Both enzymes cleaved ptRNAGly at the canonical site (+1/−1). However, whereas the E. coli holoenzyme promoted substantial miscleavage of the mutant substrates at −1/−2, the hybrid enzyme containing B. subtilis P RNA did not significantly miscleave ptRNAG74 or ptRNAG75 (Fig 2A,B). These findings show that hybrid RNase P holoenzymes consisting of B. subtilis P RNA and E. coli P protein form stable complexes in vivo that can withstand biochemical purification. The results obtained with the mutant substrates further illustrate in vitro differences in substrate recognition between RNase P enzymes harbouring type A versus type B RNA subunits.
Figure 1.
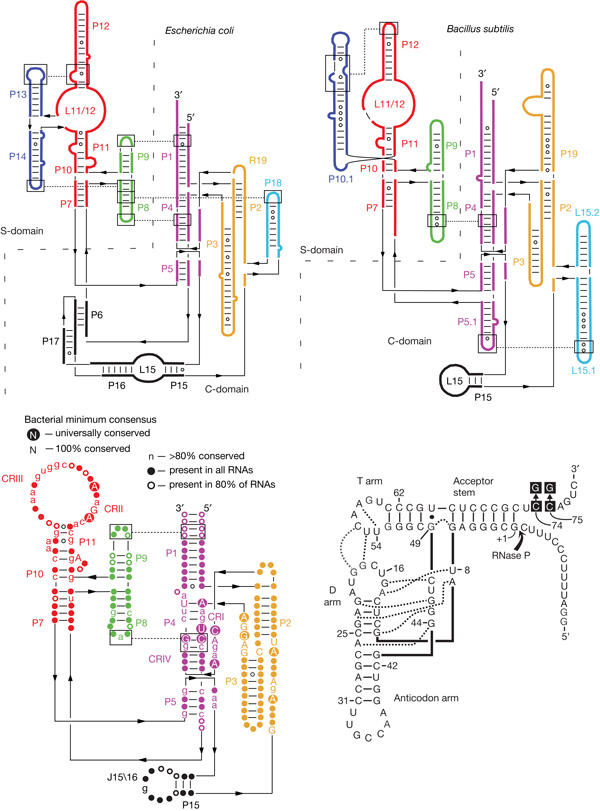
Secondary structure illustrations (Tsai et al, 2003) of Escherichia coli (type A, top left) and Bacillus subtilis (type B, top right) ribonuclease P (RNase P) RNAs, and a phylogenetic minimum consensus RNase P RNA secondary structure (bottom left; Marquez et al, 2005); C-domain, catalytic domain; CR, conserved region; S-domain, specificity domain. Bottom right: Thermus thermophilus ptRNAGly; the arrow indicates the canonical RNase P cleavage site between nucleotides −1 and +1; the point mutations in 3′-CCA of ptRNAGly are highlighted by black squares; tertiary interactions inferred from the crystal structure of yeast tRNAPhe are indicated by dotted lines (adapted from Heide et al, 2001).
Figure 2.
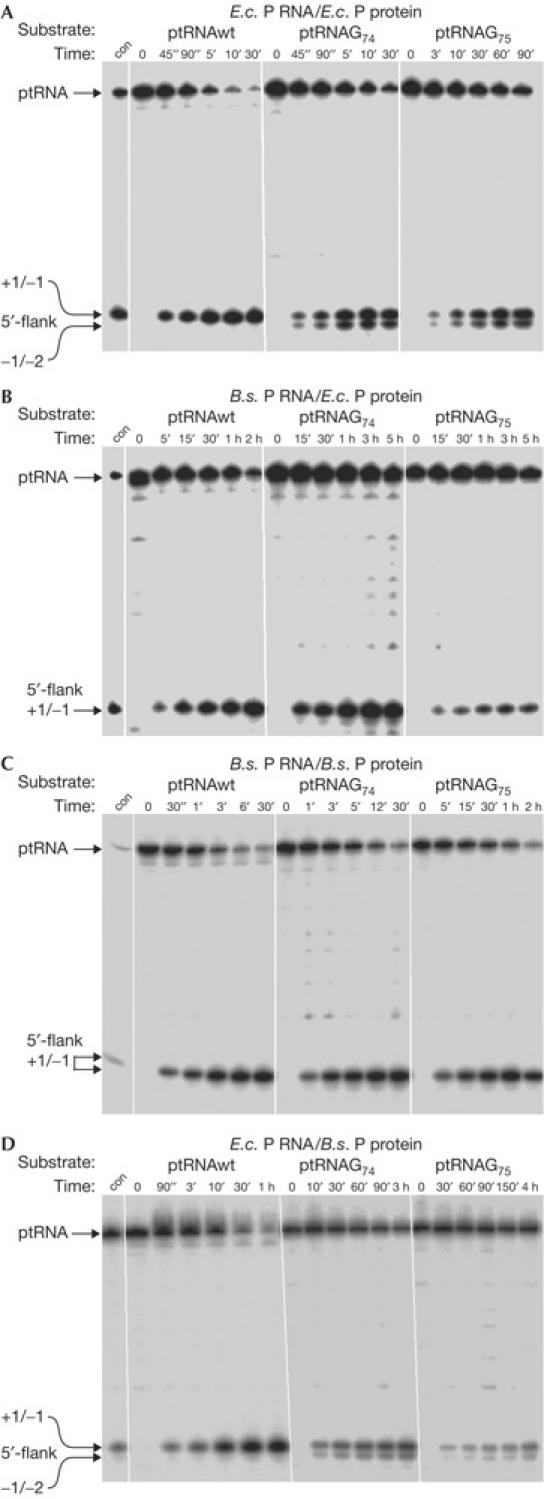
Processing and cleavage site selection of hybrid ribonuclease P (RNase P) holoenzymes isolated from Escherichia coli and Bacillus subtilis complementation strains. (A,B) RNase P partially purified from E. coli DW2 or (C,D) B. subtilis SSB318; holoenzymes contained E. coli P RNA in (A,D) or B. subtilis P RNA in (B,C). con, control cleavage by E. coli RNase P RNA. For details, see Methods. 5′-Cleavage products are indicated on the left (canonical cleavage site at +1/−1, miscleavage at −1/−2). Although the amount of extract added to processing assays was equalized in (A–D), based on absorption at 260 nm, holoenzyme concentrations are probably identical only within one panel; E.c., E. coli; B.s., B. subtilis.
Complementation in B. subtilis
For complementation studies in B. subtilis, we constructed strain SSB318. This strain expresses the chromosomal B. subtilis rnpB gene under control of the spac promoter, and cell survival depends on the presence of IPTG. The conditionally lethal phenotype cannot be rescued by overexpression of the B. subtilis protein subunit (rnpA) from a plasmid (data not shown), indicating that small quantities of P RNA that may persist owing to spac promoter leakiness cannot be coaxed into activity by increased amounts of the protein subunit. SSB318 grows slower in the presence of 1 mM IPTG (doubling time 67 min; Table 2) than the wild-type strain W168 (57 min), suggesting that the fully induced spac promoter is not as strong as the native B. subtilis rnpB promoter and that P RNA is limiting for cell growth under these conditions. This is supported by the observation that the SSB318-derived strain SSB318EcrnpB (see below) has about 3.5-fold less B. subtilis P RNA in the presence of IPTG than W168 (Fig 3, lane 9 versus 17).
Table 2.
Complementation of Bacillus subtilis SSB318 RNase P mutant strains by Escherichia coli rnpB (cell doubling times given in minutes)
| rnpB gene source | Promoter* | IPTG | No IPTG |
|---|---|---|---|
|
B. subtilis W168 (wild type) | |||
| W168 |
B.s. native |
57±2 |
56±2 |
| W168+pMAP65‡ |
B.s. native |
62±2 |
65±2 |
| |
|
|
|
|
B. subtilis SSB318 (Pspac:rnpB) | |||
| SSB318 |
spac |
67±3 |
— |
| SSB318+pMAP65‡ |
spac |
81±6 |
— |
| |
|
|
|
|
Multicopy | |||
| pHY300 (empty plasmid) |
None |
69±4 |
— |
| pHY300-BsrnpB |
B.s. native |
49±1 |
49±2 |
| pHY300-EcrnpB |
E.c. native |
52±2 |
60±2 |
| pHY300-EcrnpB |
B.s. native |
58±3 |
63±3 |
| |
|
|
|
|
Single copy | |||
| pDG364§ |
None |
63±3 |
— |
| SSB318EcrnpB∥ |
B.s. native |
52±1 |
57±1 |
| SSB318EcrnpB+pMAP65‡ | B.s. native | 56±2 | 64±4 |
| RNase P, ribonuclease P; IPTG, isopropyl-β-D-thiogalactoside; —: no growth at all; for determination of cell doubling times, see the supplementary information online. | |||
| *Promoter used for the respective rnpB gene; B.s. native and E.c. native, native rnpB promoters from B. subtilis and E. coli, respectively. | |||
| ‡Plasmid pMAP65 (pUB110 lacI; Petit et al, 1998) was used to overexpress the lac repressor to fully silence expression from the spac promoter in strain SSB318. | |||
| §Vector used for chromosomal integration into the amyE locus. | |||
| ∥E. coli rnpB integrated into the amyE locus under control of the native B. subtilis rnpB promoter and terminator. | |||
Figure 3.
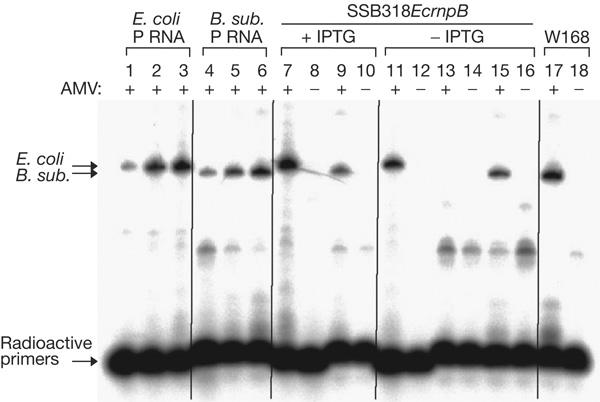
Radioactive reverse transcription PCR (RT–PCR) analysis of the SSB318-derived strain, SSB318EcrnpB. PCR products were analysed on an 8% polyacrylamide/8 M urea gel; lanes 1–3: 0.2, 1 or 5 pg, respectively, of in vitro-transcribed Escherichia coli ribonuclease P RNA (P RNA) added to 0.5 μg Bacillus subtilis W168 (wild type) total cellular RNA; lanes 4–6: 0.2, 1 or 5 pg, respectively, of in vitro-transcribed B. subtilis P RNA added to 0.5 μg of total cellular RNA from E. coli DW2 grown at 30°C; lanes 7–16: total RNA from SSB318EcrnpB grown in the presence (lanes 7–10) or absence (lanes 11–16) of 1 mM isopropyl-β-D-thiogalactoside (IPTG); lanes 7,8,11,12: RT–PCR with primers specific to E. coli P RNA; lanes 9,10,13–16: RT–PCR with primers specific to B. subtilis P RNA; lanes 15,16: the total RNA preparation was supplemented with 1 pg of in vitro-transcribed B. subtilis P RNA; lanes 17,18: total RNA from the B. subtilis W168 wild-type strain using primers specific for B. subtilis P RNA. AMV + or − indicates the presence (+) or absence (−) of reverse transcriptase. For details on RT–PCR, see the supplementary information online. Lanes 1–6 document that the amount of PCR product is sensitive to RNA template concentration (arrows on the left indicate the E. coli and B. subtilis P RNA-specific amplification products); lane 15 shows that the RNA preparation (−IPTG) does not inhibit the RT–PCR reaction with B. subtilis P RNA-specific primers. For all RT–PCR reactions, the total amount of RNA was 0.5 μg.
When expressed from a multicopy plasmid, under control of either its own or the B. subtilis rnpB promoter, the heterologous rnpB gene from E. coli fully rescued the conditionally lethal phenotype of SSB318 (Table 2). The generation time of the strain containing the B. subtilis rnpB-expressing plasmid was shorter (49 min) than that containing the E. coli rnpB genes (60 and 63 min for the constructs driven by the E. coli and B. subtilis promoters, respectively). Although this experiment might suggest that E. coli rnpB is not as efficient at restoring optimal growth as its B. subtilis counterpart, it is also possible that this was because of unrelated effects on plasmid copy number or stability. To eliminate complications of gene dosage from the plasmid-derived rnpB genes and differences in expression due to varying promoter strength, we constructed strain SSB318EcrnpB, which carries a single copy of the E. coli rnpB gene under control of the B. subtilis rnpB promoter in the amyE locus on the B. subtilis chromosome. Constitutive expression of E. coli rnpB in this strain was confirmed by radioactive reverse transcription–PCR (RT–PCR; Fig 3, lanes 7,11). The single-copy chromosomal E. coli rnpB gene efficiently rescued the lethal phenotype in the absence of IPTG, even when the lac repressor was overproduced from plasmid pMAP65 to tighten repression of the spac promoter (Table 2). The growth rates of SSB318EcrnpB and SSB318EcrnpB pMAP65 (57 and 64 min, respectively) were indistinguishable from those of the wild-type strains W168 and W168 pMAP65 (56 and 65 min, respectively), suggesting that E. coli P RNA not only carries out all the essential RNase P functions in the heterologous B. subtilis host, but is also highly efficient at catalysing any of its potential minor functions (see below). It is also interesting to note that expression of both P RNA types in the same cell, whether in single copy or on a plasmid, did not pose any problem for B. subtilis growth (Table 2).
In experiments similar to those performed with the E. coli DW2 strain, we partially purified holoenzymes from B. subtilis SSB318 transformed with E. coli or B. subtilis rnpB and grown in the absence of IPTG. In agreement with the complementary data in Fig 2A,B, the B. subtilis holoenzyme did not significantly miscleave ptRNAG74 or ptRNAG75, whereas the hybrid enzyme containing E. coli P RNA promoted substantial miscleavage of the mutant substrates (Fig 2C,D).
Discussion
Our study shows that type A and B RNase P RNAs are interchangeable in vivo under standard growth conditions, indicating that the hybrid enzymes can carry out all essential cellular functions of RNase P. These findings are surprising for several reasons. First, the lower stability of hybrid holoenzyme complexes (Day-Storms et al, 2004) evidently does not pose a problem for holoenzyme assembly in vivo, as a single E. coli rnpB copy can provide full RNase P function in B. subtilis SSB318. Second, Buck et al (2005) determined that the native B. subtilis holoenzyme has a 1,600-fold preference for ptRNA compared with that for mtRNA, whereas this selectivity is reduced twofold for the hybrid enzyme containing E. coli P RNA. Our results show that this change in the relative affinities for ptRNA and mtRNA does not abrogate RNase P function in vivo. Third, the E. coli protein reduces the Mg2+ concentration requirement for tertiary structure formation of P RNA and increases the melting temperature at physiologically relevant Mg2+ concentrations (Buck et al, 2005). In contrast, the B. subtilis protein does not exert such stabilizing effects on its cognate or E. coli P RNA (Buck et al, 2005). As E. coli P RNA can provide RNase P function in B. subtilis (Table 2), these differences in their biochemical and biophysical properties apparently do not have important consequences for hybrid RNase P function in vivo.
B. subtilis RNase P can form symmetric dimers consisting of two RNA and two protein subunits, whereas E. coli P RNA tends to form aggregates (Fang et al, 2001). Buck et al (2005) reinvestigated dimerization by B. subtilis and E. coli RNase P, and observed weak dimer formation by the E. coli holoenzyme in addition to higher order aggregation. The KD of dimer formation was about 50–100 nM for B. subtilis and 500–1000 nM for E. coli RNase P, with the dimerization properties mainly determined by the type of P RNA subunit (Buck et al, 2005). Unlike the B. subtilis enzyme, dimers containing E. coli P RNA readily dissociated to monomers in the presence of mtRNA, which correlated with the high-affinity binding of mtRNA by holoenzymes containing E. coli P RNA. As the mtRNA concentration in an E. coli cell is estimated to be 500 μM in the presence of 2 μM (Hansen et al, 2001) or lower (Buck et al, 2005) concentrations of cellular RNase P, the biological relevance of dimer formation by holoenzymes containing E. coli P RNA is questionable. Our observation that E. coli P RNA functions in B. subtilis RNase P mutant strains also makes it unlikely that RNase P dimer formation exerts an important regulatory role in B. subtilis.
The maturation pathways of the primary P RNA transcripts differ in E. coli and B. subtilis. The E. coli pathway requires RNase E, an essential enzyme that is not found in B. subtilis, whereas processing of B. subtilis P RNA may be at least partly autocatalytic. The results presented here indicate that not only are the P RNAs interchangeable, but so too are their maturation pathways, assuming the P RNA primary transcripts have to undergo processing for proper function.
Finally, E. coli RNase P is known to process several non-tRNA substrates in vivo (Li & Altman, 2003), whereas non-tRNA substrates have not yet been identified in B. subtilis. As B. subtilis P RNA provided full RNase P function in E. coli, hybrid holoenzymes containing this RNA subunit are able to process at least the essential non-tRNA substrates in E. coli, such as 4.5S RNA (Brown & Fournier, 1984). The fact that B. subtilis strains expressing single-copy E. coli or B. subtilis rnpB genes have the same doubling time suggests that, should B. subtilis RNase P also have alternative functions, the E. coli hybrid enzyme is highly efficient at performing them.
In vivo analyses of the kind reported here provide a framework to test the functional relevance of the numerous biochemical, biophysical and enzymatic differences that have been observed between bacterial type A and B RNase P enzymes. Our results show that, despite these differences, the heterologous hybrid enzymes are capable of carrying out the essential functions of RNase P in vivo, although potential defects under particular stress conditions cannot be excluded at present. This poses the question as to which conserved features of bacterial RNase P RNA and protein subunits are essential for in vivo function. Both the E. coli and B. subtilis P proteins increase the affinity of P RNA for ptRNA by factors of 700–1400 for hybrid and native enzymes containing E. coli P RNA, and 13,000–33,000 for those containing B. subtilis P RNA (Buck et al, 2005). As the E. coli P RNA/B. subtilis P protein complex permitted rescue of the mutant phenotype of B. subtilis strain SSB318, we can conclude that a 700-fold increase in ptRNA affinity conferred by the protein subunit is sufficient to support in vivo function. This increase in affinity for ptRNA is linked to an increase in the affinity for key metal ions involved in ptRNA binding and catalysis (Kurz & Fierke, 2002), probably achieved by the protein's stabilization of conserved local structure in the catalytic core of type A and B RNase P RNAs. These central functions are evidently conserved across bacterial species barriers. In the course of evolution, type A and B RNase P enzymes have developed RNA subunits with different architecture and individually tailored protein subunits, optimized for their specific cellular milieu. It is thus fascinating to observe that these RNase P enzymes still retain their biological function when their RNA subunit is exchanged with one representing a different architectural type.
Methods
Strains. E. coli strain DW2/pDW160 was used for complementation studies (see text). For construction of the B. subtilis conditional RNase P mutant strain SSB318, its derivative harbouring a single copy of E. coli rnpB, as well as complementation plasmids, see the supplementary information online.
In vitro transcription and 5′-end labelling. Runoff transcription with bacteriophage T7 RNA polymerase and 5′-end labelling was performed as described previously (Busch et al, 2000). The ptRNAGly substrate, and G74 and G75 variants (Fig 1; for construction, see the supplementary information online) were transcribed from plasmid pSBpt3′HH, and B. subtilis and E. coli P RNA from plasmids pDW66 and pJA2′, respectively (Busch et al, 2000).
Partial purification of RNase P. Cells resuspended in about 4 ml of buffer A (60 mM NH4Cl, 10 mM Mg[OAc]2, 6 mM dithiothreitol, 50 mM Tris–HCl, pH 7.2–7.5) per 600 mg cell pellet were lysed by sonication (Branson Sonifier 250, output 20, duty cycle 30–40%, 10 min on ice), followed by centrifugation for 45 min at 4°C and 10,000g. For the preparation of DEAE fractions, the supernatant was loaded onto a DEAE fast-flow Sepharose column (Aekta basic, GE Healthcare Europe, Munich, Germany). Elution was carried out by applying a continuous NH4Cl gradient from 60 to 550 mM NH4Cl. Active fractions eluted at about 300–400 mM NH4Cl.
Processing assays. For RNase P-catalysed processing of Thermus thermophilus ptRNAGly (and G74 or G75 variants; Fig 1), active DEAE fractions (0.25–1.0 μl; equal amounts based on A260 measurements) were incubated in buffer A (10 mM Mg[OAc]2, 6 mM dithiothreitol, 50 mM Tris–HCl, pH 7.5) containing 100 mM NH4Cl for 10 min at 37°C. Trace amounts (<1 nM) of 5′-end-labelled substrate were preincubated under the same conditions for 5 min at 55°C and 25 min at 37°C. Processing reactions were started by combining enzyme and substrate solutions and assayed at 37°C. For control cleavage by E. coli RNase P RNA (Fig 2), trace amounts (<1 nM) of 5′-end-labelled ptRNAGly were incubated with 5 μM E. coli RNase P RNA for 5 s at 37°C in 50 mM PIPES, 0.1 M Mg[OAc]2 and 1 M NH4OAc (pH 7.0 at 37°C).
Supplementary information is available at EMBO reports online (http://www.nature.com/embor/journal/vaop/ncurrent/extref/7400641-s1.pdf).
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie. C.C. was supported by funds from the Centre National de la Recherche, Université de Paris 7 and ACI Jeunes Chercheurs from the Ministère de la Recherche Française.
References
- Barrera A, Pan T (2004) Interaction of the Bacillus subtilis RNase P with the 30S ribosomal subunit. RNA 10: 482–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera A, Fang X, Jacob J, Casey E, Thiyagarajan P, Pan T (2002) Dimeric and monomeric Bacillus subtilis RNase P holoenzyme in the absence and presence of pre-tRNA substrates. Biochemistry 41: 12986–12994 [DOI] [PubMed] [Google Scholar]
- Brown S, Fournier MJ (1984) The 4.5S RNA gene of Escherichia coli is essential for cell growth. J Mol Biol 178: 533–550 [DOI] [PubMed] [Google Scholar]
- Buck AH, Dalby AB, Poole AW, Kazantsev AV, Pace NR (2005) Protein activation of a ribozyme: the role of bacterial RNase P protein. EMBO J 24: 3360–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch S, Kirsebom LA, Notbohm H, Hartmann RK (2000) Differential role of the intermolecular base-pairs G292-C(75) and G293-C(74) in the reaction catalyzed by Escherichia coli RNase P RNA. J Mol Biol 299: 941–951 [DOI] [PubMed] [Google Scholar]
- Day-Storms JJ, Niranjanakumari S, Fierke CA (2004) Ionic interactions between PRNA and P protein in Bacillus subtilis RNase P characterized using a magnetocapture-based assay. RNA 10: 1595–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang XW, Yang XJ, Littrell K, Niranjanakumari S, Thiyagarajan P, Fierke CA, Sosnick TR, Pan T (2001) The Bacillus subtilis RNase P holoenzyme contains two RNase P RNA and two RNase P protein subunits. RNA 7: 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S (1983) The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857 [DOI] [PubMed] [Google Scholar]
- Hall TA, Brown JW (2001) The ribonuclease P family. Methods Enzymol 341: 56–77 [DOI] [PubMed] [Google Scholar]
- Hansen A, Pfeiffer T, Zuleeg T, Limmer S, Ciesiolka J, Feltens R, Hartmann RK (2001) Exploring the minimal substrate requirements for trans-cleavage by RNase P holoenzymes from Escherichia coli and Bacillus subtilis. Mol Microbiol 41: 131–143 [DOI] [PubMed] [Google Scholar]
- Harris ME, Christian EL (2003) Recent insights into the structure and function of the ribonucleoprotein enzyme ribonuclease P. Curr Opin Struct Biol 13: 325–333 [DOI] [PubMed] [Google Scholar]
- Heide C, Busch S, Feltens R, Hartmann RK (2001) Distinct modes of mature and precursor tRNA binding to Escherichia coli RNase P RNA revealed by NAIM analyses. RNA 7: 553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz JC, Fierke CA (2002) The affinity of magnesium binding sites in the Bacillus subtilis RNase P × pre-tRNA complex is enhanced by the protein subunit. Biochemistry 41: 9545–9558 [DOI] [PubMed] [Google Scholar]
- Kurz JC, Niranjanakumari S, Fierke CA (1998) Protein component of Bacillus subtilis RNase P specifically enhances the affinity for precursor-tRNAAsp. Biochemistry 37: 2393–2400 [DOI] [PubMed] [Google Scholar]
- Li Y, Altman S (2003) A specific endoribonuclease, RNase P, affects gene expression of polycistronic operon mRNAs. Proc Natl Acad Sci USA 100: 13213–13218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria A, Pan T (2000) The 3′ substrate determinants for the catalytic efficiency of the Bacillus subtilis RNase P holoenzyme suggestautolytic processing of the RNase P RNA in vivo. RNA 6: 1413–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg U, Altman S (1995) Processing of the precursor to the catalytic RNA subunit of RNase P from Escherichia coli. RNA 1: 327–334 [PMC free article] [PubMed] [Google Scholar]
- Marquez SM, Harris JK, Kelley ST, Brown JW, Dawson SC, Roberts EC, Pace NR (2005) Structural implications of novel diversity in eucaryal RNase P RNA. RNA 11: 739–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit M-A, Dervyn E, Rose M, Entian K-D, McGovern S, Ehrlich DS, Bruand C (1998) PcrA is an essential DNA helicase of Bacillus subtilis fulfilling functions both in repair and rolling-circle replication. Mol Microbiol 29: 261–273 [DOI] [PubMed] [Google Scholar]
- Schön A (1999) Ribonuclease P: the diversity of a ubiquitous RNA processing enzyme. FEMS Microbiol Rev 23: 391–406 [DOI] [PubMed] [Google Scholar]
- Tallsjö A, Kirsebom LA (1993) Product release is a rate-limiting step during cleavage by the catalytic RNA subunit of Escherichia coli RNase P. Nucleic Acids Res 21: 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HY, Masquida B, Biswas R, Westhof E, Gopalan V (2003) Molecular modeling of the three-dimensional structure of the bacterial RNase P holoenzyme. J Mol Biol 325: 661–675 [DOI] [PubMed] [Google Scholar]
- Waugh DS, Pace NR (1990) Complementation of an RNase P RNA (rnpB) gene deletion in Escherichia coli by homologous genes from distantly related eubacteria. J Bacteriol 172: 6316–6321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke JM, Held R, Busch S, Hartmann RK (1999) Role of metal ions in the hydrolysis reaction catalyzed by RNase P RNA from Bacillus subtilis. J Mol Biol 290: 433–445 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information