

Abstract
The serine/threonine kinase HIPK2 phosphorylates the p53 protein at Ser 46, thus promoting p53-dependent gene expression and subsequent apoptosis. Here, we show that DNA damaging chemotherapeutic drugs cause degradation of endogenous HIPK2 dependent on the presence of a functional p53 protein. Early induced p53 allows caspase-mediated cleavage of HIPK2 following aspartic acids 916 and 977. The resulting C-terminally truncated HIPK2 forms show an enhanced induction of the p53 response and cell death, thus allowing the rapid amplification of the p53-dependent apoptotic program during the initiation phase of apoptosis by a regulatory feed-forward loop. The active HIPK2 fragments are further degraded during the execution and termination phase of apoptosis, thus ensuring the occurrence of HIPK2 signaling only during the early phases of apoptosis induction.
Keywords: apoptosis, caspases, HIPK2, p53
Introduction
The tumor suppressor protein p53 is a sequence-specific transcription factor that is stabilized and activated in response to a variety of stressful conditions such as DNA-damage, osmotic shock, hypoxia and oncogene activation (Vousden, 2000; Oren, 2003; Yee and Vousden, 2005). Active p53 binds to its DNA target sequences and reprograms gene expression to mediate either cell cycle arrest or apoptosis (Fridman and Lowe, 2003; Hofseth et al, 2004; Harris and Levine, 2005). p53 activity is regulated at several levels, the key event being the stabilization of the p53 protein. In unstimulated cells, p53 is constitutively associated with Mdm2, Pirh2 and COP1, which function as ubiquitin E3 ligases to promote p53 ubiquitination, thus allowing its constant destruction by the proteasome (Haupt et al, 1997; Kubbutat et al, 1997; Leng et al, 2003; Dornan et al, 2004). In response to p53 activating signals, the ubiquitin ligases dissociate from p53 allowing the subsequent stabilization of p53. In the case of Mdm2, the molecular mechanisms mediating this dissociation are quite well understood, as phosphorylation of the N-terminal phosphoacceptor sites at Ser 15, Thr 18 and Ser 20 impairs the affinity of p53 to Mdm2 (Shieh et al, 1997; Chehab et al, 1999). Furthermore, phosphorylation of Ser 33, Thr 81 and Ser 315 allows the peptidyl-prolyl isomerase Pin1 to generate a conformational change in p53 that facilitates its dissociation from Mdm2 (Zacchi et al, 2002; Berger et al, 2005). In turn p53 stimulates transcription of Mdm2, Pirh2 and COP1, ensuring the effective shutdown of the p53 response and establishing a negative feedback loop. Phosphorylation of p53 at further amino acids and additional modifications including neddylation, sumoylation and acetylation regulate the intensity, duration and amplitude of the transcriptional response (Fingerman and Briggs, 2004). Therefore, distinct p53 modification patterns control the subset of induced target genes, which determine whether p53 mediates cell cycle arrest or apoptosis. Induction of the transcription-dependent cell death program requires p53 phosphorylation at Ser 46, which is necessary for the development of apoptosis (Oda et al, 2000a) and determines whether apoptosis is attenuated or amplified (Mayo et al, 2005). The p53 protein can trigger apoptosis either via direct effects on target gene expression or alternatively by transcription-independent mechanisms via binding and inactivating prosurvival proteins such as Bcl-xL at the outer mitochondrial membrane (Dumont et al, 2003; Mihara et al, 2003). Among the genes mediating apoptosis or apoptosis-like cell death are the multidomain Bcl-2 family members Bax (Miyashita and Reed, 1995) and the ‘BH3 only' members such as PUMA (Nakano and Vousden, 2001) or Noxa (Oda et al, 2000b). Moreover, activation of p53 also upregulates and activates the effector caspase-6 (MacLachlan and El Deiry, 2002), caspase-10 (Rikhof et al, 2003) and PIDD, which associates with the adapter protein RAIDD to activate caspase-2 (Tinel and Tschopp, 2004).
Phosphorylation of p53 at Ser 46 critically depends on the serine/threonine kinase HIPK2 (D'Orazi et al, 2002), which enables subsequent CBP (CREB-binding protein)-mediated p53 acetylation at lysine 382 (Hofmann et al, 2002). These modifications depend on the presence of the PML protein and enhance the transcriptional activity of p53 to promote apoptosis (D'Orazi et al, 2002; Hofmann et al, 2002). HIPK2 and the highly homologous kinase HIPK1 were found to interact with p53 (D'Orazi et al, 2002; Hofmann et al, 2002; Kondo et al, 2003) and the p53 family member p73 (Kim et al, 2002). There is recent evidence that p53 Ser 46 phosphorylation also involves axin, which can bind to HIPK2 and p53. The stimulatory effect of Axin on p53-dependent transcriptional activity is abrogated in the presence of kinase inactive HIPK2. Removal of the Axin-interacting domain from HIPK2 (HIPK2ΔAxin) stimulates p53 transactivation even more dramatically than wild-type HIPK2. These data suggest that Axin physically occupies a putative autoinhibitory domain of HIPK2 located in the C-terminus of the kinase between amino acids 935 and 1050, thus stimulating its activity (Rui et al, 2004). In addition, HIPK2 expression leads to accumulation of p53 by decreasing the amount of Mdm2 (Wang et al, 2001). HIPK2 also antagonizes Mdm2-mediated nuclear export and ubiquitination of p53 (Di Stefano et al, 2004).
Here, we show that early p53 activation results in the caspase-dependent removal of the autoinhibitory domain in the C-terminus of HIPK2, allowing the rapid amplification of the apoptotic response. At later time points during ongoing apoptosis, the cleaved HIPK2 fragments are eliminated by further proteolytic degradation.
Results
p53-mediated gene expression induces HIPK2 degradation
In the course of experiments investigating the influence of HIPK2 on p53 functions, we noticed that treatment of human U2OS osteosarcoma cells with the DNA-damaging chemotherapeutic agent adriamycin caused apoptosis and resulted in the time-dependent disappearance of the HIPK2 protein that was paralleled by increased amounts of p53 (Figure 1A). To investigate whether HIPK2 decay is due to the impaired transcription of the HIPK2 gene, the effect of adriamycin on HIPK2 mRNA production was measured by RT–PCR (Figure 1B). These experiments revealed constant HIPK2 expression under conditions that lead to the disappearance of the HIPK2 protein, suggesting that DNA damage does not affect transcription of HIPK2. Indirect immunfluorescence showed that adriamycin induced degradation of the endogenous HIPK2 did not involve its relocalization from the subnuclear HIPK domains (Figure 1C). We next asked whether HIPK2 stability would also be affected by adriamycin doses leading to cell cycle arrest rather than apoptosis. Treatment of U2OS cells with low doses of adriamycin caused a cell cycle arrest at the G2/M phase, but did not change the amount of HIPK2 protein (Figure 1D). Does DNA-damage induced HIPK2 degradation depend on p53 activation or is it mediated by one of the numerous other pathways triggered by adriamycin (Norbury and Zhivotovsky, 2004)? To address this question, the effects of adriamycin on HIPK2 were tested in p53-deficient human H1299 lung carcinoma cells (Figure 2A). DNA damage failed to trigger HIPK2 decay in these cells, suggesting the importance of p53 for HIPK2 decline. Treatment of H1299 cells with exceedingly high concentrations of adriamycin had no effect on the relative amount of HIPK2 within the first 24 h (Figure 2B). A significant decrement of HIPK2 protein levels was only seen after 2 days in the presence of a very high dose of adriamycin in almost completely dead cells. Thus, it can be concluded that the HIPK2 protein is largely stable in p53 deficient cells, unless cells receiving exceedingly high concentrations of adriamycin are analyzed at late time points. Reconstitution of p53 expression in H1299 cells was sufficient to induce HIPK2 elimination in a dose-dependent fashion (Figure 2C), providing a direct proof for a causative role of p53 for HIPK2 cleavage. Elimination of HIPK2 was concomitant with the occurrence of two smaller bands of approximately 120 kDa (designated HIPK2*) and 105 kDa (designated HIPK2**). The antibody recognizes the N-terminal Flag epitope fused to HIPK2, indicating that the two fragments represent C-terminally cleaved HIPK2 fragments. To test whether cleavage of the endogenous HIPK2 protein results in the generation of the HIPK2* and HIPK2** fragments, HIPK2 cleavage was analyzed in adriamycin treated U2OS cells. These experiments showed that also the endogenous full-length kinase is converted to the HIPK2* and HIPK2** fragments. The rabbit αHIPK2 antibody used for these experiments also recognizes two nonspecific bands migrating almost similar to the cleavage products (Supplementary Figure 1). As HIPK2 serves as a p53 kinase, we tested whether its kinase function is required for p53 induced degradation. A kinase inactive HIPK2 point mutant (K221A) was degraded upon p53 activation, indicating that the kinase function is not decisive for HIPK2 clearance. Similarly, also HIPK2 K25A, which is point mutated in the SUMO-1 acceptor lysine (Gresko et al, 2005), was efficiently removed after p53 expression (Supplementary Figure 2). To distinguish whether p53 triggers HIPK2 degradation via transcription-dependent pathways, we tested the effects of specific p53 mutants. Expression of wild-type p53 efficiently induced the processing of HIPK2, while the naturally occuring DNA-binding domain point mutants p53 H175 and H273 remained without impact on HIPK2 protein levels (Figure 2D). Also, a p53 double point mutant (p53 22/23) with intact DNA-binding capacity but deficient in transactivation failed to trigger HIPK2 degradation, suggesting that induction of the p53 dependent transcription program is a prerequisite for its ability to cause HIPK2 processing. A p53 variant where Ser 46 was mutated to alanine was still able to trigger HIPK2 degradation, indicating that HIPK2-mediated p53 phosphorylation is not required for the induction of HIPK2 proteolysis (Supplementary Figure 3). Similarly, p53 variants lacking the C-terminal HIPK2 interaction domain and a p53 mutant lacking a growth suppression domain (p53 Δpro) (Walker and Levine, 1996) mediated HIPK2 destruction, as summarized in Figure 2E.
Figure 1.
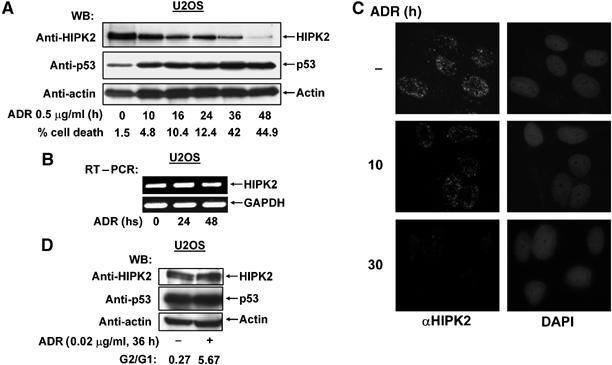
DNA damage triggers HIPK2 degradation. (A) U2OS cells were treated for the indicated periods with adriamycin (ADR). Equal amounts of proteins contained in cell extracts were separated by SDS–PAGE and analyzed by Western blotting (WB) for the occurrence of endogenous HIPK2, p53 and the loading control β-actin as shown. Apoptosis in these cells was quantified by FACS analysis, typical values are shown. (B) U2OS cells were treated with adriamycin (0.5 μg/ml) as shown. Relative mRNA levels of HIPK2 and the control GAPDH were determined by RT–PCR with specific primers, an ethidium bromide-stained agarose gel is displayed. (C) U2OS cells were treated with adriamycin (0.5 μg/ml) as shown and the localization of endogenous HIPK2 was revealed by indirect immunefluorescence. Nuclear DNA was stained with DAPI. (D) U2OS cells remained untreated or received 0.02 μg/ml adriamycin for 36 h. Cells were analyzed by FACS analysis for cell cycle distribution and by Western blotting for the occurrence of HIPK2, p53 and β-actin.
Figure 2.
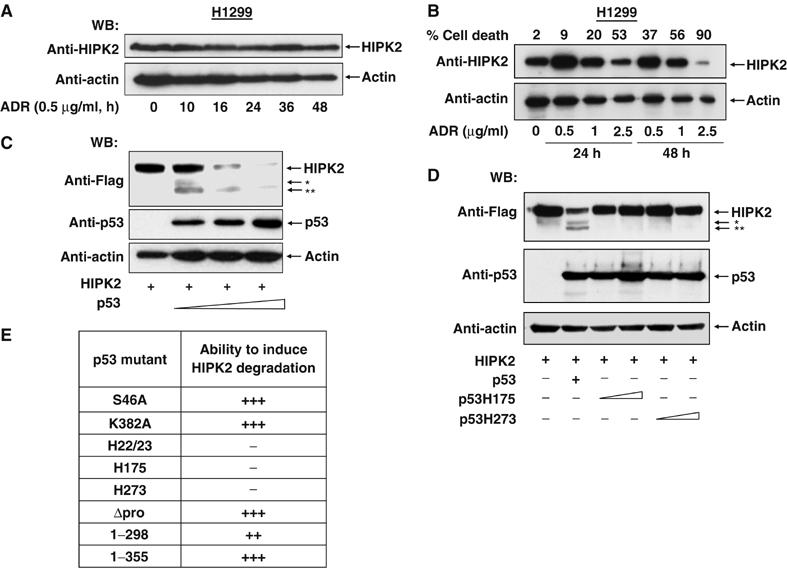
p53 activation triggers HIPK2 degradation. (A) p53-deficient H1299 cells were treated and analyzed as in (Figure 1A). (B) H1299 cells were treated with increasing concentrations of adriamycin for 24 or 48 h as shown. Cells were analyzed for apoptosis and by Western blotting for HIPK2 and the loading control β-actin. Note that cells showing massive apoptosis also contain less of the loading control. (C) H1299 cells were transiently transfected to express a constant amount of Flag-tagged HIPK2 and increasing concentrations of p53. Cell extracts were prepared and the occurrence of HIPK2 and p53 was detected by immunoblotting. The positions of HIPK2* and HIPK2** cleavage products are indicated. (D) H1299 cells were transfected to express Flag-HIPK2 in the absence or presence of p53 or escalating doses of the indicated p53 point mutants. After 24 h, cell extracts were prepared and analyzed by Western blotting. (E) Summary of the experiments measuring the impact of p53 mutants on their ability to trigger HIPK2 processing (+++ complete cleavage, ++ cleavage less efficient, − no cleavage).
p53 induces caspase-dependent HIPK2 cleavage
To investigate which class of p53-inducible proteases mediates HIPK2 proteolysis, adriamycin induced HIPK2 degradation was tested in the absence or presence of various inhibitors including those specific for Omi/HtrA2, calpains, cathepsins, serine proteases and the irreversible broad spectrum caspase inhibitor zVAD-fmk. Of all inhibitors tested, only zVAD-fmk significantly reduced the decay of endogenous HIPK2 (Figure 3A) and the generation of HIPK2* and HIPK2** fragments (data not shown), thus revealing the importance of caspases for this process. Also, p53-induced processing of HIPK2 was largely inhibited by zVAD-fmk (Figure 3B). HIPK2 cleavage was as well prevented by the zVEID-fmk peptide that specifically inhibits the function of caspase-6, although minor amounts of the HIPK2* were still detectable. The caspase-3 inhibitor Ac-DMQD-CHO inhibited the generation of the HIPK2** band but allowed the accumulation of the HIPK2* fragment, indicating that caspase-3 is not involved in the generation of HIPK2*. A quantitative analysis of band intensities demonstrated a substantial inhibition of HIPK2 cleavage by the caspase inhibitors used (Figure 3B), pointing to a prominent involvement of caspases for the p53 induced processing of HIPK2.
Figure 3.
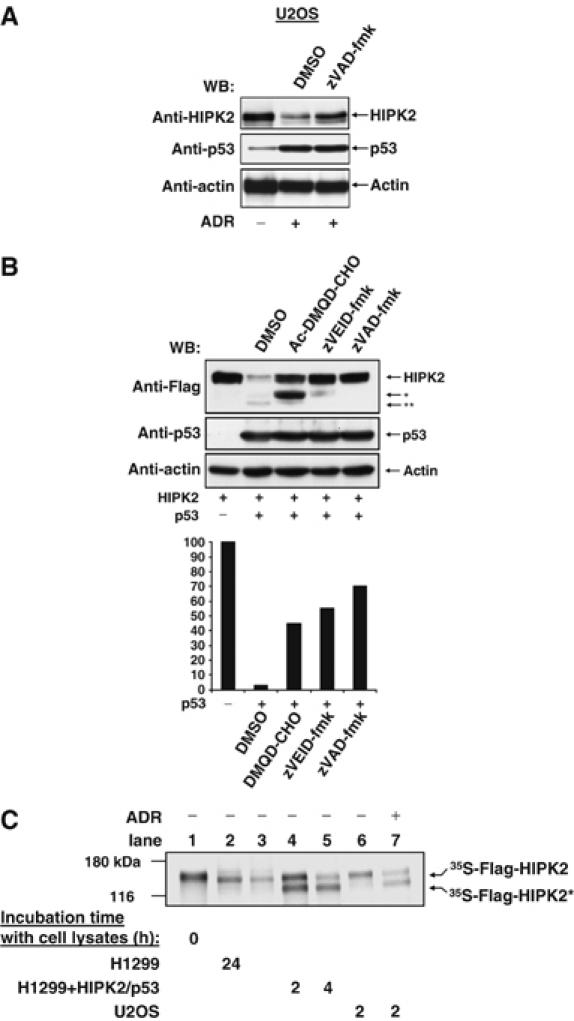
Caspase-dependent HIPK2 cleavage. (A) U2OS cells were treated for 24 h with adriamycin (0.5 μg/ml) and incubated in the presence of zVAD-fmk (25 μM) or DMSO as a solvent control as shown. Immunoblotting revealed the occurrence of HIPK2 and p53. (B) Upper: H1299 cells transiently expressing HIPK2 and p53 were incubated with Ac-DMQD-CHO, zVEID-fmk or zVAD-fmk (25 μM each) and analyzed for protein expression by immunoblotting. The positions of the full length and processed forms of HIPK2 are indicated. Lower: quantitative analysis of Western blot signal intensities from the full-length HIPK2 bands using Quantity One software (Biorad Inc.). The band intensity for HIPK2 expressed in the absence of p53 was arbitrarily set as 100%. (C) Cells were transfected with p53/HIPK2 or treated with adriamycin as shown, followed by the preparation of cell extracts and incubation with in vitro translated [35S]methionine-labeled HIPK2 at 37°C for 2 or 4 h. Reactions were analyzed by SDS–PAGE and autoradiography. The positions of marker proteins, full-length HIPK2 and the HIPK2* cleavage product are indicated.
Lysates from apoptotic cells can be used for in vitro experiments that allow further studies on substrate cleavage. HIPK2 was radioactively labelled by in vitro transcription/translation in the presence of [35S]methionine and incubated with lysates from control cells and apoptotic cells (Figure 3C). Incubation of HIPK2 with lysates from untreated H1299 cells did not result in any cleavage of HIPK2. The faint mobility shift between the control and HIPK2 in the lysate (Figure 3C, compare lanes 1 and 2) is due to the dephosphorylation of the heavily autophosphorylated kinase (Hofmann et al, 2000) and can be prevented in the presence of phosphatase inhibitors (data not shown). In contrast, incubation of radiolabelled HIPK2 with extracts of H1299 cells transfected to express p53 and HIPK2 resulted in the occurrence of a cleaved HIPK2* form that shows the same molecular weight as the 120 kDa HIPK2* form detected in vivo. The same HIPK2 fragment was observed when lysates from adriamycin-treated U2OS cells were used (Figure 3C, compare lanes 6 and 7). These results show that the HIPK2* cleavage product is generated regardless whether apoptosis is induced by DNA damage or upon p53/HIPK2 expression. However, this in vitro system did not allow to recapitulate the production of the HIPK2** fragment.
HIPK2 is cleaved by p53 induced caspase-6 following aspartic acid 977
Next, we addressed the question whether the in vitro generated HIPK2 degradation product corresponds to the previously described HIPK2* fragment with an intact N-terminus. Radiolabeled HIPK2 was incubated with control lysates or lysates from apoptotic cells for a period that allows the detection of HIPK2 in its full-length and fragmented forms, and was subsequently immunoprecipitated with αFlag or control antibodies. Analysis of immunoprecipitates by SDS–PAGE and autoradiography showed efficient precipitation of the full-length and truncated HIPK2 versions (Figure 4A). This shows that both proteins retained their N-terminal Flag epitope and strongly suggests the identity of the in vitro cleavage product with the HIPK2* form detected in vivo. To narrow down the HIPK2 cleavage site, various mutants lacking different parts of HIPK2 were produced by in vitro transcription/translation, incubated with apoptotic cell lysates and tested for the occurrence of cleavage products. While HIPK2 variants lacking different parts of the C-terminus (HIPK2Δ686–1014 and HIPK2 1–520) remained intact, a fragment encompassing the C-terminal half (551–1191) was cleaved (Figure 4B). The molecular weights of the resulting cleavage products suggest that caspases target a region between amino acids 869 and 980. This region contains seven putative caspase cleavage sites with aspartic acids at the P1 positions (Supplementary Figure 4). Mutation of aspartic acid at position 977 (but not at position 888) to alanine completely protected HIPK2 from in vitro cleavage (Figure 4C), showing that this site is crucial for the generation of the HIPK2* fragment. HIPK2* is generated upon cleavage after the sequence VECD, which matches to the reported preference of caspase-6 for valine in the P4 and glutamic acid in the P3 position (Thornberry et al, 1997). Accordingly, the caspase-6 inhibitor zVEID-fmk completely prevented in vitro HIPK2 cleavage (Figure 5A). Furthermore, the HIPK2* fragment was not generated in extracts of adriamycin-treated caspase-6 deficient cells (Figure 5B). A direct cleavage of HIPK2 by caspase-6 was tested with recombinant, active caspase-6. After 30 min of incubation, purified caspase-6 had completely converted the full-length form of the kinase to the HIPK2* cleavage product, and also allowed the detection of the 105 kDa HIPK2** fragment that was not produced in apoptotic lysates (Figure 5C). In contrast, recombinant caspase-3 was much less efficient in HIPK2 degradation. Incubation of radiolabelled HIPK2 D977A with caspase-3 or caspase-6 completely precluded the formation of the HIPK2* fragment, but allowed the detection of HIPK2**, albeit to a lesser extent (Figure 5D). We then compared the kinetics of HIPK2 degradation with the activation of caspase-6 in adriamycin treated U2OS cells (Figure 5E). The cleaved and thus activated form of caspase-6 was detectable already 14 h after induction of DNA damage and preceded the decay of endogenous HIPK2. The increase in the processed form of caspase-6 was not paralleled by a decrease of the caspase-6 precursor, presumably via the reported p53-induced resynthesis of this caspase (MacLachlan and El Deiry, 2002). The extracts were also tested for the degradation of the caspase-3 substrate poly(ADP ribose) polymerase (PARP), which is among the earliest cleaved substrates in the apoptotic process. At the latest time point analyzed, HIPK2 was almost completely processed while PARP was not fully converted to the cleaved fragment. Similarly, the coexpression of HIPK2 and p53 resulted also in caspase-6 cleavage (Supplementary Figure 5). To address the question whether the kinase function of HIPK2 affects caspase-6 activation, H1299 cells were transfected to express p53 along with HIPK2 or kinase inactive HIPK2 K221A. p53 induced activation of caspase-6 was further augmented by HIPK2, while HIPK2 K221A showed a reduced ability to trigger activation of this proteolytic enzyme (Figure 5F). The slight induction of caspase-6 activity in the absence of p53 Ser 46 phosphorylation by HIPK2 K221A shows that this p53 modification is not a prerequisite for caspase-6 activation. Collectively these data—which are supported by the notion that the caspase-2 inhibiting baculoviral p35 protein failed to prevent induced HIPK2 degradation (data not shown)—argue for the relevance of caspase-6 for HIPK2 degradation at position 977.
Figure 4.
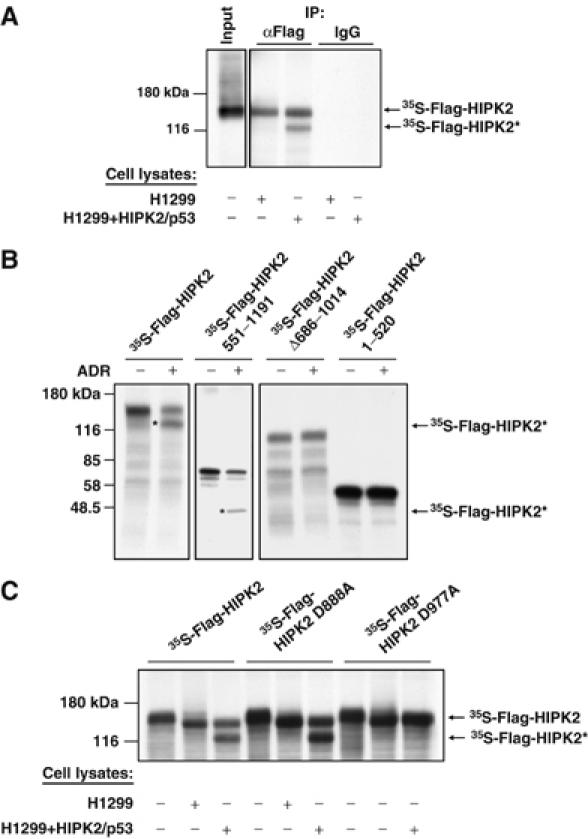
Caspase-mediated fragmentation of HIPK2 using in vitro cleavage assays. (A) Radiolabelled HIPK2 was incubated with extracts from p53/HIPK2 expressing or control cells. One aliquot was used for immunoprecipitation (IP) with αFlag antibodies, while isotype matched control antibodies were used for immunoprecipitation of another aliquot. Immunoprecipitates were analyzed by SDS–PAGE and autoradiography. (B) The indicated HIPK2 mutants were radiolabelled by in vitro transcription/translation and incubated for 2 h with extracts from control or adriamycin-treated U2OS cells. Cleavage was analyzed by SDS–PAGE and autoradiography. The positions of marker proteins are given, and cleavage products are highlighted by stars. (C) The indicated HIPK2 variants were produced by in vitro translation and incubated with extracts from control and p53/HIPK2 expressing cells. The reaction was further analyzed as in (B).
Figure 5.
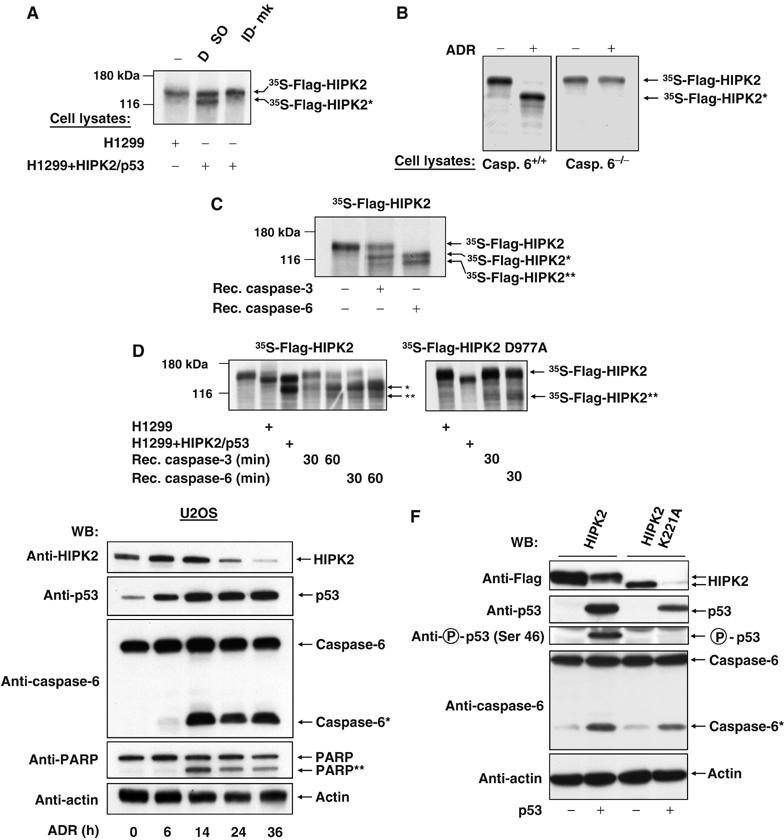
Direct involvement of caspase-6 in HIPK2 cleavage. (A) [35S]methionine-labeled HIPK2 was tested for in vitro cleavage in the absence or presence of zVEID-fmk (25 μM) as shown on the displayed autoradiogram. (B) Caspase-6 deficient DT40 cells or control cells were treated with adriamycin as shown. Cell lysates were incubated with radiolabelled HIPK2 for 2 h and analyzed for HIPK2 processing by SDS–PAGE and autoradiography. (C) HIPK2 was incubated for 30 min with 1 U of recombinant caspase-3 and -6, respectively, followed by SDS–PAGE and autoradiography. (D) Each of the recombinant caspases was incubated for the indicated periods with radiolabelled HIPK2 or HIPK2 D977A. Autoradiograms of dried SDS gels are shown. (E) U2OS cells were treated for the indicated periods with adriamycin (0.5 μg/ml) and equal amounts of protein contained in cell extracts were analyzed for the cleavage of caspase-6, PARP and HIPK2 and for the occurrence of p53 and β-actin by Western blotting. (F) H1299 cells were transfected to express the indicated HIPK2 proteins in the presence or absence of p53 as shown. After 30 h, cells were lyzed and extracts were further used for Western blot experiments.
HIPK2 is cleaved following aspartic acid 977 and 916 in vivo
In order to map the second caspase cleavage site in the C-terminus of HIPK2, we mutated further aspartic acids contained in the appropriate region (Supplementary Figure 4). HIPK2 or HIPK2 mutants bearing single and double point mutations in the putative caspase cleavage sites were expressed in the absence or presence of p53 (Figure 6A). The HIPK2 D977A mutant was significantly more stable and was cutted to yield only HIPK2**. Of all point mutants tested, only the HIPK2 D916A protein showed no p53 induced processing to HIPK2** and was cutted to yield exclusively the HIPK2* fragment, thus mapping the second cleavage site to aspartic acid 916. The double point mutant DD916/977AA was completely stable and did not allow detection of the two HIPK2 cleavage products. While the single point mutants showed no changes in their ability to phosphorylate p53 at Ser 46, the HIPK2 double point mutant displayed an increased capacity to mediate this phosphorylation when analyzed 30 h after transfection. Of note, the HIPK2 DD916/977AA mutant was only stabilized within the first 30 h after p53 expression or induction of DNA damage. At later time points during apoptosis, also this mutant kinase was eliminated further (Supplementary Figure 6). In order to characterize the HIPK2 cleavage sites utilized in vivo in more detail, we compared the electrophoretic mobility of HIPK2* and HIPK2** fragments resulting from HIPK2 cleavage with HIPK2 proteins derived from expression vectors encoding Flag-tagged HIPK2 1–916 or HIPK2 1–977 (Figure 6B). While HIPK2 1–916 exactly migrated like HIPK2**, the HIPK2 1–977 protein was indistinguishable from HIPK2*. The coexpression of p53 allowed further processing of HIPK2 1–977 to yield the HIPK2** fragment, while the HIPK2 1–916 fragment remained largely stable in the presence of p53.
Figure 6.
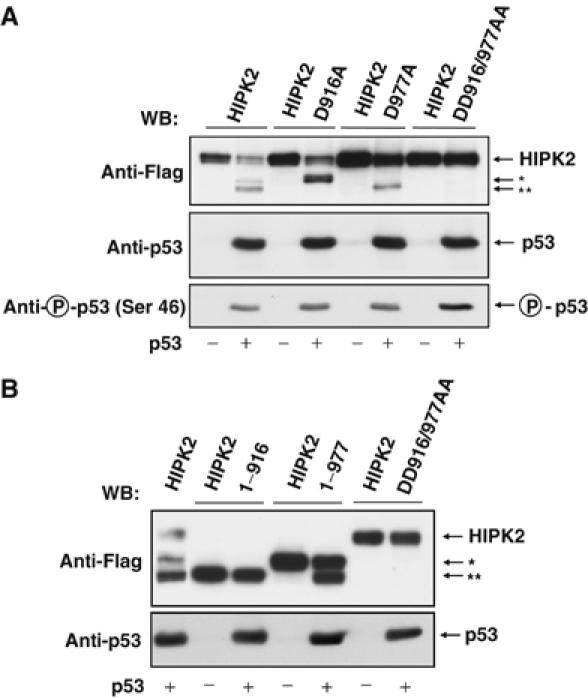
Identification of HIPK2 cleavage sites used in vivo. (A) H1299 cells were transfected with vectors encoding different Flag tagged wild type or aspartic acid to alanin HIPK2 point mutants in the absence or presence of p53 as shown. At 30 h after transfection, cells extracts were prepared and further analyzed by Western blotting. (B) H1299 cells were transfected with vectors encoding Flag tagged HIPK2, HIPK2 DD916/977AA, HIPK2 1–977 or HIPK2 1–916 in the absence or presence of p53. The next day, cells were lyzed and analyzed for HIPK2 processing and migration of the cleavage products as shown.
Early HIPK2 cleavage following aspartic acids 916 and 977 stimulates its proapoptotic activity
To check whether HIPK2 cleavage coincides with p53 phosphorylation and the onset of apoptosis, H1299 cells were transfected to express HIPK2 and p53. After 16 h, cells showing signs of beginning apoptosis such as rounding up and detaching were collected and compared to nonapoptotic and fully adherent cells. Immunoblotting showed that only the apoptotic cells displayed HIPK2 processing and concomitant p53 phosphorylation (Figure 7A). To test the physiological consequences of HIPK2 cleavage, we compared HIPK2 DD916/977AA (which cannot be cleaved to yield the active fragments) with HIPK2* and HIPK2** for their ability to trigger p53 Ser 46 phosphorylation at various time points. The fragments corresponding to the processed HIPK2 protein were more active in p53 phosphorylation early after transfection (Figure 7B), indicating that caspase-mediated removal of the autoinhibitory domain that was mapped between amino acids 935 and 1050 (Rui et al, 2004) renders the kinase more active. In contrast, 30 h after transfection during ongoing apoptosis, the differences in p53 Ser 46 phosphorylation were lost. The control blot shows that expression of the HIPK2* fragment (HIPK2 1–977) is expectedly paralleled by the production of HIPK2**, implicating that in such an experiment the activities of HIPK2* and HIPK2** are measured simultaneously. Furthermore, p53-induced expression from the proapoptotic bax promoter was triggered much better by the HIPK2* and HIPK2** fragments, when compared to the uncleavable HIPK2 DD916/977AA protein during the initiation phase of apoptosis, but not at later time points (Figure 7C). Accordingly, HIPK2 DD916/977AA is less active in bax induction and p53 phosphorylation when compared to the cleavage products generated upon HIPK2 processing (Supplementary Figure 7A and B), corroborating the finding that the HIPK2 cleavage products display higher activities. Similarly, also real-time PCR and RT–PCR confirmed a stronger ability of HIPK2* (Figure 7D) and HIPK2** (data not shown) to induce transcription of the endogenous Bax gene when compared to the noncleavable HIPK2 DD916/977AA protein. A comparison between HIPK2 and HIPK2 DD916/977AA for the enhancement of early p53 induced apoptosis revealed a similar picture. Specifically during the initiation phase of apoptosis, the uncleavable HIPK2 mutant was less active than the wild-type HIPK2 protein, which was processed to yield HIPK2* and HIPK2** (Figure 8A). We then tested whether activation of endogenous p53 by DNA damage allows the correct processing of HIPK2. HCT116 cells containing wild-type p53 and expressing either HIPK2, HIPK2 1–916, HIPK2 1–977 or HIPK2 DD916/977AA were exposed for 24 h to adriamycin to induce the p53 response. Immunoblotting showed the stability of the double point mutant and the correct processing of HIPK2 to its cleavage products (Figure 8B). The basal HIPK2 fragmentation in untreated cells can be attributed to the caspase-6 inducing activity of overexpressed HIPK2 (see Figure 5F and Supplementary Figure 5). Subsequently, we determined the abilities of the various HIPK2 forms to trigger apoptosis in response to DNA damage using p53 containing cells. HCT116 cells were transfected to express the noncleavable HIPK2 DD916/977AA mutant or each of the cleavage products, followed by induction of apoptosis upon incubation with adriamycin for short and long periods. The cleavage products caused higher apoptosis rates when compared to HIPK2 DD916/977AA exclusively during the apoptosis initiation phase, but not 30 h after apoptosis induction (Figure 8C).
Figure 7.
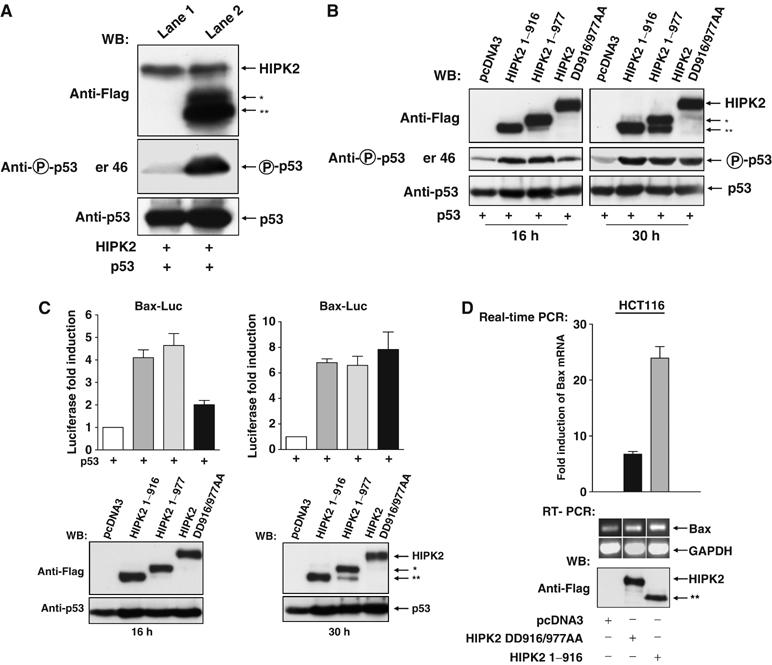
Functional analysis of HIPK2 cleavage at aspartic acids 916 and 977. (A) At 16 h after transfection of H1299 cells with expression vectors encoding HIPK2 and p53, dishes were gently rocked and the detached and swimming cells showing signs of ongoing apoptosis were collected by centrifugation (lane 2), while the adherent cells (lane 1) were collected after scraping with a rubber. Cell extracts were analyzed by immunoblotting for the phosphorylation of p53 and the processing of HIPK2 as shown. (B) H1299 cells were transfected to express p53 in combination with the HIPK2 noncleavable mutant or vectors encoding HIPK2* or HIPK2** and harvested after 16 or 30 h. Extracts were analyzed for the phosphorylation of p53 and the expression of HIPK2 variants by immunoblotting as shown. (C) H1299 cells were transfected with a luciferase reporter gene controlled by the human bax promoter and a p53 expression vector together with the indicated combinations of vectors encoding HIPK2 DD916/977AA, HIPK2 1–977 or HIPK2 1–916. Upper: Luciferase activity was analyzed 16 and 30 h after transfection, gene induction by p53 alone was arbitrarily set as 1. Lower: Cell extracts were analyzed for the occurrence of p53 and HIPK2 as shown. (D) HCT116 cells were transfected with empty vector or plasmids encoding noncleavable HIPK2 or HIPK2 1–916 as shown, followed by the extraction of RNA and subsequent real time PCR or alternatively RT–PCR for Bax and GAPDH. Upper: Quantification of normalized Bax mRNA expression, fold induction relative to pcDNA3 transfected cells is shown. Error bars show standard deviations from two independent experiments performed in triplicate. Lower: The ethidium bromide-stained agarose gel shows the products of the RT–PCR, the Western blot in the lower part ensures correct and comparable expression of HIPK2 variants.
Figure 8.
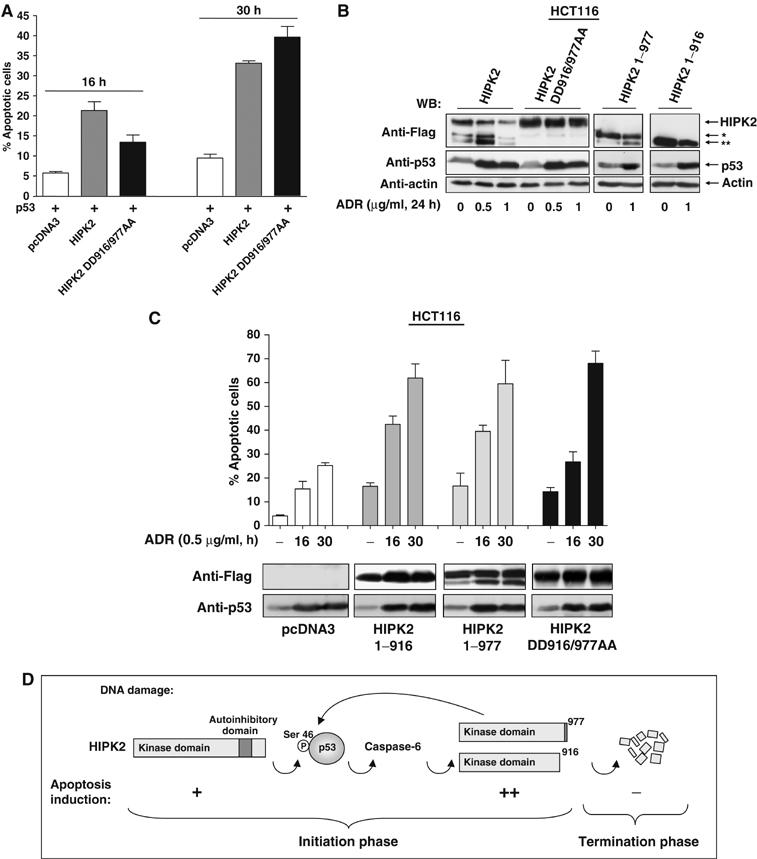
Functional analysis of apoptotic HIPK2* and HIPK2** activities. (A) H1299 cells were transfected with the indicated combinations of p53 and HIPK2 or HIPK2 DD916/977AA and analyzed for apoptosis after 16 and 30 h. Error bars show standard deviations. (B) HCT116 cells were transfected to express the indicated HIPK2 variants. The next day, cells were exposed for 24 h to adriamycin as shown. Cell extracts were analyzed by immunoblotting for HIPK2 processing and p53 accumulation. (C) HCT116 cells transfected to express the indicated HIPK2 proteins were treated for 16 or 30 h with adriamycin and then assayed for apoptosis, bars show standard deviations obtained from several independent experiments. Control blots ensured adequate expression of HIPK2 proteins and p53 induction. (D) Schematic diagram illustrating the pathways mediating p53 phosphorylation and activation, HIPK2 cleavage and apoptosis induction.
Discussion
The p53 circuit is highly linked to other signalling systems including the Wnt/β-catenin, Rb-E2F and p14/19 ARF pathways. This high connectivity poses the need for a tight and highly integrated control of p53 activity, which is ensured by seven negative and three positive feedback loops that have been described thus far (Harris and Levine, 2005). This study adds two new regulatory loops to the p53 system: a positive feedforward loop during the initiation of apoptosis serves to amplify the p53 response. The caspase dependent removal of the C-terminal autoinhibitory domain results in the generation of highly active HIPK2 fragments. Later during the execution and termination phase of apoptosis, a negative feedback loop ensures complete degradation and elimination of the HIPK2 response, as schematically displayed in Figure 8D. HIPK2 exerts a nonredundant function for p53, as downmodulation of HIPK2 by siRNA strongly impaired p53 Ser 46 phosphorylation and p53 induced bax expression (data not shown).
Caspase-dependent HIPK2 activation allows a rapid amplification of the apoptotic process. Accordingly, an HIPK2 fragment lacking the C-terminal autoinhibitory domain was found to be more active for p53 Ser 46 phosphorylation and p53 dependent gene expression than the wild-type kinase (Rui et al, 2004). This study shows that caspase-generated HIPK2* and HIPK2** fragments display strongly enhanced abilities to trigger p53 phosphorylation at Ser 46, bax expression and apoptosis. However, it is well possible that hyperactive HIPK2 fragments also amplify apoptosis by p53-independent pathways as HIPK2* shows an increased ability to trigger JNK activation (Supplementary Figure 8), and may possibly also affect phosphorylation of the cell death regulator Daxx (Ecsedy et al, 2003; Hofmann et al, 2003) or promote proteolysis of cMyb and CtBP (Kanei-Ishii et al, 2004; Zhang et al, 2005). Activation of proapoptotic kinase signaling by caspase-mediated cleavage also occurs for several other kinases including LIMK1 (Tomiyoshi et al, 2004), Mst1 (de Souza et al, 2002) and RIP (Lin et al, 1999).
Various experimental approaches indicate that caspase-6 is prominently involved in HIPK2 cleavage at aspartic acid 977. The contribution of further caspases cannot be ruled out as minor amounts of the HIPK2* fragment are detected in the presence of the caspase-6 specific inhibitor zVEID-fmk and also recombinant caspase-3 has the ability to process HIPK2 at this position. The nature of the caspase that cleaves after aspartic acid 916 remains less well defined, as this site is not efficiently processed in apoptotic lysates for unknown reasons and only generated upon incubation with recombinant caspases. The generation of the HIPK2** fragment might involve the activity of p53 induced caspases-2 and -10 (Rikhof et al, 2003; Tinel and Tschopp 2004), but a direct cleavage seems to be unlikely as caspase-10 displays a different cleavage site specificity and is exclusively found in the cytosol (Shikama et al, 2001), while the nuclear localization of HIPK2 is unchanged during apoptosis. As low concentrations of zVAD-fmk strongly prevent HIPK2 degradation but fail to efficiently inhibit caspase-2 (Thornberry et al, 1997), this nuclear caspase seems not to be of major importance for HIPK2 cleavage.
The elimination of a regulatory kinase by a p53-induced apoptotic program is also seen for ERK2, which is inactivated in order to prevent ERK-mediated cell proliferation in the presence of activated p53 (Marchetti et al, 2004). The long list of proteolytically inactivated regulators of cell proliferation and viability also includes the HIPK2 interacting protein CBP. This acetyl transferase is eliminated during neurodegeneration upon caspase-6-mediated cleavage, thus obviating its neuroprotective function (Rouaux et al, 2003). Also the PML protein, which is essential for the formation and function of PML nuclear bodies as platforms for p53 modification, is subject to proteolytic regulation after addition of the chemotherapeutic agent arsenic trioxide (Lallemand-Breitenbach et al, 2001). During ongoing apoptosis, the HIPK2* and HIPK2** fragments do not accumulate and are further degraded by mechanisms that remain to be investigated in future studies. This ensures the termination of the HIPK2 response and accordingly p53 phosphorylation at Ser 46 vanishes in the execution phase of DNA damage induced apoptosis, while phosphorylation at Ser 15 and 20 still occurs (Oda et al, 2000b). Termination of the p53 response is ensured by several mechanisms including the p53-dependent upregulation of the p53 inhibitors Mdm2, Pirh2 and COP1 (Shmueli and Oren, 2004) and also the p53-mediated destruction of the p53 activator HIPK2 described in this study. Adequate termination of the p53 response is very essential, as hyperactivation of this key pathway has been associated with degenerative diseases such as arthritis, multiple sclerosis (Wosik et al, 2003) and neuropathies (Mattson et al, 2001). It is tempting to speculate that the impaired apoptotic response induced by the p53 H175 mutant that frequently occurs in cancer is not only attributable to suppressed activation of caspase-3 (Tsang et al, 2003), but also to a lack of HIPK2 processing.
Materials and methods
Cell culture, transfections and stimulations
Human U2OS cells, p53-deficient H1299 and HCT116 cells, and caspase-6 deficient cells (Rouaux et al, 2003) were grown in Dulbecco's modified Eagle medium supplemented with 10% (v/v) fetal calf serum, 2 mM L-glutamine and 1% (v/v) penicillin/streptomycin in an humidified incubator at 37°C and 5% CO2. Cells were plated out 24 h prior to transfection and transiently transfected using Rotifect (Roth), according to the manufacturer's instructions. DNA amounts in each transfection were kept constant upon addition of empty expression vector. DNA damage was induced upon the addition of adriamycin (0.5 μg/ml).
Antisera, plasmids and reagents
Antibodies recognizing Flag (M2), p53 (DO-1) and β-actin were obtained from Santa Cruz Inc., the phospho p53–Ser 46 antibody from Cell Signalling Technology. The polyclonal αHIPK2 antibodies were previously described (Hofmann et al, 2002), the caspase-6 antibody was from MBL (clone 3E8) and PARP antibodies were from Biomol Research Laboratories Inc. The luciferase reporter gene controlled by the human bax promoter and expression vectors encoding p53 or HIPK2 and its mutants as well as the caspase-6 expression vector were published (Shikama et al, 2001; Hofmann et al, 2002). HIPK2 point mutants were produced by the QuickChange Site-Directed Kit (Stratagene). The pSUPER-HIPK2 vector was produced based on the pSUPER vector (Brummelkamp et al, 2002). The HIPK2 1–916 and HIPK2 1–977 vectors were cloned via PCR using standard procedures. The caspase inhibitors zVAD-fmk (BD Biosciences), zVEID-fmk and DMQD-CHO were from Alexis or Calbiochem.
Immunoprecipitation, cell extracts and Western blotting
Samples were preincubated for 60 min on a rotator with 800 μl IP buffer (50 mM Tris/HCl pH 7.5, 150 mM NaCl, 1 mM phenylmethylsulfonylfluoride, leupeptine (10 μg/ml), aprotinin (10 μg/ml), 1% (v/v) NP-40 and 10% (v/v) glycerol) and 20 μl of protein A/G plus agarose (Santa Cruz Inc.). After centrifugation, 1.5 μg of Flag antibody and 25 μl of protein A/G plus agarose were added to the supernatant, followed by rotation for 4 h on a spinning wheel at 4°C. After washing five times with 1 ml of IP buffer, the immunoprecipitates were boiled in 1 × SDS sample buffer, separated by SDS–PAGE and exposed to an X-ray film. Cell extracts were prepared by resuspending cell pellets directly in 1 × SDS lysis buffer, heating for 5 min at 95°C and sonification of chromosomal DNA. Equal amounts of protein were further analyzed by reducing SDS–PAGE and Western blotting onto a PVDF membrane (Millipore, Bedford, MA). The membrane was blocked and then incubated in TBST containing the primary antibody and 2% (w/v) milk powder. The respective proteins were incubated with an appropriate secondary antibody coupled to horseradish peroxidase and visualized by enhanced chemiluminescence according to the instructions of the manufacturer (NEN).
Indirect immunefluorescence
Cells were seeded on coverslips and treated for various periods with adriamycin. Cells were fixed for 1 min at −20°C with methanol/aceton (1:1), air-dried, rehydrated with PBS, blocked with goat serum and stained with αHIPK2 antibodies. Alexa 488-coupled goat α-mouse (Molecular Probes) was used as a secondary antibody, chromosomal DNA was visualized by DAPI staining.
PCR
Cells were lyzed and total RNA was extracted using the RNAeasy kit (Qiagen). RNA (1 μg) was used as a template for the generation of cDNAs from Oligo (dT)20 primers using the Superscript first strand synthesis system (Invitrogen). HIPK2 was detected with the specific primers 5′-agtccacgactccccctact and 5′-atggtgggagtgatgtaggc, Bax with 5′-cccttttgcttcagggtttc and 5′-cagttgaagttgccgtcaga and GAPDH with 5′-atgttcgtcatgggtgtgaa and 5′-acctggtgctcagtgtagcc primers. Real Time PCR was performed using LightCycler® FastStart DNA Master SYBR Green according to the manufacturer's instruction (Roche), followed by quantification, normalization to GAPDH and statistical analysis. RT–PCR was performed for 32 cycles and PCR products were detected after electrophoresis on an ethidium bromide-stained agarose gel.
Determination of cell viability and the cell cycle profile
Early apoptosis was assessed via the detection of mitochondrial depolarization visualized by JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) staining and subsequent flow cytometry: red fluorescence (FL-2 channel) of JC-1 (J-aggregates) indicated intact mitochondria, whereas green fluorescence (FL-1 channel) showed monomeric JC-1 due to the breakdown of ΔΨm at the early stages of apoptosis. Late apoptosis was measured by the uptake of 7-AAD (7-amino-actinomycin D) using flow cytometry (FL3 channel). In addition to these methods, cell viability and apoptosis was also determined by transfection of cells with the indicated plasmids and a vector allowing expression of the GFP protein in order to identify the transfected cells. After induction of apoptosis, pictures from GFP positive cells were taken and cell viability was assessed by the examining cell morphology of the GFP positive cells. At least 300 GFP expressing cells were analyzed from each sample. Cell cycle distribution was measured by incubating ethanol-fixed cells with 0.05 mg/ml propidium iodide and 400 Kuniz units of RNAse A in PBS containing 0.1% glucose for 30 min at room temperature and analyzing the DNA content in a flow cytometer.
In vitro caspase cleavage assays
The HIPK2 proteins were labelled with [35S]methionine (Amersham Lifescience) using the coupled TnT in vitro transcription/translation system from rabbit reticulocyte lysates, according to the instructions of the manufacturer (Promega Inc.). Control and apoptotic cells (grown on 6 cm culture dishes) were lysed in 30 μl of freshly prepared NP-40 lysis buffer containing 50 mM Tris–HCl (pH 8), 5 mM EDTA, 150 mM NaCl, 0.5% NP-40, 0.5 mM PMSF, 10 μg/ml Aprotinin, 10 μg/ml Leupeptin. For in vitro cleavage assays, 15 μl of freshly prepared cleavage buffer containing 20 mM HEPES pH 7.4, 2 mM DTT and 10% glycerol (v/v) were mixed with 5 μl of cell lysate (or 1 U of corresponding recombinant caspase) and 3 μl of [35S] proteins at 37°C for the periods specified in the figure legends. After separation by SDS–PAGE, the gel was fixed (10% (v/v) acetic acid, 20% (v/v) methanol). The radioactive signals were enhanced using Amplify® (Amersham) prior to drying and autoradiography.
Luciferase reporter gene assays
Harvested cells were lyzed in reporter lysis buffer (25 mM Tris-Phosphate, 2 mM DTT, 2 mM CDTA, 10% (v/v) glycerol, 1% (v/v) Triton X-100). Luciferase activity was determined in a luminometer (Duo Lumat LB 9507, Berthold) by injecting 20 μl of assay buffer (40 mM Tricine, 2.14 mM (MgCO3)4Mg(OH)2 × 5 H2O, 5.34 mM MgSO4, 0.2 mM EDTA, 66.6 mM DTT, 540 μM CoA, 940 μM luciferin, 1.06 mM ATP) and measuring light emission for 10 s.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8
Acknowledgments
We are grateful to B Earnshaw (Edinburgh) for caspase-6 deficient cells, M Ruthardt (Frankfurt) for HCT116 cells, M Calzado (Bern) for help with real time PCR, and A Levine (Princeton) for the p53 22/23 mutant. We also thank J Stein (Bern) for help with flow cytometry, Jürg Tschopp (Lausanne) for helpful discussions and T Miyashita (Tokyo) for GFP-caspase-6. Our laboratory is supported by grants from Oncosuisse, Schweizerischer Nationalfonds, Deutsche Forschungsgemeinschaft, the EU FP6 program and the Association for International Cancer Research AICR.
References
- Berger M, Stahl N, Del Sal G, Haupt Y (2005) Mutations in proline 82 of p53 impair its activation by Pin1 and Chk2 in response to DNA damage. Mol Cell Biol 25: 5380–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553 [DOI] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD (1999) Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci USA 96: 13777–13782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, Piaggio G, Fanciulli M, Appella E, Soddu S (2002) Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol 4: 11–19 [DOI] [PubMed] [Google Scholar]
- de Souza PM, Kankaanranta H, Michael A, Barnes PJ, Giembycz MA, Lindsay MA (2002) Caspase-catalyzed cleavage and activation of Mst1 correlates with eosinophil but not neutrophil apoptosis. Blood 99: 3432–3438 [DOI] [PubMed] [Google Scholar]
- Di Stefano V, Blandino G, Sacchi A, Soddu S, D'Orazi G (2004) HIPK2 neutralizes MDM2 inhibition rescuing p53 transcriptional activity and apoptotic function. Oncogene 23: 5185–5192 [DOI] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O'Rourke K, Koeppen H, Dixit VM (2004) The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429: 86–92 [DOI] [PubMed] [Google Scholar]
- Dumont P, Leu JI, Della PA III, George DL, Murphy M (2003) The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 33: 357–365 [DOI] [PubMed] [Google Scholar]
- Ecsedy JA, Michaelson JS, Leder P (2003) Homeodomain-interacting protein kinase 1 modulates Daxx localization, phosphorylation, and transcriptional activity. Mol Cell Biol 23: 950–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingerman IM, Briggs SD (2004) p53-mediated transcriptional activation: from test tube to cell. Cell 117: 690–691 [DOI] [PubMed] [Google Scholar]
- Fridman JS, Lowe SW (2003) Control of apoptosis by p53. Oncogene 22: 9030–9040 [DOI] [PubMed] [Google Scholar]
- Gresko E, Möller A, Roscic A, Schmitz ML (2005) Covalent modification of human homeodomain interacting protein kinase 2 by SUMO-1 at lysine 25 affects its stability. Biochem Biophys Res Commun 329: 1293–1299 [DOI] [PubMed] [Google Scholar]
- Harris SL, Levine AJ (2005) The p53 pathway: positive and negative feedback loops. Oncogene 24: 2899–2908 [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387: 296–299 [DOI] [PubMed] [Google Scholar]
- Hofmann TG, Mincheva A, Lichter P, Droge W, Schmitz ML (2000) Human homeodomain-interacting protein kinase-2 (HIPK2) is a member of the DYRK family of protein kinases and maps to chromosome 7q32–q34. Biochimie 82: 1123–1127 [DOI] [PubMed] [Google Scholar]
- Hofmann TG, Möller A, Sirma H, Zentgraf H, Taya Y, Droge W, Will H, Schmitz ML (2002) Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol 4: 1–10 [DOI] [PubMed] [Google Scholar]
- Hofmann TG, Stollberg N, Schmitz ML, Will H (2003) HIPK2 regulates transforming growth factor-beta-induced c-Jun NH(2)-terminal kinase activation and apoptosis in human hepatoma cells. Cancer Res 63: 8271–8277 [PubMed] [Google Scholar]
- Hofseth LJ, Hussain SP, Harris CC (2004) p53: 25 years after its discovery. Trends Pharmacol Sci 25: 177–181 [DOI] [PubMed] [Google Scholar]
- Kanei-Ishii C, Ninomiya-Tsuji J, Tanikawa J, Nomura T, Ishitani T, Kishida S, Kokura K, Kurahashi T, Ichikawa-Iwata E, Kim Y, Matsumoto K, Ishii S (2004) Wnt-1 signal induces phosphorylation and degradation of c-Myb protein via TAK1, HIPK2, and NLK. Genes Dev 18: 816–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Park JS, Um SJ (2002) Identification and characterization of HIPK2 interacting with p73 and modulating functions of the p53 family in vivo. J Biol Chem 277: 32020–32028 [DOI] [PubMed] [Google Scholar]
- Kondo S, Lu Y, Debbas M, Lin AW, Sarosi I, Itie A, Wakeham A, Tuan J, Saris C, Elliott G, Ma W, Benchimol S, Lowe SW, Mak TW, Thukral SK (2003) Characterization of cells and gene-targeted mice deficient for the p53-binding kinase homeodomain-interacting protein kinase 1 (HIPK1). Proc Natl Acad Sci USA 100: 5431–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH (1997) Regulation of p53 stability by Mdm2. Nature 387: 299–303 [DOI] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V, Zhu J, Puvion F, Koken M, Honore N, Doubeikovsky A, Duprez E, Pandolfi PP, Puvion E, Freemont P, de The H (2001) Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11S proteasome recruitment, and As2O3-induced PML or PML/retinoic acid receptor alpha degradation. J Exp Med 193: 1361–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S (2003) Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112: 779–791 [DOI] [PubMed] [Google Scholar]
- Lin Y, Devin A, Rodriguez Y, Liu ZG (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13: 2514–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLachlan TK, El Deiry WS (2002) Apoptotic threshold is lowered by p53 transactivation of caspase-6. Proc Natl Acad Sci USA 99: 9492–9497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti A, Cecchinelli B, D'Angelo M, D'Orazi G, Crescenzi M, Sacchi A, Soddu S (2004) p53 can inhibit cell proliferation through caspase-mediated cleavage of ERK2/MAPK. Cell Death Differ 11: 596–607 [DOI] [PubMed] [Google Scholar]
- Mattson MP, Duan W, Pedersen WA, Culmsee C (2001) Neurodegenerative disorders and ischemic brain diseases. Apoptosis 6: 69–81 [DOI] [PubMed] [Google Scholar]
- Mayo LD, Rok SY, Jackson MW, Smith ML, Rivera G Jr, Korgaonkar CK, Donner DB (2005) Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified. J Biol Chem 280: 25953–25959 [DOI] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11: 577–590 [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299 [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694 [DOI] [PubMed] [Google Scholar]
- Norbury CJ, Zhivotovsky B (2004) DNA damage-induced apoptosis. Oncogene 23: 2797–2808 [DOI] [PubMed] [Google Scholar]
- Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y (2000a) p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102: 849–862 [DOI] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N (2000b) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058 [DOI] [PubMed] [Google Scholar]
- Oren M (2003) Decision making by p53: life, death and cancer. Cell Death Differ 10: 431–442 [DOI] [PubMed] [Google Scholar]
- Rikhof B, Corn PG, El Deiry WS (2003) Caspase 10 levels are increased following DNA damage in a p53-dependent manner. Cancer Biol Ther 2: 707–712 [PubMed] [Google Scholar]
- Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP, Boutillier AL (2003) Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J 22: 6537–6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui Y, Xu Z, Lin S, Li Q, Rui H, Luo W, Zhou HM, Cheung PY, Wu Z, Ye Z, Li P, Han J, Lin SC (2004) Axin stimulates p53 functions by activation of HIPK2 kinase through multimeric complex formation. EMBO J 23: 4583–4594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91: 325–334 [DOI] [PubMed] [Google Scholar]
- Shikama Y, Mami U, Miyashita T, Yamada M (2001) Comprehensive studies on subcellular localizations and cell death-inducing activities of eight GFP-tagged apoptosis-related caspases. Exp Cell Res 264: 315–325 [DOI] [PubMed] [Google Scholar]
- Shmueli A, Oren M (2004) Regulation of p53 by Mdm2: fate is in the numbers. Mol Cell 13: 4–5 [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem 272: 17907–17911 [DOI] [PubMed] [Google Scholar]
- Tinel A, Tschopp J (2004) The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304: 843–846 [DOI] [PubMed] [Google Scholar]
- Tomiyoshi G, Horita Y, Nishita M, Ohashi K, Mizuno K (2004) Caspase-mediated cleavage and activation of LIM-kinase 1 and its role in apoptotic membrane blebbing. Genes Cells 9: 591–600 [DOI] [PubMed] [Google Scholar]
- Tsang WP, Chau SP, Fung KP, Kong SK, Kwok TT (2003) Modulation of multidrug resistance-associated protein 1 (MRP1) by p53 mutant in Saos-2 cells. Cancer Chemother Pharmacol 51: 161–166 [DOI] [PubMed] [Google Scholar]
- Vousden KH (2000) p53: death star. Cell 103: 691–694 [DOI] [PubMed] [Google Scholar]
- Walker KK, Levine AJ (1996) Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc Natl Acad Sci USA 93: 15335–15340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Debatin KM, Hug H (2001) HIPK2 overexpression leads to stabilization of p53 protein and increased p53 transcriptional activity by decreasing Mdm2 protein levels. BMC Mol Biol 2: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wosik K, Antel J, Kuhlmann T, Bruck W, Massie B, Nalbantoglu J (2003) Oligodendrocyte injury in multiple sclerosis: a role for p53. J Neurochem 85: 635–644 [DOI] [PubMed] [Google Scholar]
- Yee KS, Vousden KH (2005) Complicating the complexity of p53. Carcinogenesis 26: 1317–1322 [DOI] [PubMed] [Google Scholar]
- Zacchi P, Gostissa M, Uchida T, Salvagno C, Avolio F, Volinia S, Ronai Z, Blandino G, Schneider C, Del Sal G (2002) The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 419: 853–857 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Nottke A, Goodman RH (2005) Homeodomain-interacting protein kinase-2 mediates CtBP phosphorylation and degradation in UV-triggered apoptosis. Proc Natl Acad Sci USA 102: 2802–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Figure 8