

Abstract
Regulation of blood coagulation is critical for maintaining blood flow, while preventing excessive bleeding or thrombosis. One of the principal regulatory mechanisms involves heparin activation of the serpin antithrombin (AT). Inhibition of several coagulation proteases is accelerated by up to 10 000-fold by heparin, either through bridging AT and the protease or by inducing allosteric changes in the properties of AT. The anticoagulant effect of short heparin chains, including the minimal AT-specific pentasaccharide, is mediated exclusively through the allosteric activation of AT towards efficient inhibition of coagulation factors (f) IXa and Xa. Here we present the crystallographic structure of the recognition (Michaelis) complex between heparin-activated AT and S195A fXa, revealing the extensive exosite contacts that confer specificity. The heparin-induced conformational change in AT is required to allow simultaneous contacts within the active site and two distinct exosites of fXa (36-loop and the autolysis loop). This structure explains the molecular basis of protease recognition by AT, and the mechanism of action of the important therapeutic low-molecular-weight heparins.
Keywords: fondaparinux, Michaelis complex, pentasaccharide, serpin, thrombosis
Introduction
Antithrombin (AT, also known as antithrombin III) is the principal inhibitor of the coagulation proteases. Its suicide-substrate mechanism of inhibition is typical of other members of the serpin family, involving the rate-limiting formation of a recognition complex (also known as a Michaelis complex), followed by entry into the proteolytic cycle (Gettins, 2002). At the acyl–enzyme intermediate stage of proteolysis, the reactive center loop (RCL) of the serpin rapidly incorporates into the central β-sheet, resulting in the 70 Å translocation of the protease from the top to the bottom of the serpin (Huntington et al, 2000). The protease is rendered inactive by the resulting distortion of the active site architecture. How the complexity of the serpin mechanism confers a regulatory advantage over the classical ‘lock-and-key'-type protease inhibitors is best illustrated by the activation mechanism of AT.
Blood coagulation is a tightly regulated process requiring rapid and localized activation of coagulation proteases at the site of vascular damage (for reviews, see among others Davie et al, 1991; Davie, 1995; Dahlback, 2000; Butenas and Mann, 2002; Schenone et al, 2004). If the hemostatic response is insufficiently robust, the result is life-threatening bleeding; however, if it is not limited, the result is life-threatening thrombosis. As AT circulates at a high concentration (2.3 μM) in the blood, its activity must be under tight regulatory control to allow appropriate clot formation while preventing thrombosis. This is achieved by exploiting the inherent conformational plasticity of the serpins (Huntington, 2003), where AT circulates in a conformation that is poorly reactive towards its targets, and only by interacting with a specific pentasaccharide sequence (for a historical review, see Petitou et al, 2003) found in therapeutic heparin and the related heparan sulfate expressed on the blood vessel walls does AT become an efficient inhibitor of the coagulation proteases (Figure 1). AT inhibition of its principal targets, factors IXa, Xa (fIXa and fXa) and thrombin, is accelerated by up to four orders of magnitude in the presence of heparin (Huntington, 2005).
Figure 1.
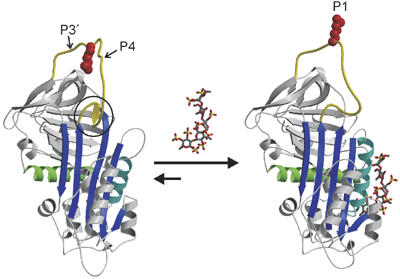
The heparin-binding mechanism of AT. The structural features of native and pentasaccharide-activated AT are illustrated by ribbon diagrams. AT is shown in the classic orientation, with β-sheet A (blue) facing, helix A (green) in the back, the principal heparin-binding helix D (cyan) to the right, and the RCL (yellow) at the top. RCL residues are numbered from the scissile P1–P1′ bond towards the N- and C-termini, respectively, and the positions of the P4 and P3′ residues are indicated. In native AT the N-terminal region of the RCL (hinge region, circle) is incorporated as strand 4 in β-sheet A, which constrains the RCL and the P1 Arg393 side chain (red space-filling). The heparin pentasaccharide (rods with gray C, red O, and yellow S) binds with contacts on helices A and D and induces local and global conformational changes in AT. Of particular relevance is the expulsion of the hinge region from β-sheet A, which predictably releases the constraints on the RCL and reorients the P1 side chain.
How AT is activated by heparin is of great medical importance, as hereditary AT deficiency is a leading cause of thrombosis (van Boven and Lane, 1997) and because heparin is the most widely used drug for the prophylaxis and treatment of thrombosis (Bussey and Francis, 2004). Partially depolymerized heparin (known as low-molecular-weight heparin) has recently surpassed unfractionated heparin as the anticoagulant drug of choice, due to a reduced risk of bleeding and of heparin-induced thrombocytopenia, while preserving effective antithrombotic activity. In addition, the AT-specific pentasaccharide has also been chemically synthesized (Petitou et al, 1986) and is now marketed (generic name fondaparinux) for similar indications as low-molecular-weight heparin (Petitou and van Boeckel, 2004). While small heparin fragments are effective antithrombotic agents, they are only capable of accelerating the rate of inhibition of fIXa and fXa by ∼500-fold and do not appreciably accelerate the inhibition of thrombin. This is because a portion of the activating effect of unfractionated heparin is conferred by a template mechanism, where both AT and protease bind to the same heparin chain. The ability of heparin to act as a template for AT and proteases depends on its length, and thus on its molecular weight. Approximately 18 monosaccharide units are minimal to bridge AT to thrombin (Bray et al, 1989), while heparin lengths of ∼30 units are required to bridge AT to fIXa and fXa (Rezaie, 1998; Rezaie and Olson, 2000; Bedsted et al, 2003). Thus, the activation of AT conferred by low-molecular-weight heparin and the synthetic pentasaccharide is fully allosteric. The pentasaccharide-induced conformational change in AT is summarized in Figure 1, and involves rearrangements in the heparin-binding region and elsewhere. Of particular relevance for the allosteric activation of AT is the expulsion of the N-terminal portion of the RCL (the hinge region) from β-sheet A, effectively relieving a constraint on the flexibility of the RCL.
The molecular basis of AT allostery is still unresolved, but two factors have become clear from mutagenesis and structural studies: the hinge region of the RCL must be expelled from β-sheet A (Langdown et al, 2004) and exosite interactions between AT and fIXa or fXa are critically involved (Olson and Chuang, 2002). We present here the crystal structure of AT in complex with the synthetic pentasaccharide and the catalytically inert S195A variant of fXa. The structure reveals extensive active site and exosite interactions and demonstrates the requirement of AT conformational change for their simultaneous engagement.
Results
Crystallization
Our initial attempts to crystallize the Michaelis complex between pentasaccharide-activated AT and S195A fXa over the last 5 years were unsuccessful. To improve our chances of success, we utilized several forms of AT (the α-glycoform of plasma AT, the recombinant β-glycoform of AT (which lacks glycosylation at Asn135), and an AT variant resistant to the latency transition (Li et al, 2004)), with both mammalian cell and Escherichia coli-derived S195A fXa, but no crystals formed. We thus decided to alter the surface properties of the largest protein in the complex, AT, in order to engender crystal contacts. The approach is to reduce the ‘surface entropy' of the protein by substituting flexible Lys and Glu residues with Ala (Derewenda, 2004). We chose the 350 loop because its location on the bottom of AT would not be expected to affect heparin binding or protease recognition (Figure 2A). AT variant E347A/K348A/K350A was thus created on the β-glycoform background S137A (prevents glycosylation at N135), and, as predicted, the mutations did not alter heparin affinity, rate of fXa inhibition, nor the dependence of fXa inhibition on pentasaccharide concentration (data not shown). Small crystals were obtained from the initial screen, which were shown to contain AT and fXa in equal amounts by SDS–PAGE (data not shown). Subsequent refinement of crystallization conditions resulted in slightly larger crystals, which diffracted poorly. Only after annealing were quality diffraction data obtained. Satisfyingly, the resulting structure revealed a crystal contact between the mutated region of one of the AT monomers with the N-terminus of the light chain of a crystallographically related fXa molecule. It is clear from the nature of the interaction, involving an Arg side chain of fXa, that replacement of the two Lys residues was essential to allow formation of the crystal contact (Supplementary Figure 1).
Figure 2.
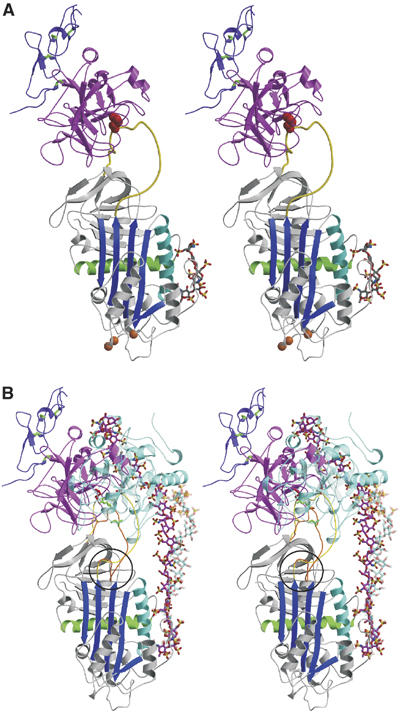
Crystal structure of the Michaelis complex between pentasaccharide-activated AT and fXa. (A) Stereo view of the complex with AT colored as before, the protease domain of fXa in magenta, and the second EGF-like domain of fXa in purple (with disulfide bonds shown in green). The orientation of fXa on AT is slightly to the left of the long axis of AT, and is rotated significantly to the left to engage in direct exosite contacts with the top surface of AT. The positions of the AT mutations which allowed crystal contacts to form are indicated by orange balls. (B) It is clear from the stereo representation of the AT–thrombin Michaelis complex superimposed on the AT–fXa complex why one is insensitive to hinge region expulsion and the other critically depends on hinge region extension (hinge region is circled). The RCL of the thrombin complex is orange, with green rods for P4 Ile390 and P1 Arg393, and the RCL from the fXa complex is yellow, with P4 and P1 residues colored red. Thrombin (semitransparent cyan) is oriented towards the front of AT and the RCL enters the active site of thrombin by the most direct line, with P4–P1 aligned along the z-axis (into the plane). In contrast, fXa is translated towards the left and rotated so that the P4–P1 enters the active site cleft along the x-axis (from right to left). The position and orientation of the active site cleft of fXa is dictated by the exosite contacts, which are evident to the left of the RCL. The forward position and orientation of thrombin permit short heparin chains (∼16mer) to bridge the heparin-binding sites of the two molecules (semitransparent cyan rods from the structure by Li et al, 2004). In contrast, based on the position and orientation of fXa observed here, a 28mer (magenta rods for the modeled heparin chain) would be predicted to be the minimal required heparin length for bridging the AT–fXa Michaelis complex.
Overall structure
Two copies of the AT–fXa–pentasaccharide complex were found in the asymmetric unit by molecular replacement. The two complexes proved to be identical, except at certain crystal contacts, suggesting a single binding mode for the activated AT–fXa Michaelis complex. A ribbon diagram of the complex in the classic orientation (for serpins the classic orientation places β-sheet A in front and the RCL on top) is shown in Figure 2A. fXa is docked onto the top of AT in a position similar to that observed for thrombin in its complex with heparin cofactor II (HCII) (Baglin et al, 2002), but the position is somewhat forward and to the right. Comparison to the position of thrombin observed in the heparin-bridged AT–thrombin Michaelis complex (Li et al, 2004) (Figure 2B) reveals a translation of 17.6 Å towards the back left-hand side of AT for fXa, accompanied by a 82° rotation in the xz plane and a 30° rotation in the yz plane (based on centroids of heavy chains, with the x-axis from left to right and the y-axis from bottom to top along the long axis of AT). The resulting orientation of fXa places the active site facing towards the right in order to engage in exosite contacts on the left. The orientation of the active site cleft of fXa requires a 15.1 Å movement of the P4 residue relative to the position adopted in the AT–thrombin structure, and a 17.0 Å displacement of P6. The P4 interaction could not occur without the expulsion and extension of the hinge region of the RCL out of and away from β-sheet A (Figure 2B). With exception to the RCL, the conformation of AT is not significantly different from that observed in the previous structure of a pentasaccharide-bound form (McCoy et al, 2003) (1E03), with an average Cα RMSD of 0.80 Å for all 405 equivalent Cα atoms (excluding RCL residues 380–400), although there is a small narrowing of the s3A–s5A distance at the top of β-sheet A (∼0.5 Å). The conformation of fXa is also very similar to that used for molecular replacement (1F0S; Maignan et al, 2000), with an average RMSD of 0.74 Å, with only small accommodating shifts in surface loops observed.
AT–pentasaccharide interactions
In the previous structure of activated AT (Jin et al, 1997; McCoy et al, 2003), the pentasaccharide used was a synthetic high-affinity variant of the natural pentasaccharide, known as idraparinux, which differs in extent and type of sulfation and by the methylation of all hydroxyl groups (Herbert et al, 1996; Petitou and van Boeckel, 2004). The present structure represents the first of AT bound to the synthetic version of the naturally occurring pentasaccharide (fondaparinux), and it is thus worthwhile to briefly describe the interactions. The density for the pentasaccharide is of high quality, as shown in Figure 3A, and the average temperature factor for the two copies of the pentasaccharide is quite low for a carbohydrate polymer (48 Å2). A schematic representation of the individual side chain interactions is given in Figure 3B, and an exhaustive list of all interactions is given in Supplementary Table 1. The pentasaccharide conformation and interactions are very similar to those previously observed for idraparinux, and corroborate the conclusion from mutagenesis data that the critical interactions involve Arg46, Arg47, Lys114, Lys125, and Arg129, with Lys114 of principal importance.
Figure 3.
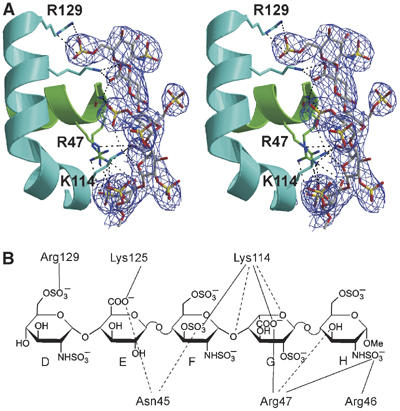
The AT–pentasaccharide interactions. (A) A stereo representation of the heparin-binding region of AT (colored as before) with 2Fo−Fc electron density contoured at one times the RMSD of the map (1σ). All side chains which interact directly with the pentasaccharide (fondaparinux) are shown as rods, with carbon atoms colored as the helix on which they are found (green for helix A and cyan for helix D). (B) Direct side-chain interactions are illustrated schematically, with salt bridges indicated by solid lines and hydrogen bonds indicated by dashed lines. Individual monosaccharide units are traditionally labeled from D to H. An exhaustive list of all interactions is given in Supplementary Table 1.
RCL interactions
Serpin specificity is typically determined by the complementarity between the RCL residues from P4–P3′ and the active site of the protease. Accordingly, the RCL contacts for the five serpin–protease Michaelis complexes solved to date are extensive, with multiple main-chain and side-chain hydrogen bonds and intimate hydrophobic contacts (Ye et al, 2001; Baglin et al, 2002; Dementiev et al, 2003, 2004; Li et al, 2004). The ability of the pentasaccharide to accelerate fXa inhibition by AT, however, is insensitive to RCL composition and largely depends on exosite interactions (outside P4–P3′) (Chuang et al, 2001a, 2001b). The paucity of interactions between the RCL of AT and the active site of fXa in our crystal structure is consistent with this observation. The interactions are illustrated in Figure 4A, and are described briefly here: Ile390 at the P4 position fits into a shallow hydrophobic groove described by Tyr99, Phe174, and Trp215 of fXa; Ala391 at P3 participates in two hydrogen bonds with the side chain of Gln192 and with Gly216 of fXa; Gly392 at the P2 position makes a single hydrogen bond to the side chain of Gln192; Arg393 at the P1 position is buried in the S1 pocket of fXa and participates in multiple interactions with both main-chain and side-chain atoms (discussed below); Ser394 at the P1′ position makes a single artifactual hydrogen bond with the side chain of His57 on fXa (this interaction would not be possible with an active site serine residue); Leu395 at P2′ participates in a single main-chain hydrogen bond with Phe41 of fXa; and Asn396 at the P3′ position makes no contacts with fXa. It is interesting to note that all of the hydrogen bonds described above, with the exception of P1 Arg393, involve only main chain atoms of AT. These hydrogen bonds are formed due to the canonical β-sheet-like conformation of the RCL, typically observed in crystal structures of other serpin–protease Michaelis complexes. The only favorable side-chain interaction observed here is the placement of the P4 Ile390 side chain into a shallow hydrophobic pocket; otherwise the RCL interactions are largely sequence independent.
Figure 4.
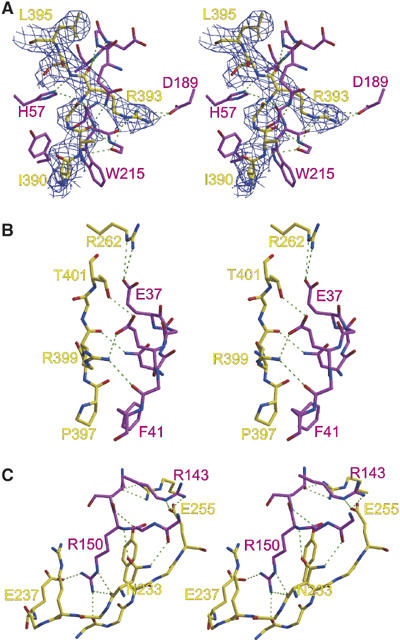
Stereo views of the interactions between AT (yellow) and fXa (magenta). (A) Interactions between the RCL (P4 I390–P2′ L395) of AT and active site residues of fXa are shown, with 2Fo−Fc electron density surrounding the RCL contoured at 1σ. The interactions are indicated by broken green lines and are generally sequence nonspecific, with the exception of the extensive P1 Arg393 side-chain interactions. (B) The first exosite on fXa includes residues from the 36-loop, from Asn35 to Phe41, and involves two important salt bridges. (C) The most important exosite on fXa is the autolysis loop, from Arg143 to Gln151. The side chain of Arg150 participates in a salt bridge with Glu237 and is neatly sandwiched between the side chains of Arg235 and Tyr253.
For all serpins, including AT, the most important RCL residue for determining inhibitory specificity is the reactive center P1 (Chuang et al, 2001a, 2001b). The S1 site of fXa is acidic and thus favors a basic P1 residue, but it is shallow relative to the S1 pocket of thrombin. Thus, the side chain of Arg393 is in a more bent conformation relative to that observed for P1 Arg in structures of serpin–thrombin Michaelis complexes (Baglin et al, 2002; Dementiev et al, 2004; Li et al, 2004), with torsion angles χ1 and χ2 differing by 101.6° and 114.9°, respectively. Nevertheless, the P1 interactions are extensive. In all, P1 Arg393 participates in nine hydrogen bonds and two ionic interactions: two main-chain oxygen hydrogen bonds to the normal oxyanion hole partners Gly193 and Ala195, and a third to Asp194; two main-chain hydrogen bonds with the side chain of His57 and the main chain of Ser214; the side-chain atoms make the remaining hydrogen bonds to the main-chain oxygens of Trp215, Gly216, and Gly219; and NH1 and NH2 participate in a salt bridge interaction with atom Oδ2 of Asp189. An exhaustive list of the interactions is provided in Supplementary Table 2, and apart from the favorable hydrophobic contacts of P4 and the extensive hydrogen bonding of the P1 Arg there is little else which might stabilize the Michaelis complex, thus explaining the requirement of exosite interactions.
Exosite interactions
The intimate nature of the interface between AT and fXa is illustrated in Figure 5. Of the 2337 Å2 of surface buried at the interface, 984 Å2 is due to active site contacts (involving AT RCL residues P4–P3′), and the remaining 1353 Å2 is accounted for by the exosites. Exosite interactions are defined here as any contact between AT and fXa outside RCL residues P4–P3′. Although it is possible to consider residues further C-terminal to P3′ as part of the RCL, the three residues between P3′ and β-sheet C, Pro397, Asn398, and Arg399, constitute an insertion loop relative to prototypical serpin α1-antitrypsin. Close analysis of the α1-antitrypsin structure shows that s1C begins at the P4′ position (Elliott et al, 2000), while in AT s1C commences at the P7′ position (Val400). Thus, the unique RCL insert of AT should be considered to be Pro397, Asn398, and Arg399, and is treated here as an exosite. The contiguous exosite of AT (Figure 5B) lies exclusively at the top left in the classic orientation (Figure 2A), and includes residues Asn233, Arg235, Glu237, Met251, Tyr253, Glu255, Arg262, Pro397, Asn398, Arg399, and Thr401. The secondary structural elements of AT which constitute the exosite are s1C (residue 401), s3C (residues 251–255), s4C (residues 233–237), s1B (residue 262), and the C-terminal insertion loop (residues 397–399). These AT residues contact two separate exosites on fXa: the acidic 36-loop, including Asn35, Glu37, Glu39, Gly40, and Phe41 (Figure 4B), accounting for 31% of the exosite surface of fXa; and the autolysis loop, including Arg143, Lys148, Arg150, and Gln151 (Figure 4C), accounting for 48% of the exosite surface of fXa. An exhaustive list of the interactions is given in Supplementary Tables 2 and 3, sorted according to either AT or fXa residues. Consistent with mutagenesis studies (Manithody et al, 2002), the most important exosite contact involves Arg150 on the autolysis loop of fXa. Arg150 is buried in a surface cavity formed by AT residues Arg235 and Tyr253. Arg150 also participates in hydrogen bonds with both the main-chain and side-chain atoms of Asn233 and a single salt bridge with Glu237 (Figure 4C). Arg150 alone accounts for 28% of the exosite surface of fXa.
Figure 5.
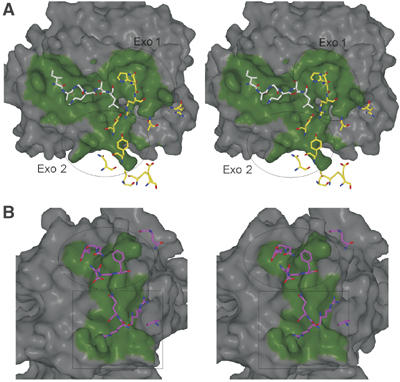
Stereo views of the contact surfaces on fXa and AT. fXa and AT surfaces within 4.5 Å of one another are colored green to illustrate the extent of the active site and exosite contacts, and contacting residues are depicted as rods. (A) The surface of fXa is shown in the standard orientation (active site facing with substrate running from left to right, from N- to C-terminus). RCL residues P4–P2′ are represented as white rods (P3′ is not shown because it makes no contact), and exosite-contacting AT residues are shown in yellow. The two exosites on fXa are indicated by the ovals; exosite 1 contains the 36-loop, and exosite 2 is the autolysis loop. (B) The top surface of AT with the RCL removed is colored as above, with contacting residues from fXa colored magenta. The oval indicates the position of the 36-loop contacts and the square indicates the position of the autolysis loop contacts. The AT residues under the oval derive from the RCL C-terminal insertion loop, s1C, and s1B, and those under the square derive from s3C and s4C.
Discussion
The crystal structure of the Michaelis complex between pentasaccharide-activated AT and fXa was long sought after, as it promised to explain the allosteric mode of action of the important anticoagulant heparin and a major effect of the natural heparan sulfate, which confers anticoagulant properties to the vascular endothelium. We were able to crystallize the complex only after mutating three residues on a remote surface of AT to engender the formation of crystal contacts, which, due to electrostatic repulsion, would not otherwise have been formed (Supplementary Figure 1). The identical nature of the two copies of the complex observed in the asymmetric unit supports a single interaction mode between pentasaccharide-activated AT and S195A fXa, consistent with an exosite-dependent recognition mechanism. Several of the residues seen to participate in exosite contacts have previously been identified by mutagenesis studies. Three residues on fXa were shown to contribute significantly to the rate of inhibition by AT in the presence of the pentasaccharide, Glu37, Glu39, and Arg150 (based on substitutions to alanine) (Quinsey et al, 2002; Manithody et al, 2002). By far the most important residue is Arg150, whose substitution to Ala reduces the rate of inhibition by nearly 10-fold. In the structure of the complex, we see that Arg150 is making more contacts with AT than any other single residue, with six of the seven contacts involving the side chain.
Similarly, mutagenesis of AT has also helped to identify the interaction interface with fXa. Based on a model of the AT–fXa complex built on the structure of the HCII–thrombin complex, a direct interaction between Lys403 and Glu39 was suggested (Huntington, 2003). Although the K403T AT variant did not inhibit the E39A fXa variant with a reduced rate, K403T AT inhibited wild-type fXa ∼3-times less efficiently than did wild-type AT (Rezaie et al, 2004). It is thus likely that Lys403 is making an interaction with fXa in the pentasaccharide-activated Michaelis complex, but that the contact does not involve Glu39 of fXa. In our structure, we are unable to model the side chain of Lys403 into electron density, but it is in position to make a salt bridge with Glu74 of fXa. Thus, although not included in the table of interactions between AT and fXa, the possible contact between Lys403 and Glu74 could explain the reduction in pentasaccharide-activated rate for the K403T AT variant. The presence of this interaction is supported by the properties of fXa variant E74A which is inhibited by pentasaccharide-activated AT with a two-fold slower rate (Chen et al, 2004). Another mutagenesis study focused on identifying the AT exosite utilizing a chimera approach, where large regions on the top of AT were replaced by the sequence found in the prototypic serpin α1-antitrypsin (Izaguirre et al, 2003). The conclusion from the study was that fXa docks further forward than predicted based on the HCII-thrombin structure, with s3C identified as the site of contact with fXa. Indeed, we observe fXa translated 15.2 Å towards the front of AT relative to the position predicted from the HCII-thrombin structure, and a 26.5° rotation of fXa extends the contact interface so that it includes s3C as well as residues from s1C, s4C and s1B. The extent of the exosite contact on AT was underestimated by the chimera study due to the sequence homology of residues now known to be of importance, with the least conservative changes from 246 to 258 belonging to s3C. Of particular relevance was the substitution of Tyr253 with Lys and Glu255 with Leu, which, together with the Gln254 to Arg mutation, would break important interactions with the autolysis loop of fXa and engender electrostatic repulsion. A mutagenesis study investigating the importance of the P′ insertion of AT is also consistent with the structure described here, with the deletion of Arg399 reducing the pentasaccharide-activated rate of inhibition by over two-fold (Rezaie, 2002).
The current structure is also consistent with the ability of only long heparin chains to further accelerate fXa inhibition by AT through the addition of a bridging effect to the allosteric mechanism. A heparin length dependence has been demonstrated, with a minimal bridging size greater than 22 and less than 35 monosaccharide units (Rezaie, 1998). The heparin-binding site of fXa has been identified by mutagenesis and is similar to the heparin-binding site of thrombin (Rezaie, 2000). Although there is no structure of fXa bound to heparin, the analogy with thrombin, whose structure with heparin has been solved (Carter et al, 2004), allows for a reasonable placement of a heparin fragment in the heparin-binding site of fXa. We thus constructed a model of the heparin-bridged AT–fXa Michaelis complex, which preserved the exosite contacts seen in the current structure (Figure 2B). Although fXa docks towards the front of AT (relative to thrombin docked on HCII), the heparin-binding site is oriented in such a way that a direct linear heparin bridge is not possible. As for the RCL of AT, which must approach the active site from an angle that requires the full extension of the hinge region (Figure 2B), the orientation of the heparin-binding sites on AT and fXa are not optimally aligned, thus requiring a longer heparin chain than suggested by the absolute distance between heparin-binding sites. Our model of the bridged complex predicts a minimal heparin chain length of ∼28 monosaccharide units, provided the AT-specific pentasaccharide sequence is located at the very reducing end of the chain. This prediction is consistent with the inability of a 22mer to bridge, with some bridging observed for the 26mer (Rezaie and Olson, 2000), as heparin fractions of this size contain a Gaussian distribution of sizes centered on the reported average size. Full activation through bridging is thus only observed for heparin fractions with average molecular weights corresponding to heparin chains of ∼30 monosaccharide units in length.
The pentasaccharide acceleration of fXa inhibition by AT and the ability of long heparin chains to further enhance the effect through the addition of a bridging mechanism are features shared by the related coagulation protease fIXa (Bedsted et al, 2003). In addition, the residues identified as participating in the interface between AT and fXa by mutagenesis studies affect the rate of fIXa inhibition in a similar fashion (Izaguirre et al, 2003; Yang et al, 2003). It is thus predicted that fIXa and fXa dock in the same way on the same surface of pentasaccharide-activated AT. To test this prediction, we created a model of the AT–fIXa Michaelis complex on the structure of the AT–fXa complex presented here, and examined the interactions. No difference in RCL-active site contacts was observed, and all of the interactions with the autolysis loop were preserved. The 36-loop, however, is significantly different between the two proteases, with 35NEENE39 of fXa replaced with 35GKVD39 in fIXa (residue 37 is missing). This sequence difference may account for the 10-fold slower pentasaccharide-activated rate of inhibition of fIXa relative to fXa. Interestingly, the predicted Lys403–Glu74 interaction is preserved for fIXa.
While it is accepted that new exosite interactions are responsible for the pentasaccharide activation of fIXa and fXa inhibition, there is some disagreement as to how the conformational change in AT allows the exosite to be engaged. Two hypotheses have been put forward: (1) that a cryptic exosite on AT is exposed upon pentasaccharide binding (Olson and Chuang, 2002); and (2) that the exosite is pre-existing and pentasaccharide binding only provides sufficient slack to the RCL to allow simultaneous engagement of the reactive center and the exosite (Huntington, 2003; Langdown et al, 2004). Now that the structure of the ternary complex between AT, the pentasaccharide, and S195A fXa has been solved, it is possible to address this issue. We observe in the complex two principal interactions involving an arginine from one protein and a corresponding acidic pocket on the other (Figure 6). It has been demonstrated that very little of the specificity of AT for fXa is conferred by the RCL sequence, apart from the binding of the reactive center residue Arg393 into the S1 pocket of fXa (Chuang et al, 2001a, 2001b) (Figure 6A). A similar interaction between an Arg side chain and an acidic pocket is observed in the exosite region, involving Arg150 of fXa and AT residues Arg235, Tyr253, and Glu237 (Figure 6B). Engagement of both the active site and exosite arginine contacts is critical for the pentasaccharide-accelerated inhibition of fXa by AT. The mechanism of allosteric activation can now focus on the accessibility of the two critical interaction sites in the native and pentasaccharide-bound states of AT. While there is some indication that the P1 side chain is making an intramolecular contact which restricts its ability to bind to proteases (Figure 1) (Jin et al, 1997), AT inhibits trypsin, fXa, and thrombin at appreciable rates in the absence of the pentasaccharide, suggesting a partially accessible reactive center (Huntington, 2005). In addition, fluorescence studies on AT variants with a fluorophore at the P1 position have been interpreted as indicating mobility for the P1 side chain in native and activated AT (Futamura et al, 2001). So, although there may be some element of exposure of the P1 Arg upon pentasaccharide binding, it exists in a relatively accessible state in both the native and activated conformations. The same can be said for the Arg150-binding pocket on AT, which is in an identical conformation in the structures of native and activated AT (Huntington, 2003), with only small changes in the position of some side chains. It is thus likely that the two principal sites of recognition on AT are individually available for interaction with fXa, and that pentasaccharide binding allows the simultaneous engagement of the two sites by liberating the hinge region from β-sheet A (Figures 1 and 6C).
Figure 6.
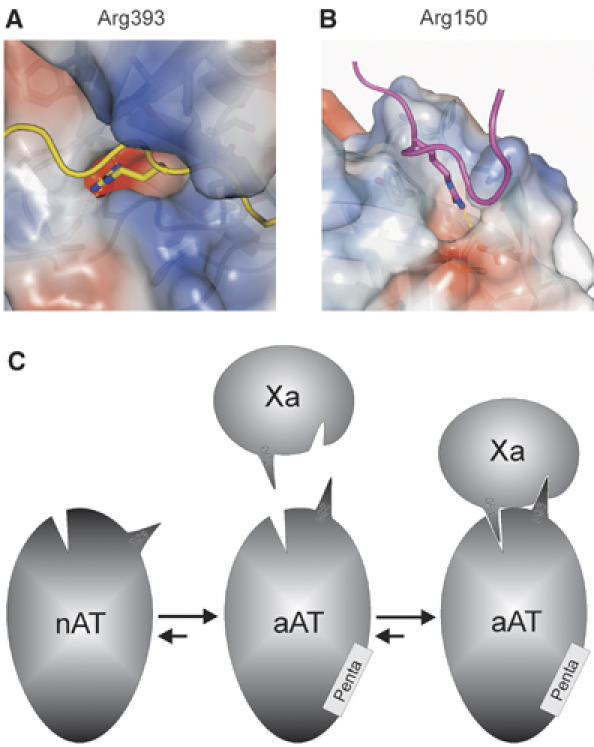
Holes and knobs that form the principal interaction sites are approximated by pentasaccharide activation. The two principal interactions between arginine side chains and corresponding acidic pockets are illustrated by coloring surfaces according to electrostatics (red for negative and blue for positive). (A) The P1 Arg393 of AT is buried deep in the S1 pocket (233 Å2 buried) of fXa, and its interactions are indicated by dashed yellow lines. (B) The principal exosite contact involves a similar burying of Arg150 from fXa into an acidic pocket on the surface between strands 3 and 4 of β-sheet C (194 Å2 buried). (C) A schematic representation is shown to illustrate the hypothesis of allosteric activation of AT towards fXa. Each molecule possesses a hole and a knob, which can interact individually but not simultaneously until native AT (nAT) binds to the specific heparin pentasaccharide (Penta). The resulting conformational change does not necessarily improve the accessibility of the hole and knob on AT, but rather repositions the knob (Arg393) to allow its engagement in the S1 pocket and the simultaneous engagement of the exosite knob of Arg150. The repositioning of Arg393 is only possible when the hinge region is freed from β-sheet A (see Figure 1).
Materials and methods
Expression and purification of proteins
The surface loop residues Glu347, Lys348, and Lys350 of AT were mutated to alanine on the recombinant β-glycoform S137A to reduce glycosylation heterogeneity as described previously (Zettlmeissl et al, 1988; Mushunje et al, 2003; Johnson and Huntington, 2004). The surface loop variant of AT had a similar heparin Sepharose elution profile to S137A AT control and the late eluting fraction was used for crystallization (Garone et al, 1996). Purified material was buffer exchanged into 20 mM Tris, pH 7.4, and stored at −80°C prior to use. The concentration of AT was determined by absorbance at 280 nm. S195A fX, consisting of the EGF2 and protease domains, was expressed in E. coli and refolded with small modifications to the previously described method (Hopfner et al, 1997). Refolded S195A fX was activated by incubation with 1:100 (w/w) RVV-X (Enzyme Research Laboratories, Swansea, UK) in the presence of 10 mM CaCl2 for 3 days at room temperature. Purification of S195A fXa was by Q-Sepharose chromatography, followed by concentration and buffer exchange into 20 mM Tris, 10% glycerol, pH 7.4.
Crystallization, data collection, refinement, analysis, and modeling
fXa and AT were mixed at a 1:1 molar ratio in the presence of a 1.2-fold molar excess of fondaparinux (kindly provided by Maurice Petitou) to give a final protein concentration of 4.2 mg/ml. Crystals were grown in hanging drops consisting of 1 μl protein solution and 1 μl 20% PEG 3350, 200 mM calcium acetate, and grew for 6 days at 21°C. Data were collected at the Daresbury Synchrotron Radiation Source (Warrington, Cheshire, UK) station 14.1 from a single crystal that had been cryoprotected in 22% PEG 3350, 20% glycerol, and 120 mM calcium acetate before flash cooling to 100 K in a nitrogen vapor stream. Crystal annealing was carried out by blocking the vapor nitrogen stream three times for 3 s. Data were processed using Mosflm, Scala, and Truncate (Leslie, 1992), and the structure was solved by molecular replacement using the program Phaser (McCoy et al, 2005). Search models were pentasaccharide-bound AT (1E03) with the RCL removed, and fXa from 1F0S, and two complexes were found in the asymmetric unit. After rigid body refinement, strict NCS was applied for several rounds of rebuilding and refinement using NCS-averaged maps. Heavy NCS restraints were subsequently used for all regions which were not demonstrably different. Refinement was conducted using the program CNS (Brunger et al, 1998) (version 1.0), and the program XtalView (McRee, 1992) was employed for rebuilding. Data processing and refinement statistics are given in Table I. The quality of the final structure and corresponding electron density were excellent, even though the resolution was limited to 3.3 Å, as seen in the figures displaying electron density. Figures were made using Bobscript (Esnouf, 1997), Raster3D (Merritt and Murphy, 1994), Pymol (www.pymol.org), and Spock. Template numbering based on chymotrypsin is used for fXa, as in molecular replacement starting model 1F0S. Coordinates and structure factors are deposited in the Protein Data Bank under PDBID code 2GD4. The contact surface area between AT and fXa was calculated using the Protein–Protein Interaction Server (www.biochem.ucl.ac.uk/bsm/PP/server/). Individual hydrogen bonding and salt bridge interactions were determined by hand using the program XtalView to measure distances and bond angles. Hydrogen bonds and salt bridges were defined by the standard bond angle and distance parameters utilized by the Protein–Protein Interaction Server; hydrogen bonds were defined by a donor–acceptor distance of less than 3.9 Å and a donor–hydrogen–acceptor angle of greater than 90°, and oppositely charged atoms were considered to be participating in a salt bridge if the intervening distance was ⩽4 Å (Barlow and Thornton, 1983). These parameters were also used in analyzing the AT–pentasaccharide interface.
Table 1.
Data processing, refinement, and model (2GD4)
| Crystal | ||
| Space group | C2 | |
| Cell dimensions (Å) | a=220.26, b=60.59, c=156.17, β=113.14 | |
| Solvent content (%) | 51.6 | |
| Data-processing statistics | ||
| Wavelength (Å) | 1.488 (SRS Daresbury, station 14.1) | |
| Resolution (Å) | 36.91–3.30 | 3.48–3.30 |
| Total reflections | 50 549 | 7380 |
| Unique reflections | 27 231 | 3981 |
| Multiplicity | 1.9 | 1.9 |
| 〈I〉/σ(〈I〉) | 5.8 | 1.9 |
| Completeness (%) | 94.6 | 95.1 |
| Rmerge | 0.175 | 0.476 |
| Model | ||
| Number of atoms modeled: | ||
| Protein | 10 763 | |
| Water | 63 | |
| Carbohydrate | 155 | |
| Ligand (fondaparinux) | 182 | |
| Calcium ions | 2 | |
| Average B-factor (Å2) | 38.9 | |
| Refinement statistics | ||
| Resolution range (Å) | 35.23–3.30 | 3.51–3.30 |
| Reflections in working/free set | 26 110/1114 | 4349/186 |
| R-factor/R-free (%) | 24.7/29.7 | 31.2/35.1 |
| RMSD of bonds (Å)/angles (deg) from ideality | 0.009/1.5 | |
| Ramachandran plot; residues in | ||
| Most favored region (%) | 68.2 | |
| Additionally allowed region (%) | 28.2 | |
| Generously allowed region (%) | 3.3 | |
| Disallowed region (%) | 0.3 | |
A model of the heparin-bridged complex between AT and fXa was created on the structure reported in the manuscript. The analogous nature of the heparin-binding sites of thrombin and fXa allowed the placement of heparin on fXa as observed in the structure of the thrombin–heparin complex (Carter et al, 2004). The intervening heparin segment was placed by hand through the rotation of glycosidic bonds of the heparin dodecasaccharide from 1HPN (Mulloy et al, 1993). For full occupancy of the heparin-binding site of fXa, a heparin length of 28 monosaccharide units is required, as well as significant deviation from the linear–helical conformation normally associated with unliganded heparin.
Supplementary Material
Supplementary Material
Acknowledgments
JAH is an MRC Senior Non-clinical Fellow, and project funding was provided by the British Heart Foundation (PG/04/115).
References
- Baglin TP, Carrell RW, Church FC, Esmon CT, Huntington JA (2002) Crystal structures of native and thrombin-complexed heparin cofactor II reveal a multistep allosteric mechanism. Proc Natl Acad Sci USA 99: 11079–11084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow DJ, Thornton JM (1983) Ion-pairs in proteins. J Mol Biol 168: 867–885 [DOI] [PubMed] [Google Scholar]
- Bedsted T, Swanson R, Chuang YJ, Bock PE, Bjork I, Olson ST (2003) Heparin and calcium ions dramatically enhance antithrombin reactivity with factor IXa by generating new interaction exosites. Biochemistry 42: 8143–8152 [DOI] [PubMed] [Google Scholar]
- Bray B, Lane DA, Freyssinet JM, Pejler G, Lindahl U (1989) Anti-thrombin activities of heparin. Effect of saccharide chain length on thrombin inhibition by heparin cofactor II and by antithrombin. Biochem J 262: 225–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, GrosseKunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Bussey H, Francis JL (2004) Heparin overview and issues. Pharmacotherapy 24: 103S–107S [DOI] [PubMed] [Google Scholar]
- Butenas S, Mann KG (2002) Blood coagulation. Biochemistry (Moscow) 67: 3–12 [DOI] [PubMed] [Google Scholar]
- Carter WJ, Cama E, Huntington JA (2004) Crystal structure of thrombin bound to heparin. J Biol Chem 280: 2745–2749 [DOI] [PubMed] [Google Scholar]
- Chen L, Manithody C, Yang L, Rezaie AR (2004) Zymogenic and enzymatic properties of the 70–80 loop mutants of factor X/Xa. Protein Sci 13: 431–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang YJ, Swanson R, Raja SM, Bock SC, Olson ST (2001a) The antithrombin P1 residue is important for target proteinase specificity but not for heparin activation of the serpin. Characterization of P1 antithrombin variants with altered proteinase specificity but normal heparin activation. Biochemistry 40: 6670–6679 [DOI] [PubMed] [Google Scholar]
- Chuang YJ, Swanson R, Raja SM, Olson ST (2001b) Heparin enhances the specificity of antithrombin for thrombin and factor Xa independent of the reactive center loop sequence. Evidence for an exosite determinant of factor Xa specificity in heparin-activated antithrombin. J Biol Chem 276: 14961–14971 [DOI] [PubMed] [Google Scholar]
- Dahlback B (2000) Blood coagulation. Lancet 355: 1627–1632 [DOI] [PubMed] [Google Scholar]
- Davie EW (1995) Biochemical and molecular aspects of the coagulation cascade. Thromb Haemost 74: 1–6 [PubMed] [Google Scholar]
- Davie EW, Fujikawa K, Kisiel W (1991) The coagulation cascade: initiation, maintenance, and regulation. Biochemistry 30: 10363–10370 [DOI] [PubMed] [Google Scholar]
- Dementiev A, Petitou M, Herbert JM, Gettins PG (2004) The ternary complex of antithrombin–anhydrothrombin–heparin reveals the basis of inhibitor specificity. Nat Struct Mol Biol 11: 863–867 [DOI] [PubMed] [Google Scholar]
- Dementiev A, Simonovic M, Volz K, Gettins PG (2003) Canonical inhibitor-like interactions explain reactivity of alpha1-proteinase inhibitor Pittsburgh and antithrombin with proteinases. J Biol Chem 278: 37881–37887 [DOI] [PubMed] [Google Scholar]
- Derewenda ZS (2004) The use of recombinant methods and molecular engineering in protein crystallization. Methods 34: 354–363 [DOI] [PubMed] [Google Scholar]
- Elliott PR, Pei XY, Dafforn TR, Lomas DA (2000) Topography of a 2.0 Å structure of alpha1-antitrypsin reveals targets for rational drug design to prevent conformational disease. Prot Sci 9: 1274–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnouf RM (1997) An extensively modified version of MolScript that includes greatly enhanced coloring capabilities. J Mol Graphics Model 15: 132. [DOI] [PubMed] [Google Scholar]
- Futamura A, Beechem JM, Gettins PG (2001) Conformational equilibrium of the reactive center loop of antithrombin examined by steady state and time-resolved fluorescence measurements: consequences for the mechanism of factor Xa inhibition by antithrombin–heparin complexes. Biochemistry 40: 6680–6687 [DOI] [PubMed] [Google Scholar]
- Garone L, Edmunds T, Hanson E, Bernasconi R, Huntington JA, Meagher JL, Fan B, Gettins PG (1996) Antithrombin–heparin affinity reduced by fucosylation of carbohydrate at asparagine 155. Biochemistry 35: 8881–8889 [DOI] [PubMed] [Google Scholar]
- Gettins PG (2002) Serpin structure, mechanism, and function. Chem Rev 102: 4751–4804 [DOI] [PubMed] [Google Scholar]
- Herbert JM, Herault JP, Bernat A, van Amsterdam RG, Vogel GM, Lormeau JC, Petitou M, Meuleman DG (1996) Biochemical and pharmacological properties of SANORG 32701. Comparison with the ‘synthetic pentasaccharide' (SR 90107/ORG 31540) and standard heparin. Circ Res 79: 590–600 [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Brandstetter H, Karcher A, Kopetzki E, Huber R, Engh RA, Bode W (1997) Converting blood coagulation factor IXa into factor Xa: dramatic increase in amidolytic activity identifies important active site determinants. EMBO J 16: 6626–6635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington JA (2003) Mechanisms of glycosaminoglycan activation of the serpins in hemostasis. J Thromb Haemost 1: 1535–1549 [DOI] [PubMed] [Google Scholar]
- Huntington JA (2005) Heparin activation of serpins. In Chemistry and Biology of Heparin and Heparan Sulfate, Garg HG, Linhardt RJ, Hales CA (eds) pp 367–398. Oxford: Elsevier [Google Scholar]
- Huntington JA, Read RJ, Carrell RW (2000) Structure of a serpin–protease complex shows inhibition by deformation. Nature 407: 923–926 [DOI] [PubMed] [Google Scholar]
- Izaguirre G, Zhang W, Swanson R, Bedsted T, Olson ST (2003) Localization of an antithrombin exosite that promotes rapid inhibition of factors Xa and IXa dependent on heparin activation of the serpin. J Biol Chem 278: 51433–51440 [DOI] [PubMed] [Google Scholar]
- Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW (1997) The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci USA 94: 14683–14688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DJ, Huntington JA (2004) The influence of hinge region residue Glu381 on antithrombin allostery and metastability. J Biol Chem 279: 4913–4921 [DOI] [PubMed] [Google Scholar]
- Langdown J, Johnson DJ, Baglin TP, Huntington JA (2004) Allosteric activation of antithrombin critically depends upon hinge region extension. J Biol Chem 279: 47288–47297 [DOI] [PubMed] [Google Scholar]
- Leslie AWG (1992) Joint CCP4 and ESF-EACMB Newsletter on Protein Crystallography. Warrington, UK: Daresbury Laboratory, p 26 [Google Scholar]
- Li W, Johnson DJ, Esmon CT, Huntington JA (2004) Structure of the antithrombin–thrombin–heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat Struct Mol Biol 11: 857–862 [DOI] [PubMed] [Google Scholar]
- Maignan S, Guilloteau JP, Pouzieux S, Choi-Sledeski YM, Becker MR, Klein SI, Ewing WR, Pauls HW, Spada AP, Mikol V (2000) Crystal structures of human factor Xa complexed with potent inhibitors. J Med Chem 43: 3226–3232 [DOI] [PubMed] [Google Scholar]
- Manithody C, Yang L, Rezaie AR (2002) Role of basic residues of the autolysis loop in the catalytic function of factor Xa. Biochemistry 41: 6780–6788 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr D 61: 458–464 [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Pei XY, Skinner R, Abrahams JP, Carrell RW (2003) Structure of beta-antithrombin and the effect of glycosylation on antithrombin's heparin affinity and activity. J Mol Biol 326: 823–833 [DOI] [PubMed] [Google Scholar]
- McRee DE (1992) A visual protein crystallographic software system for X11/XView. J Mol Graphics 10: 44–46 [Google Scholar]
- Merritt EA, Murphy MEP (1994) Raster3d version-2.0—a program for photorealistic molecular graphics. Acta Crystallogr D 50: 869–873 [DOI] [PubMed] [Google Scholar]
- Mulloy B, Forster MJ, Jones C, Davies DB (1993) N.m.r. and molecular-modelling studies of the solution conformation of heparin. Biochem J 293 (Part 3): 849–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mushunje A, Zhou A, Carrell RW, Huntington JA (2003) Heparin-induced substrate behavior of antithrombin Cambridge II. Blood 102: 4028–4034 [DOI] [PubMed] [Google Scholar]
- Olson ST, Chuang YJ (2002) Heparin activates antithrombin anticoagulant function by generating new interaction sites (exosites) for blood clotting proteinases. Trends Cardiovasc Med 12: 331–338 [DOI] [PubMed] [Google Scholar]
- Petitou M, van Boeckel CA (2004) A synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew Chem Int Ed Engl 43: 3118–3133 [DOI] [PubMed] [Google Scholar]
- Petitou M, Casu B, Lindahl U (2003) 1976–1983, a critical period in the history of heparin: the discoveryof the antithrombin binding site. Biochimie 85: 83–89 [DOI] [PubMed] [Google Scholar]
- Petitou M, Duchaussoy P, Lederman I, Choay J, Sinay P, Jacquinet JC, Torri G (1986) Synthesis of heparin fragments. A chemical synthesis of the pentasaccharide O-(2-deoxy-2-sulfamido-6-O-sulfo-alpha-D-glucopyranosyl)-(1–4)-O-(beta-D-glucopyranosyluronic acid)-(1–4)-O-(2-deoxy-2-sulfamido-3, 6-di-O-sulfo-alpha-D-glucopyranosyl)-(1–4)-O-(2-O-sulfo-alpha-L-idopyranosyluronic acid)-(1–4)-2-deoxy-2-sulfamido-6-O-sulfo-D-glucopyranose decasodium salt, a heparin fragment having high affinity for antithrombin III. Carbohydr Res 147: 221–236 [DOI] [PubMed] [Google Scholar]
- Quinsey NS, Whisstock JC, Le Bonniec B, Louvain V, Bottomley SP, Pike RN (2002) Molecular determinants of the mechanism underlying acceleration of the interaction between antithrombin and factor Xa by heparin pentasaccharide. J Biol Chem 277: 15971–15978 [DOI] [PubMed] [Google Scholar]
- Rezaie AR (1998) Calcium enhances heparin catalysis of the antithrombin-factor Xa reaction by a template mechanism. Evidence that calcium alleviates Gla domain antagonism of heparin binding to factor Xa. J Biol Chem 273: 16824–16827 [DOI] [PubMed] [Google Scholar]
- Rezaie AR (2000) Identification of basic residues in the heparin-binding exosite of factor Xa critical for heparin and factor Va binding. J Biol Chem 275: 3320–3327 [DOI] [PubMed] [Google Scholar]
- Rezaie AR (2002) Partial activation of antithrombin without heparin through deletion of a unique sequence on the reactive site loop of the serpin. J Biol Chem 277: 1235–1239 [DOI] [PubMed] [Google Scholar]
- Rezaie AR, Olson ST (2000) Calcium enhances heparin catalysis of the antithrombin-factor Xa reaction by promoting the assembly of an intermediate heparin–antithrombin-factor Xa bridging complex. Demonstration by rapid kinetics studies. Biochemistry 39: 12083–12090 [DOI] [PubMed] [Google Scholar]
- Rezaie AR, Yang L, Manithody C (2004) Mutagenesis studies toward understanding the mechanism of differential reactivity of factor Xa with the native and heparin-activated antithrombin. Biochemistry 43: 2898–2905 [DOI] [PubMed] [Google Scholar]
- Schenone M, Furie BC, Furie B (2004) The blood coagulation cascade. Curr Opin Hematol 11: 272–277 [DOI] [PubMed] [Google Scholar]
- van Boven HH, Lane DA (1997) Antithrombin and its inherited deficiency states. Semin Hematol 34: 188–204 [PubMed] [Google Scholar]
- Yang L, Manithody C, Olson ST, Rezaie AR (2003) Contribution of basic residues of the autolysis loop to the substrate and inhibitor specificity of factor IXa. J Biol Chem 278: 25032–25038 [DOI] [PubMed] [Google Scholar]
- Ye S, Chech AL, Belmares R, Bergstrom RC, Tong Y, Corey DR, Kanost MR, Goldsmith EJ (2001) The structure of a Michaelis serpin–protease complex. Nat Struct Biol 8: 979–983 [DOI] [PubMed] [Google Scholar]
- Zettlmeissl G, Wirth M, Hauser H, Kupper HA (1988) Efficient expression system for human antithrombin III in baby hamster kidney cells. Behring Inst Mitt 82: 26–34 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material