

Abstract
Mutations in the SEPN1 gene encoding the selenoprotein N (SelN) have been described in different congenital myopathies. Here, we report the first mutation in the selenocysteine insertion sequence (SECIS) of SelN messenger RNA, a hairpin structure located in the 3′ untranslated region, in a patient presenting a classical although mild form of rigid spine muscular dystrophy. We detected a significant reduction in both mRNA and protein levels in the patient's skin fibroblasts. The SECIS element is crucial for the insertion of selenocysteine at the reprogrammed UGA codon by recruiting the SECIS-binding protein 2 (SBP2), and we demonstrated that this mutation abolishes SBP2 binding to SECIS in vitro, thereby preventing co-translational incorporation of selenocysteine and SelN synthesis. The identification of this mutation affecting a conserved base in the SECIS functional motif thereby reveals the structural basis for a novel pathological mechanism leading to SEPN1-related myopathy.
Keywords: SECIS structure, selenoprotein, SEPN1-related myopathy
Introduction
The selenoprotein N (SelN) was the first member of the selenoprotein family shown to be involved in a genetic disorder (Moghadaszadeh et al, 2001), and the identification of disease-causing mutations in the SEPN1 gene encoding SelN delineated a novel entity of early-onset muscular disorders (Ferreiro et al, 2004). This SEPN1-related myopathy now regroups four autosomal recessive disorders: rigid spine muscular dystrophy (RSMD1, Online Mendelian Inheritance in Man (http://www.ncbi.nlm.nih.gov/Omim/) [MIM *602771]; Dubowitz, 1973; Moghadaszadeh et al, 2001), the ‘classical form' of multiminicore disease (MmD [MIM *607552]; Ferreiro et al, 2002), rare cases of desmin-related myopathy with Mallory body-like inclusions (MB-DRM [MIM 253850]; Ferreiro et al, 2004) and a recessive form of congenital fibre-type disproportion (CFTD; Clarke et al, in press). All these phenotypes are characterized by early-onset generalized hypotonia and weakness, with predominant axial muscle impairment leading to neck weakness and scoliosis, a variable degree of spinal rigidity and life-threatening respiratory failure. Recent studies demonstrated that SelN is a glycoprotein targeted to the endoplasmic reticulum and expressed at an early developmental stage (Lescure et al, 1999; Petit et al, 2003). Selenium is co-translationally incorporated into the nascent polypeptide as a selenocysteine residue (Sec), the 21st amino acid of the genetic code, and is encoded by a reprogrammed UGA codon within the open reading frame (Hatfield & Gladyshev, 2002; reviewed by Lescure et al, 2002). Recognition of UGA as a selenocysteine instead of a termination codon requires a selenocysteine insertion sequence (SECIS), a cis-acting stem–loop structure located in the 3′ untranslated region (UTR; reviewed by Krol, 2002). This structural motif is recognized by the SECIS-binding protein 2 (SBP2), which in turn recruits the complex formed between the specialized elongation factor mSelB/eFSec and a specific Sec transfer RNA (Zavacki et al, 2003). Among the 26 selenoproteins identified in humans (Kryukov et al, 2003; Castellano et al, 2004), those that are functionally characterized are enzymes involved in oxidation–reduction reactions (Driscoll & Chavatte, 2004; Johansson et al, 2005). Nevertheless, the function of many selenoproteins, including SelN, remains unknown.
So far, all the pathological mutations identified in SEPN1 are localized in the 5′UTR and the coding sequence. They consist of micro-deletions or insertions leading to frameshift, splice site mutations leading to aberrant pre-messenger RNA splicing or single-nucleotide changes giving rise to missense mutations, essentially centred around the potential catalytic site. Here, we report a novel mutation in SEPN1 causing RSMD1. Interestingly, this is the first mutation identified in the 3′UTR of the gene, more specifically within the SECIS element. Furthermore, we demonstrated that this point mutation in the functional core of the SECIS element disables the binding of the SBP2 protein and probably selenocysteine incorporation, leading to a drastic reduction of mRNA and protein levels.
Results
A mild rigid spine muscular dystrophy phenotype
The propositus is a Syrian 45-year-old women (II-2) whose parents, both healthy, are distant relatives (Fig 1A). Briefly, during early infancy, her parents noted that her neck muscles were weak; she achieved independent ambulation before 18 months of age but presented poor sports performance during childhood. After her first pregnancy at the age of 32 years, she complained of persisting difficulties in climbing stairs and exercise intolerance. Two years later, after her second pregnancy, a significant restrictive respiratory insufficiency was diagnosed. Non-invasive nocturnal ventilation was implemented and proved beneficial in terms of exercise intolerance and weakness. At age 45 years, her physical examination showed a marked weakness and wasting in the neck, trunk flexors and pelvic muscles. She had significant rigidity of the cervical and dorsal spine as well as a typical although moderate scoliosis, with dorsal lordosis and lateral trunk deviation. The patient is at present ambulant and can walk long distances unaided provided that she has been properly ventilated at night, but needs some support in climbing stairs. Two sisters were also reported to present muscle weakness and scoliosis.
Figure 1.
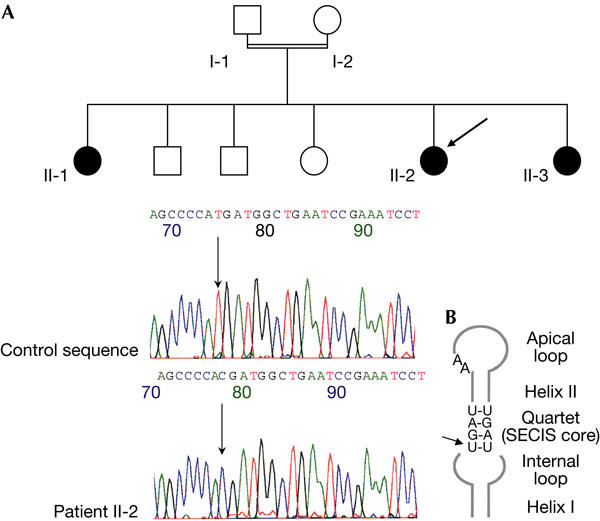
Pedigree of the family and SEPN1 mutation in the selenocysteine insertion sequence (SECIS) element. (A) The propositus (II-2) is indicated by an arrow. Affected individuals are indicated by black symbols. No DNA sample was available for individual II-3. Electrophoregrams of nucleotide sequences flanking the mutation in the SECIS element in the 3′ untranslated region of SEPN1. The upper and lower panels represent the control and patient sequences, respectively. The arrow indicates the position of the nucleotide change g.17195T>C (numbering according to SEPN1 genomic sequence from Moghadaszadeh et al, 2001; GenBank accession number NM_020451). (B) Schematic representation of the human selenoprotein N SECIS element showing the hairpin structure composed of two helices (I and II) interrupted by an internal loop and terminated by an apical loop. The position of the mutation affecting the highly conserved non-Watson–Crick base-pair quartet UGAN/NGAN is indicated by an arrow.
A disease-causing mutation in the 3′UTR of SelN mRNA
Genetic analyses and sequencing identified a single-nucleotide change at the homozygous state in the 3′UTR of SelN mRNA, more precisely located within the SECIS element. This mutation consists of a T to C transition at position 17195 (g.17195T>C; GenBank accession number NM_020451; Fig 1A). Interestingly, this mutation affects the invariant 5′U in the quartet of non-Watson–Crick base pairs UGAN/NGAN, the main functional motif of the SECIS element (Fig 1B). None of the polymorphisms identified in the SEPN1 gene (Table 1) was present in patient II-2. The same homozygous mutation was found in the affected sister (II-1).
Table 1.
Nucleotide changes in the SEPN1 gene
Mutation in the SECIS element impairs SelN expression
The consequences of this mutation were examined at both the protein and mRNA levels in human skin fibroblasts by western blotting and quantitative reverse transcription–PCR, respectively. The 70 kDa band corresponding to SelN was indeed detected in a control individual and in an RSMD1 patient with a c.1386A>G mutation changing the selenocysteine codon into a glycine (U462G; Fig 2). In contrast, using the carboxy-terminal 137 antibody, a similarly faint band was observed in patient II-2 (30 or 50 μg of proteins) and in five other RSMD1 or MmD patients with various homozygous frameshift mutations: c.A1>G affecting the initiation codon, c.23–32dup10bp in exon 1, c.713–714insA in exon 5 or c.1446delC in exon 11 (Fig 2). Similar results were observed after prolonged exposure (data not shown). In addition, identical results were obtained with the more proximal R168 antibody, but no truncated protein corresponding to the recognition of the selenocysteine codon as a premature termination codon (PTC) was detected (data not shown).
Figure 2.
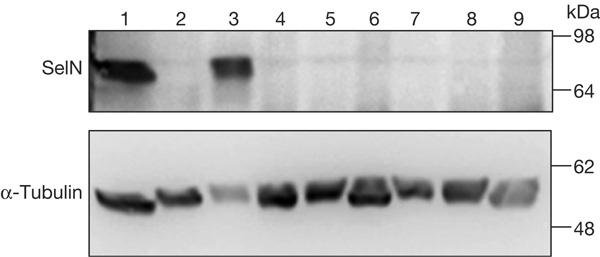
Protein levels in cultured skin fibroblasts. Total proteins were extracted from fibroblasts of a control (lane 1) and seven RSMD1 (rigid spine muscular dystrophy) and MmD patients with homozygous SEPN1 mutations (lanes 2–9). Total proteins (50 μg) were resolved on 7% SDS–polyacrylamide gel electrophoresis and analysed by western blotting with an anti-selenoprotein N (SelN) R137 antibody. Lane 1: control; lanes 2 and 9: patient II-2, 50 and 30 μg, respectively; lanes 3 and 4: patients with the c.1386A>G and c.1446delC mutations (Moghadaszadeh et al, 2001); lane 5: c.A1>G mutation affecting the initiation codon; lanes 6 and 7: two patients with c.713–714insA mutation (Ferreiro et al, 2002); lane 8: c.23–32dup10 bp mutation. A monoclonal antibody against α-tubulin was used as a loading control.
The mRNA levels were studied by real-time quantitative PCR on total RNA extracted from cultured fibroblasts. SelN mRNA levels from patient II-2 were significantly reduced to 25% (median of seven experiments) compared with the control individual (Fig 3). In fibroblasts from the patient with the c.1446delC mutation, SelN mRNA levels decreased markedly to 4% of the control (median of three experiments; Fig 3). These results indicated a destabilization of the mRNA in both patients. However, in the patient with the missense change in the UGA selenocysteine codon SelN, mRNA levels were reduced to a much lesser extent (37% of control levels; Fig 3), and were nearly normal in the patient with the mutation in the initiation codon (Fig 3).
Figure 3.
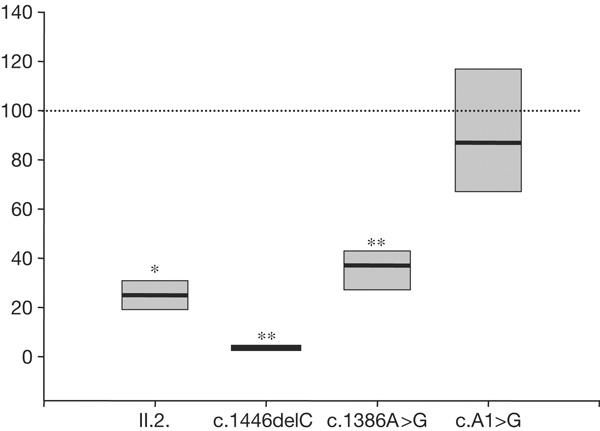
Selenoprotein N messenger RNA levels normalized to HPRT (hypoxanthine phosphoribosyltransferase) mRNA. Median values of several quantitative reverse transcription–PCR (n=7 for II-2; n=3 for the patients harbouring the c.1446delC, c.1386A>G and c.A1>G mutations) are presented ±deviation to 25–75%. Statistical significance was determined by a Mann–Whitney rank sum test (*P=0.001; **P=0.024).
The SECIS mutation abolishes SBP2 binding in vitro
The effect of the SECIS mutation on the selenoprotein-specific synthesis machinery was investigated. As it was previously shown that the non-Watson–Crick quartet is the primary binding site for SBP2, the interaction between SBP2 and an SECIS RNA carrying the U to C mutation was tested in vitro and compared with the wild type. As depicted in Fig 4, no complex between SBP2 and the SECIS harbouring the patient mutation could be detected even in the presence of the highest amount of SBP2, a concentration for which most of the wild-type SECIS RNA was retarded (Fig 4, compare lanes 5 and 10). It is impressive that this single point mutation totally prevents formation of the SBP2–SECIS complex in the in vitro assay.
Figure 4.
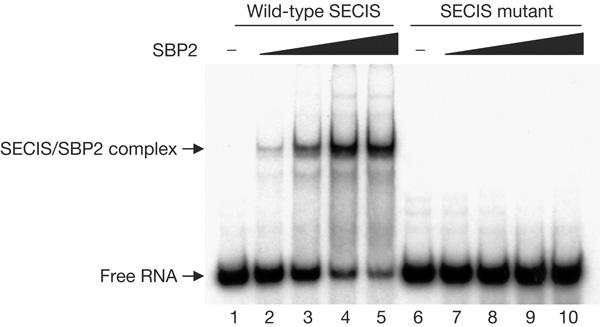
The binding activity of selenocysteine insertion sequence (SECIS)-binding protein 2 (SBP2) to the SECIS element harbouring the patient mutation. A DNA fragment containing the selenoprotein N SECIS element (corresponding to position 17187–17245 of genomic DNA) was PCR amplified and cloned downstream of a T7 RNA polymerase transcription start site. The U to C mutation was introduced by site-directed mutagenesis. Both wild-type and mutated SECIS RNAs were produced and radiolabelled by T7 transcription in the presence of [α-32P]ATP. Either wild-type SECIS RNA (lanes 1–5) or mutated (lanes 6–10) 32P-labelled SECIS RNA was incubated in the presence of increasing amounts of recombinant SBP2 protein (200 ng, lanes 2 and 7; 400 ng, lanes 3 and 8; 800 ng, lanes 4 and 9; 1,000 ng, lanes 5 and 10). The protein/RNA complexes were resolved on a 6% non-denaturing polyacrylamide gel and visualized by autoradiography. Migration was compared with the signal obtained in the absence of SBP2 (lanes 1 and 6). Migration positions of the free SECIS RNA and SBP2/SECIS complexes are shown.
Discussion
Here, we identified the first mutation in the 3′UTR of the SEPN1 gene leading to a typical form of RSMD1 characterized by an early-onset and relatively mild phenotype until the patient reached her 30s, worsened by the pregnancies. The mutation, g.17195T>C, is located in the SECIS element of SelN mRNA, a stem–loop structure, crucial for the co-translational incorporation of a selenocysteine residue at the in-frame UGA codon in exon 10. The mutation affects the invariant 5′U in the quartet of non-Watson–Crick base pairs UGAN/NGAN, the main functional motif of SECIS elements that are found in all selenoprotein mRNAs (Walczak et al, 1996, 1998; Fagegaltier et al, 2000). We found that the SEPN1 mutation induced a drastic reduction of SelN in the patient's skin fibroblasts and that in vitro the U>C SECIS RNA could no longer form a complex with SBP2, a necessary interaction for selenocysteine incorporation (Copeland & Driscoll, 1999; Fletcher et al, 2001).
Interestingly, mutations of this conserved U residue in other SECIS elements have been previously shown to impair the formation of the SBP2–SECIS RNA complex in vitro and to abolish selenoprotein synthesis (Lesoon et al, 1997; Fletcher et al, 2001). Further, a structure-guided strategy corroborated these findings by identifying putative contact sites between the human SPB2 and SECIS RNA (Allmang et al, 2002). In that study, mutations of specific amino-acid residues of SBP2 abolished RNA-binding activities and, interestingly, the mutants that had the most drastic effects contain residues that are in close contact with the U in the SECIS RNA.
Interestingly, a 1125G>A polymorphism in the functional SECIS element of the human 15 kDa selenoprotein has been reported and shown to influence selenocysteine incorporation into this protein and its response to selenium supplementation (Kumaraswamy et al, 2000). However, it occurs in the apical helix that does not bear sequence conservation, and the consequences of this polymorphism on SBP2 binding to the SECIS are unknown. The frequency of this polymorphism varies with ethnicity, and it might contribute to cancer risk (Jun Hu et al, 2001).
The binding activity of SBP2 to the SECIS element is primarily involved in the redefinition of the UGA as a selenocysteine codon, therefore preventing termination at this position (Copeland et al, 2000). As the U to C mutation in the SECIS RNA disables SBP2 binding, the UGA codon is probably recognized as a premature termination codon and, as a consequence, the mRNA is destabilized. The most common mechanism leading to PTC-bearing mRNA degradation is the nonsense-mediated decay pathway (NMD; reviewed by Maquat, 2004). Although the putative termination codon reported here is the last codon of exon 10 and thus does not follow the ‘position-of-an-exon-exon-junction' rule, involvement of the NMD pathway cannot be excluded, as previously described for the T-cell receptor-β mRNA (Carter et al, 1996; Wang et al, 2002). Recently, another mechanism for mRNA degradation was described, involving a binding site to the Staufen1 protein in the 3′UTR of the mRNA (SMD; Kim et al, 2005). Indeed, in patient II-2, we cannot rule out that either of these mechanisms is involved in the degradation of the mRNA.
Although this patient is not the mildest case in our series, her respiratory insufficiency was diagnosed later than in some other patients carrying various homozygous frameshift mutations also leading to an absence of SelN. Indeed, most of these cases showed a more severe phenotype necessitating respiratory assistance during the second decade of life. Even among patients with an identical SEPN1 mutation, a high inter-individual variability was described (Ferreiro et al, 2004; reviewed by Rederstorff et al, 2006), suggesting the involvement of other genetic or environmental factors in the severity of the phenotype. Indeed, SBP2 was shown to be part of a large protein complex of 500 kDa in vivo (Copeland et al, 2000; Kinzy et al, 2005); it is thus likely that other factors from this complex contribute to the stabilization of the SBP2–SECIS interaction in vivo, and therefore may allow residual synthesis of SelN protein, particularly in muscle. In addition, some basal level of readthrough cannot be excluded (Howard et al, 2005). Identification of other patients with SECIS element mutations would help to further clarify the phenotypic consequences of this type of mutations.
In conclusion, we report here the first homozygous mutation in the SECIS RNA element of SEPN1. This U to C transition lies within the non-Watson–Crick quartet, thus affecting the main functional region of the SECIS RNA. In accordance with structural mapping of SBP2–SECIS interaction sites and mutational studies, we showed that this mutation abolished the interaction between SBP2 and SECIS RNA in vitro, preventing the co-translational incorporation of selenocysteine in the nascent polypeptide. This report is the first in vivo demonstration of the importance of a specific SECIS mutation for SelN function in humans.
Methods
Genetic analysis. Analyses were carried out on genomic DNA extracted from leucocytes of the propositus (II-2) and her sister (II-1) after informed written consent was obtained according to the ethical committee regulations of Assistance Publique-Hôpitaux de Paris. All 12 coding exons of SEPN1 were amplified with primers located in the intronic flanking regions and the SECIS element with primers located in the 3′UTR domain. Mutation screening was carried out by DNA single-strand conformation polymorphism analysis (Ferreiro et al, 2002) at two temperatures (7 and 20°C) on 10% polyacrylamide gels, and bands were shown by silver staining (Bio-Rad, Marnes-la-Coquette, France). All fragments with an abnormal profile were sequenced on both strands with the Big Dye terminator sequencing kit (Applera, Courtaboeuf, France) and run on an ABI3100 sequencer (Applera).
Immunoblotting. Total proteins from cultured fibroblasts obtained from skin biopsies were extracted in a 10% SDS-containing buffer. Following fractionation under reducing conditions by SDS–polyacrylamide gel electrophoresis, proteins were transferred onto PVDF membranes (Immobilon-P, Millipore, Saint-Quentin-en-Yvelines, France) that were hybridized with the polyclonal antibody R137 against SelN, as described previously (Petit et al, 2003). A monoclonal antibody against α-tubulin (clone B-5-1-2; Sigma, Saint-Quentin-en-Yvelines, France) was used as a loading control.
RNA extraction and real-time reverse transcription–PCR. Total RNA was extracted from cultured fibroblasts using Trizol, as recommended by the manufacturer. RNA quality control and quantification was performed using the Bioanalyzer apparatus (Agilent, Massy, France). First-strand complementary DNA was prepared from 1 μg of total RNA using oligo(dT)18 and Superscript II Reverse Transcriptase as recommended by the manufacturer (Invitrogen, Cergy Pontoise, France). Quantitative real-time PCR was carried out on equal amounts of cDNAs, using the LightCycler real-time PCR machine (Roche Diagnostics, Mannheim, Germany), and SelN mRNA levels were quantified relative to the mRNA of the housekeeping gene HPRT (hypoxanthine phosphoribosyltransferase). In each experiment, results were expressed relative to control fibroblasts normalized to 100. Because the equal variance test failed, a Mann–Whitney rank sum test was performed on data medians with SigmaStat (Systat, Erkrath, Germany).
Band-shift assay. DNA fragments, corresponding to either wild-type or mutated SECIS element, were PCR amplified and cloned downstream of a T7 promoter. RNA were synthesized by in vitro T7 transcription and incubated in the presence of increasing amounts of SBP2 protein, as described previously (Allmang et al, 2002). The recombinant SBP2 protein used in this experiment corresponds to the C-terminal two-thirds part of the protein, which contains the specific RNA-binding domain. The RNA–protein complex was separated by electrophoretic shift assay.
Acknowledgments
We are grateful to the family for its participation in this study. We thank A. Mégarbané (Beirut) for the extraction of DNA from patient II-1. This work was supported by funds from the Institut National de la Santé et de la Recherche Médicale, the Association Française contre les Myopathies, the GIS-Institut des Maladies Rares and the Assistance Publique-Hôpitaux de Paris.
References
- Allmang C, Carbon P, Krol A (2002) The SBP2 and 15.5 kD/Snu13p proteins share the same RNA binding domain: identification of SBP2 amino acids important to SECIS RNA binding. RNA 8: 1308–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter MS, Li S, Wilkinson MF (1996) A splicing-dependent regulatory mechanism that detects translational signals. EMBO J 15: 5965–5975 [PMC free article] [PubMed] [Google Scholar]
- Castellano S, Novoselov SV, Kryukov GV, Lescure A, Blanco E, Krol A, Gladyshev VN, Guigo R (2004) Reconsidering the evolution of eukaryotic selenoproteins: a novel nonmammalian family with scattered phylogenetic distribution. EMBO Rep 5: 71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke NF et al. (2005) SEPN1: Associated with congenital fiber type disproportion & insulin resistance. Ann Neurol 19 Dec [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Copeland PR, Driscoll DM (1999) Purification, redox sensitivity, and RNA binding properties of SECIS-binding protein 2, a protein involved in selenoprotein biosynthesis. J Biol Chem 274: 25447–25454 [DOI] [PubMed] [Google Scholar]
- Copeland PR, Fletcher JE, Carlson BA, Hatfield DL, Driscoll DM (2000) A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. EMBO J 19: 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll DM, Chavatte L (2004) Finding needles in a haystack. In silico identification of eukaryotic selenoprotein genes. EMBO Rep 5: 140–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubowitz V (1973) Rigid spine syndrome: a muscle syndrome in search of a name. Proc R Soc Med 66: 219–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagegaltier D, Lescure A, Walczak R, Carbon P, Krol A (2000) Structural analysis of new local features in SECIS RNA hairpins. Nucleic Acids Res 28: 2679–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreiro A et al. (2002) Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet 71: 739–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreiro A et al. (2004) Desmin-related myopathy with Mallory body-like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol 55: 676–686 [DOI] [PubMed] [Google Scholar]
- Fletcher JE, Copeland PR, Driscoll DM, Krol A (2001) The selenocysteine incorporation machinery: interactions between the SECIS RNA and the SECIS-binding protein SBP2. RNA 7: 1442–1453 [PMC free article] [PubMed] [Google Scholar]
- Hatfield DL, Gladyshev VN (2002) How selenium has altered our understanding of the genetic code. Mol Cell Biol 22: 3565–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MT, Aggarwal G, Anderson CB, Khatri S, Flanigan KM, Atkins JF (2005) Recoding elements located adjacent to a subset of eukaryal selenocysteine-specifying UGA codons. EMBO J 24: 1596–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson L, Gafvelin G, Arner ES (2005) Selenocysteine in proteins—properties and biotechnological use. Biochim Biophys Acta 1726: 1–13 [DOI] [PubMed] [Google Scholar]
- Jun Hu Y et al. (2001) Distribution and functional consequences of nucleotide polymorphisms in the 3′-untranslated region of the human Sep15 gene. Cancer Res 61: 2307–2310 [PubMed] [Google Scholar]
- Kim YK, Furic L, DesGroseillers L, Maquat LE (2005) Mammalian Staufen1 recruits Upf1 to specific mRNA 3′UTRs so as to elicit mRNA decay. Cell 120: 195–208 [DOI] [PubMed] [Google Scholar]
- Kinzy SA, Caban K, Copeland PR (2005) Characterization of the SECIS binding protein 2 complex required for the co-translational insertion of selenocysteine in mammals. Nucleic Acids Res 33: 5172–5180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol A (2002) Evolutionarily different RNA motifs and RNA–protein complexes to achieve selenoprotein synthesis. Biochimie 84: 765–774 [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN (2003) Characterization of mammalian selenoproteomes. Science 300: 1439–1443 [DOI] [PubMed] [Google Scholar]
- Kumaraswamy E et al. (2000) Structure–expression relationships of the 15-kDa selenoprotein gene. Possible role of the protein in cancer etiology. J Biol Chem 275: 35540–35547 [DOI] [PubMed] [Google Scholar]
- Lescure A, Gautheret D, Carbon P, Krol A (1999) Novel selenoproteins identified in silico and in vivo by using a conserved RNA structural motif. J Biol Chem 274: 38147–38154 [DOI] [PubMed] [Google Scholar]
- Lescure A, Fagegaltier D, Carbon P, Krol A (2002) Protein factors mediating selenoprotein synthesis. Curr Protein Peptide Sci 3: 143–151 [DOI] [PubMed] [Google Scholar]
- Lesoon A, Mehta A, Singh R, Chisolm G, Driscoll D (1997) An RNA-binding protein recognizes a mammalian selenocysteine insertion sequence element required for cotranslational incorporation of selenocysteine. Mol Cell Biol 17: 1977–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquat LE (2004) Nonsens-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat Rev Mol Cell Biol 5: 89–99 [DOI] [PubMed] [Google Scholar]
- Moghadaszadeh B et al. (2001) Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat Genet 29: 17–18 [DOI] [PubMed] [Google Scholar]
- Petit N, Lescure A, Rederstorff M, Krol A, Moghadaszadeh B, Wewer UM, Guicheney P (2003) Selenoprotein N: an endoplasmic reticulum glycoprotein with an early developmental expression pattern. Hum Mol Genet 12: 1045–1053 [DOI] [PubMed] [Google Scholar]
- Rederstorff M, Krol A, Lescure A (2006) Understanding the importance of selenium and selenoproteins in muscle function. Cell Mol Life Sci 63: 52–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak R, Westhof E, Carbon P, Krol A (1996) A novel RNA structural motif in the selenocysteine insertion element of eukaryotic selenoprotein mRNAs. RNA 2: 367–379 [PMC free article] [PubMed] [Google Scholar]
- Walczak R, Carbon P, Krol A (1998) An essential non-Watson–Crick base pair motif in 3′UTR to mediate selenoprotein translation. RNA 4: 74–84 [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hamilton JI, Carter MS, Li S, Wilkinson MF (2002) Alternatively spliced TCR mRNA induced by disruption of reading frame. Science 297: 108–110 [DOI] [PubMed] [Google Scholar]
- Zavacki AM, Mansell JB, Chung M, Klimovitsky B, Harney JW, Berry MJ (2003) Coupled tRNASec-dependent assembly of the selenocysteine decoding apparatus. Mol Cell 11: 773–781 [DOI] [PubMed] [Google Scholar]