

Abstract
In two-metal catalysis, metal ion A has been proposed to activate the nucleophile and metal ion B to stabilize the transition state. We recently reported crystal structures of RNase H–RNA/DNA substrate complexes obtained at 1.5–2.2 Å. We have now determined and report here structures of reaction intermediate and product complexes of RNase H at 1.65–1.85 Å. The movement of the two metal ions suggests how they may facilitate RNA hydrolysis during the catalytic process. Firstly, metal ion A may assist nucleophilic attack by moving towards metal ion B and bringing the nucleophile close to the scissile phosphate. Secondly, metal ion B transforms from an irregular coordination in the substrate complex to a more regular geometry in the product complex. The exquisite sensitivity of Mg2+ to the coordination environment likely destabilizes the enzyme–substrate complex and reduces the energy barrier to form product. Lastly, product release probably requires dissociation of metal ion A, which is inhibited by either high concentrations of divalent cations or mutation of an assisting protein residue.
Keywords: Ca2+, Mg2+, Mn2+, RNA/DNA, two-metal-ion catalysis
Introduction
RNase H (RNase HI) is an endonuclease that binds RNA/DNA hybrids and hydrolyzes the RNA strand in a sequence nonspecific manner. It is present in all forms of life from bacteria to animals. The main function of RNase H is to remove RNA primers from Okazaki fragments during DNA replication (Kogoma and Foster, 1998). Knockout of RNase H results in failure of mitochondrial DNA replication and embryonic lethality in mice (Cerritelli et al, 2003). An RNase H domain is also present at the C-terminus of retroviral reverse transcriptase and plays an essential role in converting a single-stranded retroviral genomic RNA into a dsDNA for integration into host chromosomes (Hughes et al, 1998).
The first crystal structures of Escherichia coli RNase H were determined in 1990 and revealed a novel fold (Katayanagi et al, 1990; Yang et al, 1990). A similar fold has since been found in other nucleic acid processing enzymes, for example, integrases, transposases, Holliday junction resolvase and RNAi slicer Argonaute (Yang and Steitz, 1995; Rice and Baker, 2001; Song et al, 2004). Last year, we reported two crystal structures of the catalytic domain of Bacillus halodurans RNase H (Bh-RNase HC) complexed with an RNA/DNA substrate (Nowotny et al, 2005). These structures showed that the RNA strand of the hybrid adopts the A-form conformation and the 2′-OH groups form close contacts with the protein. Interestingly, the DNA strand adopts the B-form conformation, which is necessary to complement the surface of RNase H. Since RNA cannot be in the B-form, the protein can only bind RNA/DNA hybrids and not dsRNA.
The active site of RNase H consists of four absolutely conserved carboxylates (D71, E109, D132 and D192 in Bh-RNase H) and requires Mg2+ or Mn2+ for catalysis. In the Bh-RNase HC–substrate complexes, two Mg2+ ions (A and B) are found ∼4 Å apart in the active site (Nowotny et al, 2005). The coordination geometry of the Mg2+ ions supports a two-metal-ion catalysis mechanism as proposed by Steitz and Steitz (1993). The two metal ions are jointly coordinated by a nonbridging oxygen of the scissile phosphate and two active-site carboxylates and are located on either side of the plane bisecting the scissile phosphate. The A-site Mg2+ coordinates a water molecule, which is aligned for an in-line attack of the scissile phosphate in an SN2-like mechanism. The B-site Mg2+ is positioned to stabilize the transition state and leaving group. Similar two-metal-ion catalysis is likely used by other RNase H-like enzymes (Nowotny et al, 2005), many nucleases (Lee et al, 2005) and all DNA and RNA polymerases (Steitz, 1998; Yang, 2005).
Biochemical and structural characterization of RNase H, however, has raised questions about the functional roles of metal ions A and B. According to the existing model, metal ion A drives the phosphoryl transfer reaction by coordinating and activating the nucleophilic attack, while metal ion B plays the auxiliary role of stabilizing the transition state. Mutation of a conserved Asp that directly coordinates metal ion B (D132N) leads to inactivation of Bh-RNase H in the presence of Mg2+ or Mn2+ ions. However, mutation of a conserved Asp that coordinates metal ion A (D192N) has a less detrimental effect, and the D192N mutant proteins retains catalytic activity in the presence of Mn2+ (Nowotny et al, 2005). Mutations of the equivalent carboxylates in E. coli and human RNases H have the similar effects (Kanaya et al, 1990; M Nowotny, S Gaidamakov, and R Crouch, unpublished data), indicating that metal ion B plays as important a role as, if nor more than, metal ion A.
The crystal structures of D132N and D192N mutant Bh-RNase HC complexed with an RNA/DNA substrate (Nowotny et al, 2005) provided some mechanistic clues but raised more questions. Structurally, the two mutant proteins are nearly identical, but the distance between the two metal ions is shorter (4.1 Å) in D192N (partially active with Mn2+) than in D132N (4.5 Å) (inactive) (Nowotny et al, 2005). What is the significance of the distance change between the two metal ions? Should the two metal ions be immobile throughout the phosphoryl transfer reactions? In the Bh-RNase HC–substrate complexes, the potential nucleophile (a water-derived hydroxide ion) is 3.4–3.8 Å away from its target phosphorus atom. In the pentacovalent transition state, both attacking and leaving oxygen need to be ∼2 Å from the phosphorus (Mildvan, 1997). How is the attacking nucleophile brought closer to the phosphorus to form the transition state? Moreover, in both Bh-RNase HC–substrate complex structures, the coordination geometry of the B-site Mg2+ is rather unusual. It has five instead of six ligands, and none of the five ligands is water. We suspected that this irregular coordination is owing to the nature of substrate complexes (Nowotny et al, 2005). If so, how is metal ion B coordinated after the phosphoryl transfer reaction occurs?
Metal ion choice and concentration present additional unresolved issues in two-metal-ion-dependent phosphoryl transfer reactions. Mg2+ is most often preferred and can be substituted by Mn2+ or even Zn2+, Cu2+ and Cd2+ (Uchiyama et al, 1994; Keck et al, 1998). In contrast, Ca2+ can replace Mg2+ in enzyme–substrate complex formation, but inhibits catalysis in most cases (Savilahti et al, 1995; Vipond and Halford, 1995; Jones and Gellert, 2002; Lee et al, 2005). To date, no obvious structural difference has been found between Ca2+- and Mg2+-bound enzyme–nucleic acid complexes (Ling et al, 2004; Viadiu and Aggarwal, 1998). Why Ca2+ ion do not support catalysis remains unanswered. Regarding metal ion concentrations, although metal ions are essential for catalysis, all RNases H studied to date are inhibited by high concentrations of Mg2+ or Mn2+, a feature known as attenuation (Keck et al, 1998). The cause of this phenomenon is uncertain. One possibility is that a nonspecific binding site induced by high metal ion concentrations may perturb the active-site geometry and inhibit RNase H activity. A second possibility is that high metal ion concentrations may reduce product release, thus leading to the attenuation of RNase H activity. Interestingly, Ala replacement of E188 in Bh-RNase H or its equivalent in E. coli RNase H (H124), which swivels in and out of the active site (Nowotny et al, 2005), lowers the enzyme activity but abolishes metal ion attenuation (Keck et al, 1998).
With the above questions in mind, we designed RNA/DNA hybrids that mimic reaction intermediates and products and determined the crystal structures of these oligonucleotides complexed with D132N, D192N and E188A Bh-RNase HC proteins in the presence of various divalent cations. Since the overall structure and the active site of Bh-RNase H are essentially unchanged throughout the reaction cycle, the structural differences observed with different metal ions are uncomplicated. We compare these structures, particularly the metal–ion coordination, with the substrate complexes and discuss the implications for the role of the two metal ions in phosphoryl transfer reactions.
Results
Design of RNA/DNA hybrids for crystallographic studies
To crystallize Bh-RNase HC in a reaction intermediate or product state with an RNA/DNA hybrid, we need to overcome two hurdles. One is to specifically place a nick of the RNA strand in the active site of the enzyme, and the other is to create a pentacovalent phosphate transition-state analog (intermediate of the reaction). Fortuitously, in the crystals of Bh-RNase HC–substrate complexes, the 12 bp blunt-ended RNA/DNA hybrids are stacked head-to-tail forming continuous fibers (Nowotny et al, 2005) (Figure 1A and B). These nucleic acid fibers are laterally connected by RNase H molecules, two per 12 bp RNA/DNA duplex (Figure 2A).
Figure 1.
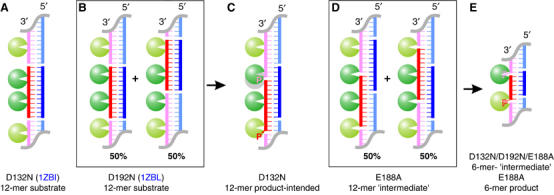
RNA/DNA hybrids used in crystallographic studies of substrate, ‘intermediate' and product complexes of Bh-RNase HC. RNAs (red) and DNAs (blue) that form hybrids for crystallization are schematically shown. The RNA/DNA hybrids are stacked head-to-tail in the crystal lattice, and symmetry mates are shown in lighter colors. RNase H molecules bound to these hybrids are shown as green spheres, and the active site is highlighted by a white triangle. (A) In the D132N–12-mer substrate complex, two protein molecules are separated by 5 bp. (B) In the D192N–substrate complex, two protein molecules are separated by 6 bp, and each RNA/DNA hybrid has two alternative positions related by a 6 bp shift. The PDB codes for previously published structures are shown in blue. (C) In the D132N–12-mer product-intended structure, the intended protein position is shown as a gray circle and the actual protein observed in the crystal structure is shown in green. The 5′-phosphorylated RNA strand is labeled with a ‘P'. (D) In the E188A–6-mer ‘intermediate' complex crystals, the hybrid has two alternative positions as in (B). (E) The final design of a 6-mer RNA/DNA hybrid forming a 5 bp duplex and two 1-nt sticky ends. Nonphosphorylated RNA gives rise to ‘intermediate' complexes, and 5′-phosphorylated RNA to product complexes.
Figure 2.
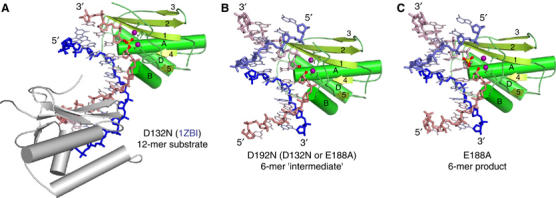
Structures of substrate, ‘intermediate' and product complexes. (A) Ribbon diagram of the substrate complex. Bh-RNase HC (D132N) with a 12-mer RNA/DNA hybrid (PDB: 1ZBI, published). The RNA is shown as pink and the DNA backbone as blue sticks. The two magnesium ions observed in the active site are shown as purple spheres, and the scissile phosphate is highlighted in ball-and-stick presentation. The secondary structure elements are labeled. (B) Ribbon diagram of the ‘intermediate' complex. Bh-RNase HC (D192N) with the nonphosphorylated 6-mer RNA/DNA hybrid. The 5′- and 3′-OH groups at the nick are highlighted as red spheres. The symmetry-related RNA and DNA are shown in lighter pink and blue, respectively. (C) Ribbon diagram of the product complex. Bh-RNase HC (E188A) with the 5′-phosphorylated 6-mer RNA/DNA. The cleavage products, 5′-phosphate and 3′-OH, are highlighted in ball-and-stick presentations.
To take the advantage of RNA/DNA fibers formed in the crystal, we first designed a 12-mer RNA/DNA hybrid with a 9 bp duplex and two 3-nucleotide (nt) sticky ends (Figure 1C). Based on the Bh-RNase HC–substrate complex structures, the nick placed in the DNA strand does not participate in protein interaction. When this RNA/DNA was crystallized with the D132N Bh-RNase HC, the 5′-phosphorylated nick was 1 nt over from the active site (Figure 1C). When a similar RNA/DNA hybrid without 5′-phosphate in the RNA strand was crystallized with the E188A Bh-RNase HC, the nick was located at the active site, but only half of the time. Each RNA/DNA fiber had two different registers relative to the crystal lattice made of RNase H molecules, and the two are related by a 6-bp shift (Figure 1D). The active site of RNase H is alternatively occupied by a nick and continuous RNA strand. The situation is reminiscent of the shift of the nucleic acid fiber observed in the D192N–substrate cocrystals (Nowotny et al, 2005) (Figure 1B). Since each Bh-RNase HC binds a 6 bp segment of RNA/DNA hybrid, we then designed a 6-mer RNA/DNA hybrid with a 5 bp duplex and two 1-nt sticky ends to avoid the ambiguity (Figure 1E). With this design, we have crystallized all three mutant Bh-RNases HC (D132N, D192N and E188A) with the nonphosphorylated RNA nick and E188A with the phosphorylated nick in the active site. The phosphorylated nick in the active site of RNase H resembles an enzyme–product complex.
To make a transition state analog, we followed the examples of structural studies of a hairpin ribozyme (Rupert et al, 2002), which showed that VO3− sandwiched between a nonphosphorylated nick can mimic the pentacovalent-linked intermediate of a phosphoryl transfer reaction. We tried crystallization of Bh-RNase HC and nonphosphorylated RNA nick in the presence of metavanadate (VO3−) and soaking such cocrystals in aluminum fluoride (AlF3), beryllium fluoride (BeF3−), magnesium fluoride (MgF3−) and tungstate (WO42−) solutions (see Materials and methods).
Crystallization of Bh-RNase HC with transition state analogs
Crystals of E188A and D192N Bh-RNase HC grew readily in the presence of the nonphosphorylated 6-nt RNA/DNA hybrid and the transition state analog VO3−. They diffracted X-rays to up to 1.65 Å on a rotating anode. Crystals of the D132N variant with VO3− and the same RNA/DNA hybrid were more difficult to grow and required microseeding. They diffracted X-rays only to 2.7 Å. Structures were solved by molecular replacement (MR) using the published substrate complex (PDB: 1ZBI). Refinement was concluded at Rfree of 20.7% for the E188A cocrystal, 25.4% for D192N and 28.8% for D132N (Table I).
Table 1.
X-ray data collection and refinement statistics
| Refinement | E188A–int.–Mg2+ | D192N–int.–Mg2+ | D192N–int.–Mn2+ | D192N–int.–Ca2+ | D132N–int.–Mg2+ | E188A–prod.–Mg2+ |
|---|---|---|---|---|---|---|
| Space group | C2 | C2 | C2 | C2 | C2 | C2 |
| Unit cell a, b, c (Å) | 82.1, 36.9, 61.8 | 82.4, 37.0, 61.8 | 82.1, 37.1, 61.8 | 82.0, 36.9, 61.9 | 83.9, 35.4,63.8 | 81.9, 36.8, 62.1 |
| α, β, γ (deg) | 90.0, 96.5, 90.0 | 90.0, 96.4, 90.0 | 90.0, 96.5, 90.0 | 90.0, 96.3, 90.0 | 90.0, 98.7, 90.0 | 90.0, 97.2, 90.0 |
| Resolution range (Å)a | 30–1.65 (1.71–1.65) | 30–1.85 (1.92–1.85) | 30–1.65 (1.71–1.65) | 30–1.65 (1.71–1.65) | 30–2.7 (2.82–2.7) | 30–1.85 (1.92–1.85) |
| Completeness (%)a | 90.6 (52.7) | 98.2 (89.6) | 93.2 (53.0) | 93.9 (57.2) | 99.0 (99.7) | 98.2 (94.9) |
| Rmergea,b | 3.5 (20.2) | 6.2 (44.6) | 5.4 (23.8) | 3.7 (23.2) | 7.4 (42.0) | 4.2 (36.2) |
| I/σ(I)a | 27.4 (3.6) | 18.3 (1.8) | 20.5 (2.6) | 33.2 (3.2) | 16.0 (2.4) | 23.0 (2.4) |
| Unique reflection | 19 921 (1967) | 14 882 (1513) | 20 416 (2014) | 20 635 (2042) | 4718 (512) | 14 810 (1504) |
| Nonhydrogen atoms | 1592 | 1613 | 1680 | 1626 | 1344 | 1494 |
| R-valuec | 18.2 | 20.6 | 17.3 | 18.2 | 22.3 | 19.8 |
| Rfreed | 20.7 | 25.4 | 20.2 | 21 | 28.8 | 24 |
| R.m.s.d. bond len. (Å) | 0.004 | 0.005 | 0.004 | 0.004 | 0.007 | 0.005 |
| R.m.s.d. bond angle (deg) | 1.13 | 1.12 | 1.11 | 1.15 | 1.3 | 1.13 |
| Ave. B-value (Å2) | 19.4 | 29.5 | 19.5 | 20.3 | 49.1 | 28.5 |
| φ, ψ distribution (%)e | 90.6, 9.4 | 92.4, 7.6 | 89.7,10.3 | 88.2,11.8 | 80.2, 19.8 | 90.6, 9.4 |
| Luzzati coord. error (Å) | 0.21 | 0.3 | 0.21 | 0.22 | 0.51 | 0.27 |
| 2 Me2+ distance (Å) |
3.65 |
3.43 |
3.63 |
6.22 |
4.47 |
4.8 |
| aData of the highest resolution shell is shown in parenthesis. | ||||||
| bRmerge=∑h∑i∣Ihi−〈Ih〉∣/∑〈Ih〉, where Ihi is the intensity of the ith observation of reflection h, and 〈Ih〉 is the average intensity of redundant measurements of the h reflections. | ||||||
| cR-value=∑∣∣Fo∣−∣Fc∣∣/∑∣Fo∣, where Fo and Fc are the observed and calculated structure–factor amplitudes. | ||||||
| dRfree is monitored with the number of reflections in parenthesis excluded from refinement. | ||||||
| eResidues in the most favored and additionally allowed regions of Ramachandran plot. | ||||||
In all three structures, the RNA/DNA duplex forms a continuous fiber and each asymmetric unit contains one RNase H and one 6-mer hybrid (Figure 2B). The nonphosphorylated nick is located in the active site. However, cocrystallization with metavanadate plus additional soakings with BeF3−, AlF3, MgF3− or WO42− did not result in any density at the nonphosphorylated nick that could be attributed to a transition state analog. Interestingly, VO3− is found in other locations in the crystal. A plausible reason for the absence of VO3−, BeF3− and AlF3 in the active site is that they are not stably bound. When used successfully as a transition state mimic, the vanadium is covalently attached to a nucleophile originating from a macromolecule. In the hairpin ribozyme structure (Rupert et al, 2002), the 2′-OH is the nucleophile and is directly linked to the vanadium in addition to the 3′- and 5′-OH. Similarly VO3− has been successfully used to mimic the transition state in a DNA enzyme, when a protein side chain forms a covalent linkage with the scissile phosphate (Davies and Hol, 2004). In the case of RNase H, a hydroxide anion is the nuleophile instead of a 2′-OH or protein side chain, and VO3− can be bonded only to the 3′- and 5′-OH of disconnected RNA strands.
The A and B metal ions are 3.5 Å apart in the ‘intermediate' complexes
To determine the number and location of metal ion binding sites, the E188A-nonphosphorylated nick crystals were soaked in magnesium salts at low and high concentrations prior to diffraction. Only the B-site was occupied by an Mg2+ ion in the crystal grown in 2.5 mM MgCl2. After soaking of the crystals in 300 mM MgCl2, the A-site also became occupied. D132N and D192N crystals were consequently soaked in 300 mM MgCl2 prior to structure determination. In these structures, two metal ions are found in the active site as well.
The structures of the three mutants—E188A, D132N and D192N—complexed with the nonphosphorylated nick, which are called ‘intermediate' to distinguish them from the substrate and product complexes, are nearly identical. In the active site, a water molecule replaces the nonbridging oxygen of the scissile phosphate that coordinates both metal ions in the substrate complexes (Figure 3A and C). Protein molecules of the ‘intermediate' and substrate complex structures were superimposed, and no discernable change was found. The only change is that the two metal ions slightly shifted towards each other in the ‘intermediate' D192N and E188A structures. In the substrate complex structures, the distances between the metal ions were 4.1 Å (D192N) and 4.5 Å (D132N). In the E188A– and D192N–‘intermediate' complexes, the metal ions are closer and only 3.4–3.6 Å apart. Interestingly, in the D132N–‘intermediate' complex, the metal ions remain 4.5 Å apart (Figure 3B). As suggested previously (Nowotny et al, 2005), since D132 coordinates both metal ions (B directly and A via water), removal of the negative charge by the D132N mutation may prevent the two metal ions from getting closer than 4.5 Å.
Figure 3.
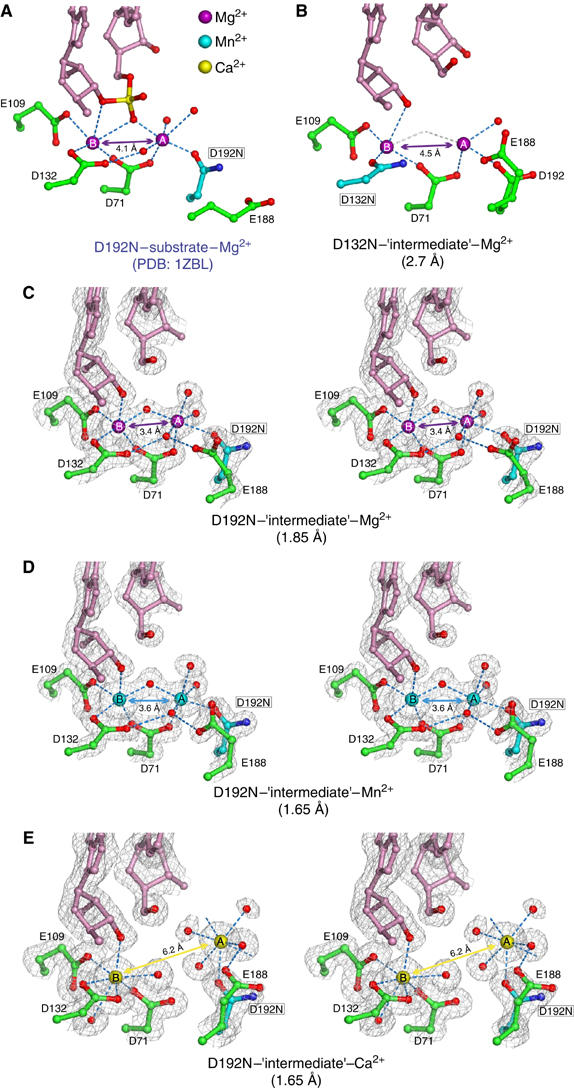
Comparison of metal ion coordination in the substrate and ‘intermediate' complexes. (A) The active site of a previously reported substrate complex (D192N, PDB: 1ZBL). The RNA is shown in pink, the scissile phosphate in yellow and red and the active-site residues in green, except for the mutated carboxylate in cyan. Oxygen atoms are in red and nitrogen in blue. Mg2+ ions are shown as large purple spheres, and water molecules as small red spheres. (B) The active site of D132N Bh-RNase HC complexed with a nonphosphorylated nick. The resolution limit of this structure is indicated in the parenthesis. Owing to lower resolution only one water molecule was assigned to coordinate metal ion A in the structure. Coordination provided by the putative water between magnesium ions is shown in gray. (C–E) Stereo views of the active site of D192N–‘intermediate' complexes with Mg2+ (C), Mn2+ (D) and Ca2+ (E) ions. 2Fo−Fc electron density maps are superimposed on the refined coordinates.
To verify the separation distance between the two metal ions, we expanded the structural studies of the substrate and ‘intermediate' state to include Mn2+ and Ca2+ ions (Supplementary data). In the E188A–substrate complex, Ca2+ ions are 4.0 Å apart just like Mg2+ in the D192N–substrate complexes. However, Ca2+ ions behave differently from Mn2+ and Mg2+ in the ‘intermediate' complexes with D192N and E188A mutant proteins. For example, crystals of the D192N Bh-RNase HC–‘intermediate' complexes were soaked in 300 mM MnCl2 or 300 mM CaCl2, and both structures were determined at 1.65 Å resolutions (Table I). The overall structures and positions of the B metal ion were superimposable among the Mg2+, Mn2+ and Ca2+ soaks (Figure 3C–E). Mn2+, which supports the RNase H activity, has the same coordination geometry as Mg2+ in both metal ion binding sites. However, Ca2+, which inhibits RNase H, is displaced from the A-site by almost 3 Å (Figure 3E), and the two Ca2+ ions are separated by 6.2 Å. The reason for this may be that Ca has larger atomic radius (2.0 Å) than Mg (1.6 Å) and Mn (1.4 Å) (Pauling, 1947). Therefore, Ca2+ ions can replace Mg2+ ions in an enzyme–substrate complex, where the two metal ions are separated by 4.0 Å, but Ca2+ ions cannot mimic the Mg2+ ions 3.5 Å apart in the ‘intermediate' state.
Structures of the product complexes and changes of metal ion coordination
Since the cleavage products by RNase H are 5′-phosphate and 3′-OH, 5′-phosphorylated RNA/DNA hybrid was used to generate crystals of product complexes. We found that D132N Bh-RNase HC preferred to bind the hybrid as a substrate and avoided the phosphorylated nick in its active site (Figure 1C). Similarly, the phosphorylated nick was avoided by D192N Bh-RNase HC when the mutant protein was crystallized with the 6-mer RNA/DNA hybrid (not shown). Only E188A Bh-RNase HC was crystallized with the product mimicry, and the 5′-phosphorylated nick of the RNA strand is located in the active site of E188A mutant protein.
To determine whether RNase H contains a third, low-affinity metal binding site, diffraction data sets were collected from the E188A–product crystals grown in the presence of 2.5 mM MgCl2 and soaked in 0 or 300 mM MgCl2. These crystals diffracted X-rays to 2.1 Å (0 mM MgCl2) and 1.85 Å (300 mM MgCl2). The structures were solved by MR and refined (Table I, Figure 2C). Whether soaked in the high or low Mg2+, the structures of the E188A–product complexes were virtually identical with an r.m.s.d. of 0.32 Å for all protein and nucleic acid atoms. Both structures contained two Mg2+ ions in the active site, and there was no sign of a third metal ion. The high-Mg2+ soaked structure was solved to a higher resolution and was therefore used for further analyses.
In comparison with the apoprotein and substrate complexes, the overall structure of RNase HC and the active-site residues remain the same with an r.m.s.d. of 0.24 Å over 120 Cα atoms. The main difference between the substrate and product complexes is the location of the scissile phosphate before and after cleavage. The phosphosugar group moves 5.9 Å away from the 3′-OH and is displaced from the active site toward the major groove (Figure 5A and D). Accompanying the phosphosugar movement, the A-site Mg2+ is shifted by 2.1 Å and the B-site Mg2+ by 0.9 Å. The two metal ions are separated by 4.8 Å, farther apart than in the substrate complex (4.1 Å) and the ‘intermediate' complex (3.5 Å). In this product complex, the A-site metal ion is coordinated by five water molecules and the side chain of D192 in a regular octahedral geometry (Figure 5C). One of the water molecules is also hydrogen bonded with the cleaved 5′-phosphate of RNA. Interestingly, the B-site metal ion is now coordinated in a near-perfect octahedral geometry by the 3′-OH of the product, side chains of D71, E109 and D132, and two water molecules. The electron densities for the 5′-phosphate, the A-site Mg2+ ion and its coordinating water molecules are much poorer than for the rest of the molecule (Figure 5A). Consequently, the B-factors are high for the phosphorus and A-site Mg2+ (∼70 Å2) in comparison with the average B-factor of 28 Å2 for the whole structure and 36 Å2 for metal ion B.
Figure 5.
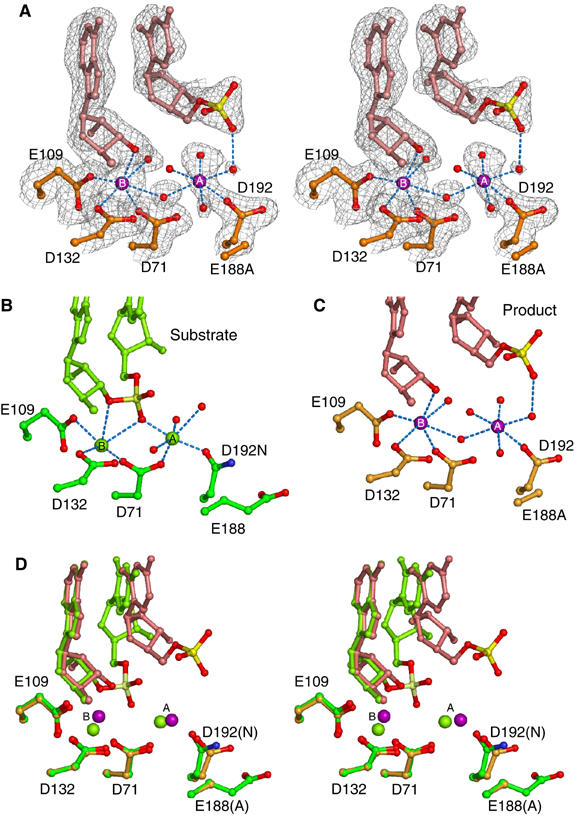
Structure of the E188A Bh-RNase HC product complex. (A) Stereo view of the active site superimposed with the 2Fo−Fc map contoured at 0.9σ. The 5′-phosphate is shown as red and yellow ball-and-sticks, Mg2+ ions as purple spheres labeled with ‘A' and ‘B', and water molecules as small red spheres. (B, C) Side-by-side comparison of metal ion coordination in the substrate and product complexes. The substrate complex (D192N, PDB: 1ZBL) is shown in green with oxygen atoms in red and nitrogen atoms in blue, and the product complex (E188A) is shown in orange. (D) Stereo view of the superposition of substrate and product complexes.
The Bh-RNase HC structures are also compared with the structures of substrate and product complexes of BamHI and EcoRV restriction endonucleases (Viadiu and Aggarwal, 1998; Horton and Perona, 2004). All three endonucleases use two-metal-ion catalysis and generate 5′-phosphate and 3′-OH products. In BamHI and EcoRV, the cleavage product 5′-phosphate is displaced relative to the scissile phosphate in the substrate complex by ∼2.5 Å, much smaller than the 6 Å observed in Bh-RNase HC. Two features of Bh-RNase HC may explain such a difference. In contrast to the flexibility of restriction endonucleases, the active site of Bh-RNase H is unchanged between the substrate and product state. Furthermore, unlike BamHI and EcoRV, which make extensive sequence-specific contacts in the DNA major groove and may constrain the movement of the 5′-phosphate, RNase H mainly contacts the minor groove (Figure 2) and leaves the major groove wide open to accommodate the displaced 5′-phosphate.
Discussion
The role of B metal ion in destabilizing RNase H–substrate complex
In the Bh-RNase HC–substrate complexes, the B-site Mg2+ is coordinated by five oxygen atoms: three from active-site carboxylates, D71, E109 and D132, and two from the RNA backbone (Figure 5B) (Nowotny et al 2005). Remarkably, the Mg2+ is devoid of any water ligand. The coordination geometry is irregular and deviates from the ideal octahedral coordination for Mg2+ ions (Harding, 2001). In the product complex reported here, where the scissile phosphate is replaced by two water molecules, the B-site Mg2+ ion is coordinated by six ligands in a near octahedral geometry (Figure 5C). Owing to the unusual structural stability of Bh-RNase HC, the observed differences in metal ion coordination cannot be attributed to protein movement. The transformation from the fully dehydrated and irregular coordination by five ligands in the substrate complex to the hydrated and octahedral coordination in the product complex suggests a previously unappreciated and perhaps more critical function of the B-site metal ion. In addition to stabilize the 3′-leaving group (Figure 5), metal ion B may destabilize the substrate–enzyme complexes and thus reduce the energy barrier between the substrate and product states.
Metal ions A and B closer than 4 Å may be essential for the pentacovalent intermediate formation
The separation of the two metal ions appears to correlate with the activity of RNase H based on comparison of nine different Bh-RNase HC–substrate complexes (data not shown). The D132N mutant Bh-RNase HC is catalytically inactive, and it is also unable to bring the two metal ions closer than 4.5 Å in the substrate or ‘intermediate' complexes. The Ca2+ ions, which do not support RNA hydrolysis by RNase H, are 4.0 Å apart in the substrate complex (Supplementary data) and 6.2 Å apart in the ‘intermediate' complex. A shortening of metal–metal distance may be necessary for the hydrolysis reaction to occur. In the enzyme–substrate complex structures, the attacking hydroxyl (nucleophile) coordinated by metal ion A is 3.4–3.8 Å away from the phosphorus atom of the scissile phosphate. In the pentacovalent transition state, the attacking oxygen must be covalently linked to and within 2.0 Å of the phosphorus (Mildvan, 1997). The shortened separation between the A and B metal ions observed in the ‘intermediate' complexes of D192N (Figure 3) and E188A may bring the nucleophile within the striking distance to the phosphorus. Although these ‘intermediate' structures contain no transition state analog, an imaginary phosphorus atom defined by the 3′- and 5′-OH groups at the nick is only 2.4 Å from the hydroxide anion coordinated by the A metal ion (Figure 4).
Figure 4.
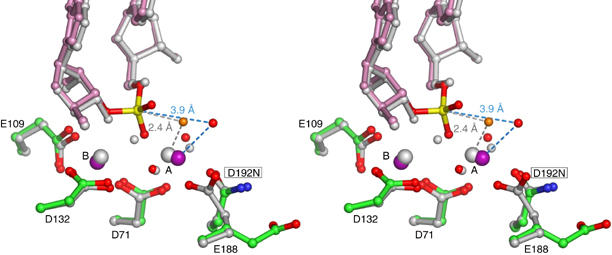
Stereo view of the superposition of D192N–substrate complex with D192N–‘intermediate' complex. The substrate complex is shown in green (protein), pink (RNA), red (oxygens), blue (nitrogens), purple (Mg2+ ions) and yellow (phosphorus). The ‘intermediate' complex is shown in gray with the potential nucleophilic water molecule in orange. The distance between the phosphorus atom and nucleophile waters in the substrate and ‘intermediate' complexes are indicated.
Choices of metal ions, Mg2+ versus Ca2+
RNase H as well as all RNA and DNA polymerases and many nucleases have a clear metal ion preference. The difference in the atomic radius may be one reason for RNase H and related enzymes to prefer Mg2+ over Ca2+ because large Ca2+ ions cannot be much closer than 4 Å. Another significant difference between Mg2+, Mn2+ and Ca2+ is their coordination geometry (Crowley et al, 2000; Maguire and Cowan, 2002). Mg2+ and Mn2+ prefer to have six ligands arranged in an octahedral geometry with the ligand-to-metal ion distance between 2.0 and 2.2 Å. Ca2+ is less particular and can be coordinated by 6, 7 or 8 ligands in variable geometries and a ligand-to-metal ion distance of 2.2–2.7 Å. For instance, the two Ca2+ ions in the D192N–‘intermediate' complex are coordinated by seven ligands, and the coordination distance is ∼2.4 Å (Figure 3E). As discussed earlier, the strict coordination requirement by Mg2+ has the potential to destabilize substrate and facilitate product formation. With the coordination flexibility, Ca2+ at the B-site is unlikely to destabilize the substrate.
Based on our structural analyses, the B-site needs to be occupied by Mg2+, while the A-site may be other divalent cations. This prediction is in agreement with the tolerance of substitution of D192 and its equivalent, which coordinates metal ion A only. In Argonaute, the equivalent of D192 is a His rather than a carboxylate (Rivas et al, 2005). In contrast, there is strong selection against amino-acid substitutions of carboxylates that coordinate metal ion B. In addition to inactivation by D132N mutation, substitution of E109 in Bh-RNase H or its equivalent E48 in E. coli RNase H, which coordinates metal ion B only, renders each enzyme inactive (Kanaya et al, 1990; Nowotny et al, 2005). The two metal ions are functionally not equal and chemically may not have to be the same.
Attenuation at high metal ion concentrations is owing to inhibition of product release
We have not found a third weak-binding metal ion in the active site of Bh-RNase HC in over a dozen substrate, ‘intermediate' and product complex structures after soaking crystals in 300 mM MgCl2 or MnCl2. Additional low-affinity metal binding sites do exist but they are distal from the substrate binding site. These observations disfavor the hypothesis that a third metal ion with low binding affinity perturbs the active site and attenuates the RNase H activity.
What is the likelihood of the alternative proposal that the attenuation is owing to inhibition of product release? We have reported that E188A mutant has lower activity than the wild-type enzyme, but its activity is not reduced in the presence of high Mg2+ concentration (Nowotny et al, 2005). Interestingly, E188A is the only mutant protein that can accommodate the phosphorylated nick in the active site. The D132N and D192N mutant proteins do not form product complexes in our crystallization trials, and their active site avoids the nick. Although the 5′-phosphate and metal ion A in the E188A–product complex is more mobile than the rest of the structure (Figure 5A), the A-site retains the Mg2+ even after soaking in a cryoprotecting solution without divalent cation. This implies that the E188A mutant protein has abnormally high affinity for the 5′-phosphate product and metal ion A. The mutant protein exhibits reduced catalytic activity probably because of the inhibition of product release. E188 has great conformational flexibility and can either participate in metal ion A coordination or swing out from the active site (Nowotny et al, 2005). Its equivalents in E. coli RNase H (H124), HIV integrase and transposases (Mandel et al, 1995; Rice et al, 1996) all have high mobility. Such mobility may be used to perturb the coordination of metal ion A and promote product release.
Interestingly, when H124 in E. coli RNase H is replaced by Ala, the enzyme activity is reduced by about 100-fold (Keck et al, 1998). The same amount of reduction was observed with the wild-type enzyme at high concentrations of divalent cations. Like the E188A Bh-RNase H, the H124A mutant RNase H cannot be further attenuated. These observations suggest that high concentrations of metal ions have the same effect on the RNase H activity as E188A or H124A substitution, and the two are redundant. When efficient product release is inhibited by an amino-acid replacement, it cannot be further attenuated by an increase of metal ion concentration. Consistent with the inhibition of product reslease, both kcat and Km of E. coli RNase H are reduced with increasing metal ion concentrations (Keck et al, 1998).
Concluding remarks
The structures described here and in our previous publication (Nowotny et al, 2005) represent different stages of the reaction catalyzed by RNase H, which are schematically depicted in Figure 6. Upon binding of an RNA/DNA hybrid, the two metal ions find their appropriate binding sites 4.0 Å apart in the active site and are positioned for catalysis. Metal ion A is coordinated in the octahedral geometry and orients and activates the water molecule for the nucleophilic attack. Metal ion B, which is coordinated in an irregular geometry, may destabilize the enzyme–substrate complex. In the next stage, a movement of two metal ions towards each other brings the nucleophile close to the phosphorus atom to form the pentacovalent transition state. The closer than 4 Å separation between the two divalent cations (3.5 Å) may efficiently neutralize the developing negative charge in the transition state. The transition state is then converted to the products, and the 5′-phosphate and 3′-OH dissociate. The release of the 5′-phosphate group allows metal ion B to relax and attain a regular octahedral coordination with two new ligands from water molecules. Normally, the cleavage products are efficiently released, but the E188A mutation hinders it. Product release might also be prevented by high concentrations of metal ions.
Figure 6.
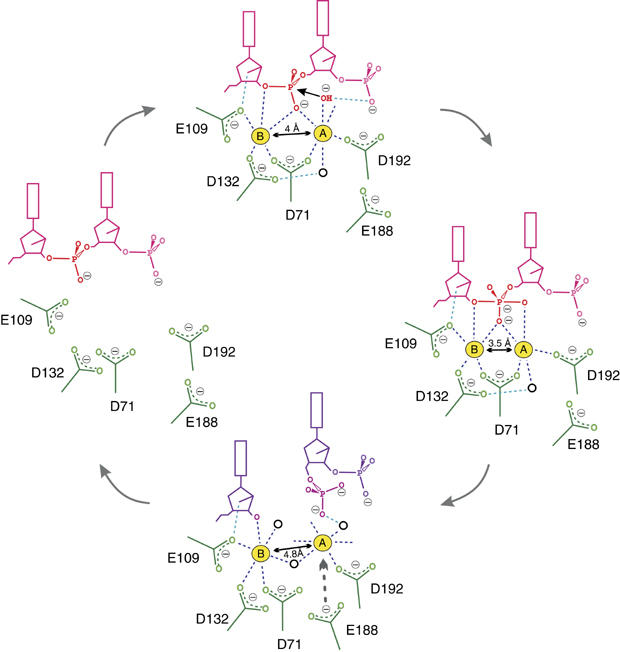
Schematic representation of the reaction steps proposed for RNase H. The substrate RNA is shown in pink and products in purple. Coordination of metal ions is highlighted in dark blue, and scissile phosphate in red. Selected hydrogen bonds are shown as blue lines. Black circles represent water molecules. The distance between the two metal ions is indicated in the enzyme–substrate, enzyme–intermediate and enzyme–product complexes.
This two-metal-ion catalysis mechanism is likely shared by DNA and RNA polymerases and many nucleases and recombinases. The high-resolution step-by-step structural studies of RNase H delineate the importance of the chemical properties of divalent cations in phosphoryl transfer reactions. The large atomic radius, variable number of ligands and flexible coordination geometry of Ca2+ are the likely reasons why it cannot support catalysis by the two-metal-ion mechanism.
Materials and methods
Crystallization
The mutant forms of Bh-RNase HC, D132N, E188A and D192, were expressed in E. coli and purified as described previously (Nowotny et al, 2005). The 5′-phosphate (5′-UUCGACACCUGA-3′) and nonphosphorylated (5′-CCUGAUUCGACA-3′) RNAs and DNA (5′-GAATCAGGTGTC-3′) were prepared according to the previously described protocol (Nowotny et al, 2005). The RNA oligos (5′-UCGACA-3′) either with or without 5′-phosphate and the DNA oligo (5′-ATGTCG-3′) (no 5′-phosphate) were purchased from Yale University (keck.med.yale.edu). The RNA was deprotected using 1.0 M solution of tetrabutylammonium fluoride in tetrahydrofuran and both RNA and DNA were desalted on NAP-25 column. The RNA and DNA were annealed at 1:1 molar ratio. For crystallization, the 6-mer hybrids were mixed with Bh-RNase HC mutant proteins (final concentration 8 mg/ml) at 1:1 molar ratio in the presence of 5 mM MgCl2. During the trials for making a transition-state analog, NaVO3 was found to improve crystal quality in general. 12–25 mM NaVO3 was therefore included in the protein–nucleic acid mixture prior to crystallization.
All crystals were obtained using the sitting drop vapor diffusion method at 4°C. Bh-RNase HC mutants with a nonphosphorylated nick were crystallized in 25–30% MPD as precipitant, 0.25 M ammonium acetate or 0.2 M NaCl, and at pH 5.6 (sodium citrate buffer), 7.0 (Tris) or 7.5 (HEPES) (for details see Supplementary Table I). D132N-nonphosphorylated nick crystals required one round of microseeding to obtain diffraction quality material. E188A mutant with phosphorylated nick was crystallized in 20% ethanol and 0.1 M Tris, pH 7.0. For soaks with high concentrations of metal ions, concentrated MgCl2, MnCl2 or CaCl2 were added directly to the crystallization drops and incubated 4–24 h. CaCl2 soaks were conducted with D192N crystals grown in the absence of NaVO3 as described below. Crystals were flash frozen directly from the drops or with cryoprotection with 30% MPD.
Preparation of transition state analogs
To make transition state analogs, 12–25 mM NaVO3 was added in the protein–nucleic acid mixture (see Supplementary Table I). The stock solution of NaVO3 was activated by adjusting the pH to above 10. After the structures of nonphosphorylated RNA/DNA hybrid were solved, vanadate was not found at the nick located in the active site. A round of multiple soaking experiments was carried out with crystals of all three mutant proteins. MgCl2 or MnCl2 were used in combination with a new set of transition state analogues—AlF3 (produced by combining AlCl3 with NaF), BeF3− (combining BeCl2 with NaF) and Na2WO4. The final concentrations of the analogs were varied from 1 to 40 mM and of metal ions between 80 and 300 mM. Again no transition state analog was observed at the nonphosphorylated nick. However, NaCl or NH4Ac at 200–300 mM were present in the crystallization/soaking solutions.
To reduce the relatively high salt concentrations, a batch of crystals of Bh-RNase H D192N with a nonphosphorylated nick was grown without NaVO3 and against 30% MPD, 0.25 M NH4Ac, 0.1 M Na citrate (pH 5.6) and in the presence of 25 mM NaF and 75 mM MgCl2 (to form transition state analog MgF3−), and the crystals were soaked in 50 mM NH4Ac and 10 mM BeF3−, but no analog density was found after these soaks either.
Data collection and structure determination
X-ray diffraction data were collected on an RUH3R rotating anode source equipped with multilayer focusing optics using CuKα radiation and a Raxis IV image plate detector at 95 K. All described crystals belong to the C2 space group and diffracted X-rays up to 1.65–2.7 Å (Table I). The data sets were processed and scaled using HKL2000 (Otwinowski and Minor, 1997). Structures were solved using the MR method with EPMR software (Kissinger et al, 1999). Several rounds of manual model building using O (Jones et al, 1991) and refinement using CNS (Brunger et al, 1998) were followed. Table I shows the statistics for the final models. Structural comparison and analyses were performed in O or MolMol (hugin.ethz.ch/wuthrich/software/molmol/). Figures were prepared using Pymol (www.pymol.org).
PDB deposition
The coordinates have been deposited in the PDB with the following accession codes for the ‘intermediate' complexes: 2G8U (D132N-Mg2+), 2G8H (D192N-Mg2+), 2G8I (D192-Mn2+), 2G8K (D192-Ca2+) and 2G8F (E188A- Mg2+), the PDB code for the E188A–product complex is 2G8V and E188A–substrate/Ca2+ complex is 2G8W.
Supplementary Material
Supplementary Information
Acknowledgments
We thank R Craigie for reading our manuscript. This research was supported fully by the Intramural Research Program of the NIH, NIDDK.
References
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54 (Part 5): 905–921 [DOI] [PubMed] [Google Scholar]
- Cerritelli SM, Frolova EG, Feng C, Grinberg A, Love PE, Crouch RJ (2003) Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol Cell 11: 807–815 [DOI] [PubMed] [Google Scholar]
- Crowley JD, Traynor DA, Weatherburn DC (2000) Enzymes and proteins containing manganese: an overview. In Metal Ions Biological Systems, Sigel A, Sigel H (eds), Vol. 37, pp 209–278. New York: Marcel Drekker Inc [PubMed] [Google Scholar]
- Davies DR, Hol WG (2004) The power of vanadate in crystallographic investigations of phosphoryl transfer enzymes. FEBS Lett 577: 315–321 [DOI] [PubMed] [Google Scholar]
- Harding MM (2001) Geometry of metal–ligand interactions in proteins. Acta Crystallogr D 57: 401–411 [DOI] [PubMed] [Google Scholar]
- Horton NC, Perona JJ (2004) DNA cleavage by EcoRV endonuclease: two metal ions in three metal ion binding sites. Biochemistry 43: 6841–6857 [DOI] [PubMed] [Google Scholar]
- Hughes SH, Arnold E, Hostomsky Z (1998) RNase H of retroviral reverse transcriptases. In Ribonucleases H, Crouch RJ and Toulme JJ (eds) pp 195–224. Paris: INSERM [Google Scholar]
- Jones JM, Gellert M (2002) Ordered assembly of the V(D)J synaptic complex ensures accurate recombination. EMBO J 21: 4162–4171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47 (Part 2): 110–119 [DOI] [PubMed] [Google Scholar]
- Kanaya S, Kohara A, Miura Y, Sekiguchi A, Iwai S, Inoue H, Ohtsuka E, Ikehara M (1990) Identification of the amino acid residues involved in an active site of Escherichia coli ribonuclease H by site-directed mutagenesis. J Biol Chem 265: 4615–4621 [PubMed] [Google Scholar]
- Katayanagi K, Miyagawa M, Matsushima M, Ishikawa M, Kanaya S, Ikehara M, Matsuzaki T, Morikawa K (1990) Three-dimensional structure of ribonuclease H from E coli. Nature 347: 306–309 [DOI] [PubMed] [Google Scholar]
- Keck JL, Goedken ER, Marqusee S (1998) Activation/attenuation model for RNase H. A one-metal mechanism with second-metal inhibition. J Biol Chem 273: 34128–34133 [DOI] [PubMed] [Google Scholar]
- Kissinger CR, Gehlhaar DK, Fogel DB (1999) Rapid automated molecular replacement by evolutionary search. Acta Crystallogr D 55 (Part 2): 484–491 [DOI] [PubMed] [Google Scholar]
- Kogoma T, Foster PL (1998) Physiological functions of E. coli RNase HI. In Ribonucleases H, Crouch RJ, Toulme JJ (eds) pp 39–66. Paris: INSERM [Google Scholar]
- Lee JY, Chang J, Joseph N, Ghirlando R, Rao DN, Yang W (2005) MutH complexed with hemi- and unmethylated DNAs: coupling base recognition and DNA cleavage. Mol Cell 20: 155–166 [DOI] [PubMed] [Google Scholar]
- Ling H, Boudsocq F, Woodgate R, Yang W (2004) Snapshots of replication through an abasic lesion; structural basis for base substitutions and frameshifts. Mol Cell 13: 751–762 [DOI] [PubMed] [Google Scholar]
- Maguire ME, Cowan JA (2002) Magnesium chemistry and biochemistry. Biometals 15: 203–210 [DOI] [PubMed] [Google Scholar]
- Mandel AM, Akke M, Palmer AG III (1995) Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J Mol Biol 246: 144–163 [DOI] [PubMed] [Google Scholar]
- Mildvan AS (1997) Mechanisms of signaling and related enzymes. Proteins 29: 401–416 [PubMed] [Google Scholar]
- Nowotny M, Gaidamakov SA, Crouch RJ, Yang W (2005) Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell 121: 1005–1016 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. In Methods in Enzymol, Carter CW, Sweet RM (eds), Vol. 276, pp 307–326. New York: Academic Press [DOI] [PubMed] [Google Scholar]
- Pauling L (1947) Atomic radii and interatomic distances in metals. J Am Chem Soc 69: 542–553 [Google Scholar]
- Rice P, Craigie R, Davies DR (1996) Retroviral integrases and their cousins. Curr Opin Struct Biol 6: 76–83 [DOI] [PubMed] [Google Scholar]
- Rice PA, Baker TA (2001) Comparative architecture of transposase and integrase complexes. Nat Struct Biol 8: 302–307 [DOI] [PubMed] [Google Scholar]
- Rivas FV, Tolia NH, Song JJ, Aragon JP, Liu J, Hannon GJ, Joshua-Tor L (2005) Purified Argonaute2 and an siRNA form recombinant human RISC. Nat Struct Mol Biol 12: 340–349 [DOI] [PubMed] [Google Scholar]
- Rupert PB, Massey AP, Sigurdsson ST, Ferre-D'Amare AR (2002) Transition state stabilization by a catalytic RNA. Science 298: 1421–1424 [DOI] [PubMed] [Google Scholar]
- Savilahti H, Rice PA, Mizuuchi K (1995) The phage Mu transpososome core: DNA requirements for assembly and function. EMBO J 14: 4893–4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JJ, Smith SK, Hannon GJ, Joshua-Tor L (2004) Crystal structure of Argonaute and its implications for RISC slicer activity. Science 305: 1434–1437 [DOI] [PubMed] [Google Scholar]
- Steitz TA (1998) A mechanism for all polymerases. Nature 391: 231–232 [DOI] [PubMed] [Google Scholar]
- Steitz TA, Steitz JA (1993) A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci USA 90: 6498–6502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama Y, Iwai S, Ueno Y, Ikehara M, Ohtsuka E (1994) Role of the Mg2+ ion in the Escherichia coli ribonuclease HI reaction. J Biochem (Tokyo) 116: 1322–1329 [DOI] [PubMed] [Google Scholar]
- Viadiu H, Aggarwal AK (1998) The role of metals in catalysis by the restriction endonuclease BamHI. Nat Struct Biol 5: 910–916 [DOI] [PubMed] [Google Scholar]
- Vipond IB, Halford SE (1995) Specific DNA recognition by EcoRV restriction endonuclease induced by calcium ions. Biochemistry 34: 1113–1119 [DOI] [PubMed] [Google Scholar]
- Yang W (2005) Portraits of a Y-family DNA polymerase. FEBS Lett 579: 868–872 [DOI] [PubMed] [Google Scholar]
- Yang W, Hendrickson WA, Crouch RJ, Satow Y (1990) Structure of ribonuclease H phased at 2 A resolution by MAD analysis of the selenomethionyl protein. Science 249: 1398–1405 [DOI] [PubMed] [Google Scholar]
- Yang W, Steitz TA (1995) Recombining the structures of HIV integrase, RuvC and RNase H. Structure 3: 131–134 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information