

Abstract
The NF-κB p50 is the N-terminal processed product of the precursor, p105. It has been suggested that p50 is generated not from full-length p105 but cotranslationally from incompletely synthesized molecules by the proteasome. We show that the 20S proteasome endoproteolytically cleaves the fully synthesized p105 and selectively degrades the C-terminus of p105, leading to p50 generation in a ubiquitin-independent manner. As small as 25 residues C-terminus to the site of processing are sufficient to promote processing in vivo. However, any p105 mutant that lacks complete ankyrin repeat domain (ARD) is processed aberrantly, suggesting that native processing must occur from a precursor, which extends beyond the ARD. Remarkably, the mutant p105 that lacks the internal region including the glycine-rich region (GRR) is completely degraded by 20S proteasome in vitro. This suggests that the GRR impedes the complete degradation of the p105 precursor, thus contributing to p50 generation.
Keywords: 20S proteasome, NF-κB, p105, processing, ubiquitination
Introduction
NF-κB dimers play a central role in orchestrating diverse biological processes through their transcriptional activity (Ghosh et al, 1998). The NF-κB family has five members, p50, p52, p65, c-Rel, and RelB. p50 homo- and heterodimers carry out broad and diverse cellular functions. All cellular p50 is generated from its precursor p105, and thus the ultimate composition of potent NF-κB dimers is controlled at the level of processing of p105 into p50. This processing event has generated significant attention since the discovery of p105 because of its unique nature. It has been well established that the proteasome is responsible for p105 processing (Coux and Goldberg, 1998; Ciechanover et al, 2001). The generation of p50 from p105 is one of the few examples in which the substrate undergoes processing instead of complete degradation by the proteasome. The other examples of proteasome-mediated processing that generate products from the precursors are the inducible processing of yeast transcription factors SPT23 and MGA2 in response to metabolic state of yeast cells and the regulated processing of Drosophila Ci transcription factor (Chen et al, 1999; Hoppe et al, 2000).
p105 is a multidomain protein consisting of four structural domains connected and flanked by flexible segments. These domains are the N-terminal domain (NTD), dimerization domain (DimD), ankyrin repeat domain (ARD), and death domain (DD) (Figure 1A). The DimD and ARD are connected by a 180-residue-long largely unstructured region, which contains the site of processing and critical elements regulating processing. The 22-amino-acid segment (351–372) immediately C-terminal to the DimD contains the nuclear localization sequence (NLS). Together the NTD, DimD, and NLS polypeptide constitute the p50 Rel homology region (RHR). A glycine-rich region (GRR) follows the RHR and ends at the site of processing (Lin and Ghosh, 1996; Orian et al, 1999). From this study, it appears that the site of processing is at or near residue 430 of mouse p105 sequence. The segment between the GRR and the ARD (roughly from residue 431 to 530) is not homologous to any known structural domain and its role in p105 processing has not been well defined. The C-terminus of p105 (residues 365–971) is also referred to as IκBγ (Inoue et al, 1992). The full-length p105 functions as an inhibitor of NF-κB transcriptional activity (Liou et al, 1992). Specific stimulus activates IκB kinase β (IKKβ), which phosphorylates p105 C-terminal serine residues 927 and 932 allowing for degradation of entire p105 and release of the bound NF-κB subunits (Lang et al, 2003). A number of studies have attempted to elucidate the mechanism of p105 processing. The accepted mechanism of p105 processing is the cotranslational processing model (Lin et al, 1998, 2000). This model proposes that the random pausing of the ribosome after synthesizing approximately 530 residues (around the beginning of ARD) leads to the generation of p50. This has been suggested to occur by proteasomal processing while the synthesized protein is still bound to the ribosome. However, when the ribosome does not pause, a complete p105 molecule is synthesized, which cannot undergo further processing. Several other studies have shown that ubiquitination was also essential for constitutive p105 processing (Coux and Goldberg, 1998; Sears et al, 1998; Ciechanover et al, 2001).
Figure 1.
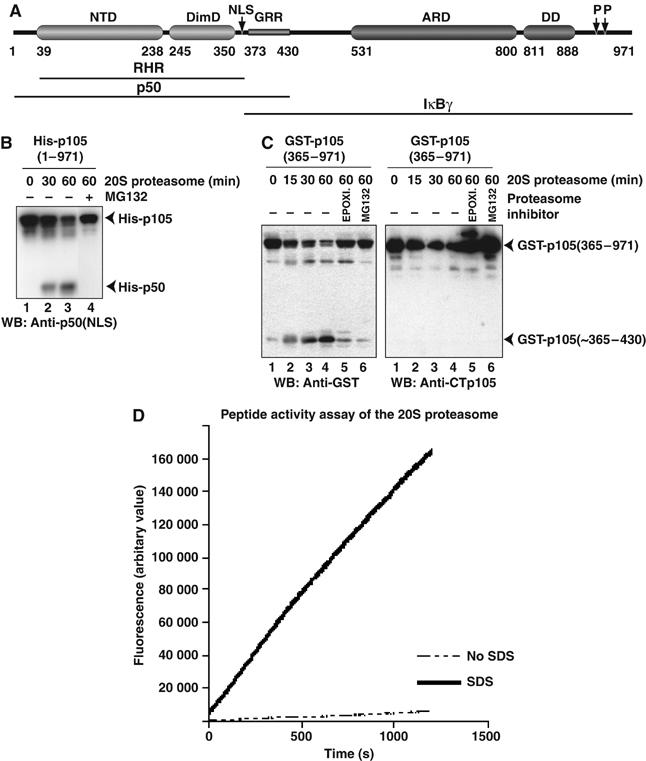
Processing of p105(1–971) and GST-p105(365–971) to p50 or p50-like product in vitro by the 20S proteasome. (A) Domain organization of mouse p105 showing NTD, DimD, ARD, and death domain (DD). Positions of NLS, GRR, and phosphorylation sites (P) are indicated. (B) Processing of His-tagged full-length p105(1–971) by 20S proteasome. Reaction products were separated by SDS–PAGE and visualized by Western blotting with p50(NLS) antibody. (C) Processing of GST-tagged p105(365–971) by the 20S proteasome. Reaction products were separated by SDS–PAGE and visualized by Western blotting with GST antibody (left panel) or CTp105 antibody (right panel). (D) Proteasome activity assays to test activity of 20S proteasome toward a fluorogenic peptide substrate. The activity with and without 0.03% SDS is indicated by solid and dotted lines, respectively.
It is generally believed that the physiologically functional proteasome is primarily the 26S proteasome and hence both constitutive processing and signal-dependent degradation of p105 are attributed to this protein complex. The 26S proteasome is composed of three large moieties: the catalytic 20S core particle and two 19S regulatory particles (Voges et al, 1999). The two gates of the barrel-like structure of the 20S proteasome are closed by interdigitating N-terminal residues of the α-type subunits restricting access for cellular proteins to the catalytic sites that reside in the inner cavity (Groll et al, 2000; Orlowski and Wilk, 2000). Two 19S regulatory particles bind to each end of the 20S core forming the 26S proteasome, which is involved in ubiquitin (Ub)-dependent and Ub-independent proteolysis. The 19S regulatory particle recognizes the target proteins, unfolds them in an energy-dependent manner, opens the entry gate of the core particle, and transfers the unfolded proteins to the sites of catalysis. A new study demonstrates a functional role for REGγ, a member of the 11S family of proteasome activators, in enhancing the Ub-independent degradation of transcriptional coactivator protein SRC-3 (Li et al, 2006). Biological activity of the 20S proteasome, independent of the regulatory subunits, did not receive significant attention until recently. An increasing number of cellular proteins including p21Cip1 as well as translation initiation factors eIF3α and eIF4G were reported to be degraded by the 20S proteasome both in vivo and in vitro (Verma and Deshaies, 2000; Touitou et al, 2001; Liu et al, 2003; Orlowski and Wilk, 2003; Baugh and Pilipenko, 2004).
In this study, we wanted to test if the 20S proteasome can process p105 into p50. In vitro results using pure proteins show that indeed the 20S proteasome can generate p50 from p105. We also show that the proteasome can function as an endoprotease, and that it preferentially degrades the entire C-terminal end of the molecule. The GRR serves as a stop signal for the proteasome and increases the stability of the processed product. We tested that p50 generation is independent of translation and does not require ubiquitination.
Results
20S proteasome processes p105 into p50
To test whether the 20S proteasome could generate p50 from p105 in vitro, we used recombinantly expressed and purified p105 and highly pure 20S proteasome for the processing reaction (Supplementary Figures 1 and 2). The reaction products were separated by SDS–PAGE and visualized by Western blotting with p50 (NLS) antibody. We observe that the amount of p105 decreased over time with concomitant increase of p50 (Figure 1B). These experiments were repeated reproducibly several times with different preparations of p105 and with proteasome from different sources: human cultured cells, bovine liver, and rabbit reticulocytes. We do not detect any products that would correspond to free C-terminus of p105, which is consistent with earlier in vivo studies, suggesting that the C-terminal region is completely degraded (data not shown). This would also imply that the isolated C-terminal region of p105 should be readily degraded by the 20S proteasome. To test this, we purified N-terminal glutathione-S-transferase (GST) fused to the C-terminus of p105, GST-p105(365–971), from Escherichia coli and tested its degradation by the proteasome. Reaction products were detected by Western blotting with GST antibody. We observe that the 20S proteasome generates a free GST-related product (Figure 1C, left panel). The same reaction products were probed with antibody against the extreme C-terminal peptide of p105. We do not detect any products corresponding to free C-terminus (Figure 1C, right panel).
To eliminate the possibility that the 20S proteasome, used in our assays, had been artificially activated during purification or freezing, we did a fluorogenic peptide activity assay. We compared proteasomal activity in the absence and presence of 0.03% SDS. If the proteasome was artificially activated, one would expect peptide degradation even in the absence of SDS with no significant enhancement in activity with SDS. Our results, however, show that the 20S proteasome used in our p105 processing assay is indeed latent (Figure 1D).
Furthermore, to confirm that the 20S proteasome did not contain other contaminating proteases, a mass spectrometric analysis was carried out. Trypsin digestion combined with LC/MS showed that the proteasome sample contained only 14 proteins, corresponding to the seven α- and seven β-subunits of mammalian 20S proteasome (Supplementary Figure 2). Thus, the activity observed in our assay must come from the 20S proteasome only. Together, these results suggest that p105 is a natural substrate of latent 20S proteasome and that p50 can be generated from the full-length precursor.
p50 generation is independent of translation
Generation of p50 from pure full-length p105 by the 20S proteasome prompted us to revisit the current cotranslational processing model, which excludes a precursor–product relationship between p105 and p50. Earlier studies by Fan and Maniatis (1991) had shown that p50 is generated from p105. Hence it appeared that the results of pulse–chase experiments had been interpreted differently. We therefore performed a similar pulse–chase radiolabeling experiment. HEK293T cells were transfected with a vector expressing full-length p105 as an N-terminal Flag fusion. Consistent with Fan and Maniatis's observation, we do see generation of p50 from the full-length p105 precursor (Figure 2A). However, even after a prolonged period of chase, only a fraction of p105 undergoes processing, suggesting that a significant pool of p105 is resistant to processing. Because unprocessed p105 serves specific function in vivo, there must be a mechanism to partially protect p105 from processing.
Figure 2.
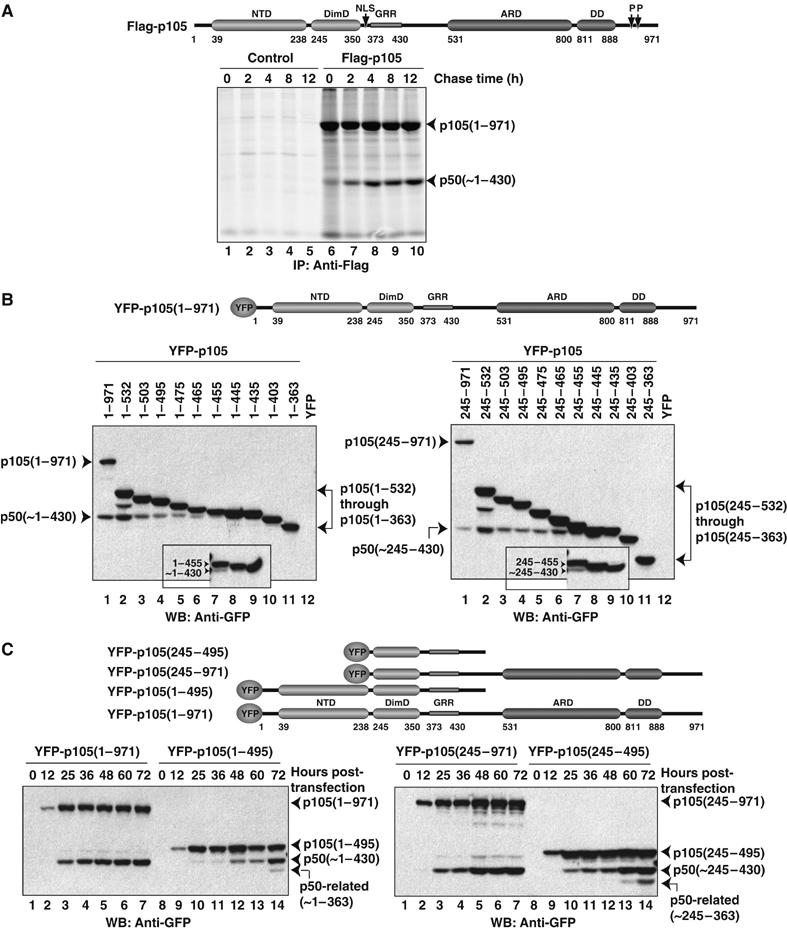
p50 generation in vivo is independent of translation and requires ankyrin repeat containing precursors for precise processing. (A) HEK293T cells transfected (right panel) or untransfected (left panel) with Flag-tagged full-length p105 were pulse-radiolabeled with 35S-Met for 30 min and chased for the indicated time. Cell lysates were immunoprecipitated with Flag antibody and separated by SDS–PAGE and visualized by fluorography. The schematic diagram for p105 is given as a residue numbering guideline. (B) HEK293T cells transfected with various YFP-tagged p105 truncations or with the empty vector. The precursors and processed products were separated on 10% (15% in insets) SDS–PAGE gel and visualized by Western blotting with GFP antibody. The schematic diagram for p105 is given as a residue numbering guideline. (C) HEK293T cells transfected with various YFP-tagged p105 constructs, and analyzed at indicated times after transfection. Levels of precursor and product proteins were assessed by Western blotting with GFP antibody. The schematic representation of constructs used is given.
We also generated a series of C-terminal deletion p105 mutants and tested if p50 is generated from these precursors by transfecting HEK293T cells with vectors expressing the truncated proteins as N-terminal yellow fluorescence protein (YFP) fusions (Figure 2B). Comparing the migration distances of the processed YFP-p50 and mutant YFP-p105(1–430) protein on SDS–PAGE gel, we concluded that the processing site appears to be at or near residue 430 (Figure 2B). Hereafter, we shall refer to 430 as the site of processing. Although we failed to resolve the unprocessed and processed bands for YFP-p105(1–445) fusion protein, the presence of the processed product from the YFP-p105(1–455) construct is clear. All proteins longer than residue 455 also undergo processing. Hence our experiments reveal that p50 can be generated from a precursor as short as ∼25 residues extended beyond the site of processing. It has been known for over 40 years, and reconfirmed recently with the elucidation of the 50S ribosome subunit crystal structure, that the peptide channel in the ribosome covers 35–50 residues of the growing polypeptide chain. Thus, such a short extension of 25 residues beyond the processing site is too small for the proteasome to make direct contact and catalyze the processing reaction while the precursor is still bound to the ribosome, which refutes the very essence of the cotranslational model. Indeed, our results are consistent with earlier observations made by Blank et al (1991) where they showed that a truncated p105 encompassing only 435 residues underwent processing. To test if the NTD has any effect in processing, we made these C-terminal deletion constructs with or without the NTD (1–244). Removal of the NTD does not affect the processing of p105, and the p50-like product, p50(∼245–430), is generated from all N-terminally truncated p105 mutants amenable for processing in vivo (Figure 2B, right panel).
Our results also show that the efficiency of processing varies between shorter and longer precursors. While processing of C-terminally truncated p105 proteins is clearly evident, the full-length p105 appeared to be the most efficient substrate for generation of the processed p50 product. We do not know the reason behind this differential processing. We and others have shown that although full-length p105 is cytoplasmic, the shorter C-terminally truncated (shorter than ∼800 residues) constructs are predominantly nuclear (Blank et al, 1991; Moorthy and Ghosh, 2003). It is possible that the proteasome that processes p105 is less abundant in the nucleus and it is only during their short stay in the cytoplasm that they get processed. Interestingly, we observe an additional cryptic, smaller p50-related product that is generated only from truncated p105 precursors, which lack complete ARD and sequences beyond ARD (Figures 2C and 4E). Based on the similar molecular weights between this smaller cleavage product and an expressed fragment ending at residue 363 and the recognition of the smaller product by the p50(NLS) antibody, the aberrant cleavage site appears to be located near but C-terminal to the NLS (data not shown). We do not observe such a cryptic product when the C-terminus of the precursor ends beyond the ARD. Thus, it appears that shorter precursors are processed aberrantly. Overall, our experiments show that even a short precursor, containing only 25 residues beyond the processing site, is sufficient for p50 generation. However, although p50 is the major product of processing from these shorter precursors, another minor aberrant product is also generated. Both these observations contradict the cotranslational model.
Figure 4.
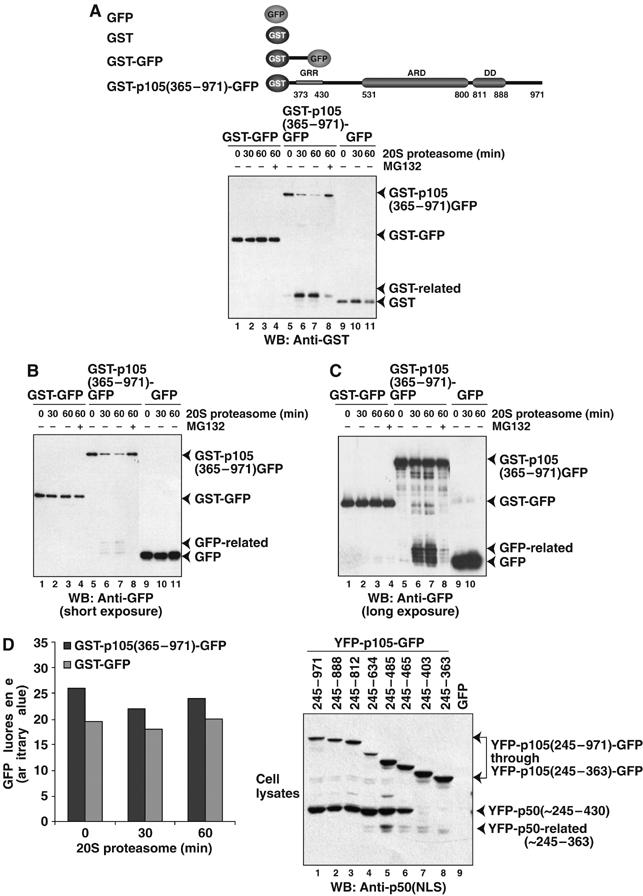
p105 processing is an endoproteolytic event. (A) 20S proteasome endoproteolytically cleaves GST-p105(365–971)-GFP in vitro as detected by generation of GST-related products by Western blotting with GST antibody. (B, C) 20S proteasome endoproteolytically cleaves GST-p105(365–971)-GFP in vitro as detected by generation of GFP-related products by Western blotting with GFP antibody. GST-GFP, GST, and GFP remain stable over the course of the reaction. The schematic diagram of proteins used is given. (D) GFP fluorescence (excitation 395 nm, emission 508 nm) of GST-p105(365–971)-GFP and GST-GFP was monitored during the 20S proteasomal reaction. (E) HEK293T cells transfected with various YFP-p105-GFP double fusion proteins as indicated schematically on top. Lane 9 represents the empty GFP vector control transfection. The precursors and processed products were visualized by Western blotting with p50(NLS) antibody.
To test if the minor processed product is also generated from the shorter precursors by the 20S proteasome in vitro, we expressed and purified p105(1–495) and p105(245–495) from E. coli. Indeed, a smaller p50-related product is generated from the shorter precursors by the 20S proteasome in vitro (Supplementary Figure 3). Together these in vitro and in vivo experiments demonstrate that accurate processing requires full-length p105 precursor or at least a precursor that extends beyond ARD. Similar observations both in vivo and in vitro reiterate two facts: firstly, the smaller cryptic product may not be a mere artifact of random proteolysis and, secondly, the 20S proteasome may indeed be the cellular proteasome involved in the constitutive processing of p105.
p105 processing is Ub-independent
Our in vitro processing reaction did not require ubiquitination. To test whether cellular constitutive processing of p105 requires ubiquitination, we used mouse ts20 cell line. These cells have a temperature-sensitive E1 Ub-activating enzyme and, at the restrictive temperature, protein degradation by the classical Ub–26S proteasome pathway is drastically reduced (Ciechanover et al, 1984; Finley et al, 1984; Chowdary et al, 1994; Salvat et al, 2000). At the permissive temperature, we observe the processing of p105 to p50. Interestingly, processing of p105 in the ts20 cell line is unaffected at the restrictive temperature (Figure 3A, top panel). As a control, the level of the transcription factor p53 was monitored at the two temperatures in the ts20 cells. At the permissive temperature, the unstable p53 is completely absent, but at the restrictive temperature, p53 is present at an appreciable level (Chowdary et al, 1994; Salvat et al, 2000) (Figure 3A, bottom panel). These results imply that ubiquitination may not be required for constitutive p105 processing. To further monitor p50 generation from p105 in these E1-deficient cells, we attempted to determine the half-life of p105. The cells were grown for different lengths of time followed by treatment with the protein synthesis inhibitor cycloheximide (CHX). Toxicity seemed to be enhanced when ts20 cells at the restrictive temperature were treated with CHX for even the relatively short time period of 4 h (Supplementary Figure 4). Because the level of p50 was already very high at the point of CHX treatment, processing of p105, if any, could not be monitored above the background level. Our attempts to carry out pulse–chase experiment to monitor endogenous processing were not successful owing to the absence of a good p50 antibody for immunoprecipitation.
Figure 3.
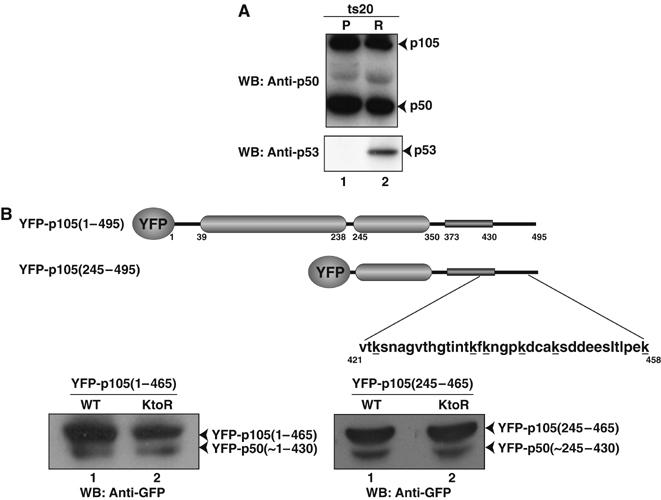
p50 generation in vivo is Ub-independent. (A) Temperature-sensitive E1 mutant ts20 cells maintained at the permissive (P) or restrictive (R) temperatures. Levels of p50 and p105 in lysates from cells grown in P and R temperatures were assessed by Western blotting with p50(NLS) antibody (top panel). Ub-dependent cellular p53 accumulated at the R temperature in ts20 cells (bottom panel). (B) HEK293T cells were transfected with expression plasmids encoding wild-type p105 (WT) and lysine to arginine mutant (KtoR) of p105 fused to YFP. The minimal processing labile p105, 1–465 and 245–465, were used. Proteins were separated on 15% (left panel) and 10% (right panel) SDS–PAGE gel and visualized by Western blotting with GFP antibody. The schematic diagram for the p105 constructs with the positions of the mutated lysines is given.
As an additional proof of Ub-independent processing of p105, we took the two processing compatible precursors, p105(1–465) and p105(245–465), and mutated lysines to arginines. It is known that in Ub-mediated proteasomal degradation, lysines located close to the flexible portions of a protein substrate are the ones that undergo ubiquitination (Prakash et al, 2004). Therefore, we mutated all ubiquitination susceptible lysines around the processing region, K423/K436/K438/K442/K446/K458, to arginines. The wild-type and mutant constructs were transfected into HEK293T cells as YFP fusions, and processing of wild type and mutants was compared. We observe that replacing the lysines did not affect the processing of p105 (Figure 3B). Not only did both the wild type and the mutants show processing, but the level of processing was comparable. Hence, p105 processing appears to be a Ub-independent event.
The 20S proteasome functions as an endoprotease
Next we addressed if p50 is generated as a result of internal cleavage of p105 polypeptide by the 20S proteasome. We created a fusion protein in which the p105 C-terminal fragment is flanked by the stable GST and green fluorescent protein (GFP) domains at the N- and C-termini, respectively, GST-p105(365–971)-GFP. As a control, we also created a GST and GFP fusion protein containing only 20 random intervening residues derived form the cloning site, GST-GFP. These proteins were expressed in E. coli and purified to near homogeneity and subjected to 20S proteasome digestion. We observed that 20S proteasome endoproteolytically cleaves GST-p105(365–971)-GFP recombinant protein and generates GST- and GFP-related products as detected by Western blotting with GST and GFP antibodies (Figure 4A–C). Although the generation of GFP-related products was not very prominent with short exposures (Figure 4B, left panel, lanes 6 and 7), we repeated the experiment, and longer exposures showed clear generation of smaller GFP-related products (Figure 4C, right panel, lanes 6 and 7). Additionally, the GFP fluorescence did not change significantly during the course of this degradation reaction (Figure 4D). This implies that, perhaps, a spectrum of C-terminal p105 polypeptides of different lengths, each retaining intact GFP, is generated during the course of this degradation reaction. The control fusion protein, GST-GFP, exhibits no cleavage and no change in GFP fluorescence in the duration of the proteasomal reaction (Figures 4A–D). We further tested if the isolated GST and GFP proteins are substrates of the 20S proteasome. As expected, no substantial degradation of free GST and GFP substrates was observed (Figures 4A–C).
To test if processing of p105 is initiated through an endoproteolytic cleavage in vivo, HEK293T cells were transfected with expression vectors encoding different C-terminally truncated p105 proteins with both termini fused to stably folded domains, YFP at the N-terminus and GFP at the C-terminus. We observe the generation of YFP-p50(∼245–430) in all cases where the C-terminus of the linker is at residue 465 or beyond (Figure 4E). However, as shown earlier, the substrate must retain at least 812 residues to avoid the generation of the cryptic processed product. The fact that p105 with ‘blocked' termini, protected by the stable domains, can undergo processing suggests that processing initiates through an endoproteolytic cleavage event.
The GRR is the proteasomal stop signal
To further test the role of the GRR and other p105 C-terminal sequence elements in processing, we have generated two additional GST fusion proteins: one lacking the C-terminal 171 residues, GST-p105(365–800), and the other lacking GRR, GST-p105(435–971). Like GST-p105(365–971), 20S proteasome efficiently utilizes GST-p105(365–800) in vitro and generates an identical stable product that is larger in size than GST (Figure 5A, left panel). This product is recognized by the GST antibody and its size appears to be equivalent to the recombinant, purified GST-GRR, which suggests that the product contains the GRR. Considering this and our in vivo results (Figure 4E), we conclude that the last 171 residues are not essential for p105 processing.
Figure 5.
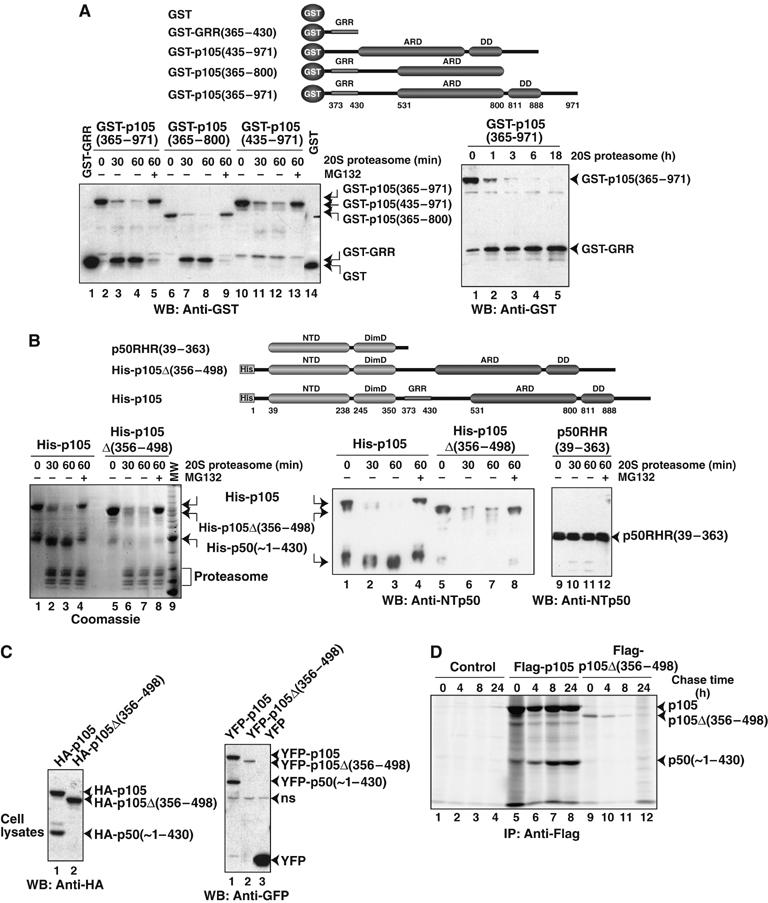
GRR protects upstream domains from degradation by the 20S proteasome. (A) The left panel shows that in vitro 20S proteasome generates stable products from GST-p105(365–971) and GST-p105(365–800), but not from GST-p105(435–971), which lacks the GRR. The size of the product generated is equivalent to the size of recombinant GST-GRR. The right panel shows that the GST-GRR generated from GST-p105(365–971) is stable over a prolonged period of incubation. Precursors and products were visualized by Western blotting with GST antibodies. The schematic diagram of proteins used is given. (B) Stable p50-like product, His-p50(∼1–430), is generated from the full-length p105, but not from mutant p105, which lacks GRR, p105Δ(356–498) (left and middle panels). RHR, p105(39–363) was stable over the same period of incubation with 20S proteasome (right panel). Proteins were visualized by both Coomassie staining and Western blotting with p50 antibody. The schematic diagram of proteins used in the in vitro 20S proteasome assay is given on top. (C) HEK293T cells were transfected with full-length p105 or p105 deletion mutant p105Δ(356–498) as HA (left panel) or YFP (right panel) fusion proteins. Cell extracts were separated by SDS–PAGE followed by Western blotting with HA and GFP antibodies. ns refers to nonspecific band. (D) HEK293T cells transfected (right and middle panels) or untransfected (left panel) with Flag-tagged full-length p105 and p105Δ(356–498) were pulse-radiolabeled with 35S-Met for 30 min and chased for the indicated time. Cell lysates were immunoprecipitated with Flag antibody and separated by SDS–PAGE and visualized by fluorography.
When GST-p105(435–971) was subjected to 20S proteasome, we observed similar reduction of the full-length protein with increasing reaction time, but no significant amount of GST accumulated during the course of the reaction (Figure 5A, left panel). It is possible that in the absence of the GRR, the entire GST-p105(435–971) is degraded by the 20S proteasome. An alternative explanation would be that in the absence of the GRR sequence, the proteasome makes multiple cleavages within GST-p105(435–971), and the amount of each of these products is too low to be detected by Western blotting. Overall, these results indicate that the GRR is not a good substrate for the proteasome and serves as an effective stop signal, protecting the NTDs from degradation. GST-GRR generated by the 20S proteasome from GST-p105(365–971) was resistant to further degradation, confirming that the GRR is a poor substrate for proteasomal degradation (Figure 5A, right panel).
To further test the role of the GRR in proteasomal degradation, we used an internal deletion mutant of p105 where the entire processing region containing GRR was removed, p105Δ(356–498) (Fu et al, 2004). We cloned p105Δ(356–498) into a bacterial expression vector and expressed it in E. coli. Unfortunately, we failed to purify this deletion mutant to homogeneity. A contaminant approximately 50 kDa remained associated with it. Based on the immunoreactivity of this 50 kDa protein, we concluded that this molecule is p105-related. Its approximate molecular weight suggests that this N-terminal fragment originated from the p105Δ(356–498) deletion mutant, probably due to nonspecific proteolysis in bacteria, and retained a large segment of roughly 50–80 amino acids starting from p105 residue 499, which was fused to residue 356. Both p105Δ(356–498) and the related product were completely degraded by the 20S proteasome in vitro (Figure 5B, left and middle panels). We also observe that isolated p50RHR (residues 39–363) is not degradable by the 20S proteasome (Figure 5B, right panel). This suggests that the presence of p105 C-terminal sequences directly linked to the RHR, in the absence of the GRR, leads to complete degradation of the entire p105Δ(356–498) protein. In contrast, stable p50 is generated from full-length p105. The ability of the 20S proteasome to degrade a relatively stably folded N-terminal p50 RHR in the case of the deletion mutant led us to believe that the proteasome also completely degraded the N-terminal GST domain in GST-p105(435–971) (Figure 5A). Altogether, our results show that the GRR plays an important role in the stabilization of processed p50 by serving as a stop signal for the proteasome.
To test if removal of the GRR from p105 results in its complete degradation in vivo, we transiently expressed N-terminal hemagglutinin (HA)- or YFP-tagged internal deletion p105 mutants, HA-p105Δ(356–498) and YFP-p105Δ(356–498), as well as the corresponding wild-type p105 in HEK293T cells. We monitored processing by Western blotting (Figure 5C). Although we observe the precursors, no processed products were detected from either of the deletion mutants.
To further confirm that the p105Δ(356–498) mutant protein indeed undergoes complete degradation, we performed radiolabeled pulse–chase experiments in HEK293T cells transfected with full-length and internally deleted p105 proteins. As expected, the full-length p105 generated the processed product but no corresponding product was observed in p105Δ(356–498) (Figure 5D). Moreover, p105Δ(356–498) underwent gradual degradation over the chase period. These results, in conjunction with our in vitro observations, show that p105Δ(356–498) mutant protein is completely degraded in vivo.
Overall, our experiments show that the cleavage site is located downstream of the GRR and the presence of GRR blocks the proteasome from degrading the N-terminus but allows for degradation of the C-terminus. Protection of the region N-terminus to the GRR is abolished when the GRR is deleted.
Discussion
20S proteasome generates p50 from p105 in a Ub-independent manner
An increasing number of proteins are implicated to be degraded by the 20S proteasome. The most well-studied, and still somewhat controversial, is the degradation of p21cip1, a natively unfolded protein in its free state that can undergo both Ub-dependent and Ub-independent degradation in vivo. Whereas its inducible degradation requires ubiquitination and the 26S proteasome, its basal degradation is mediated by the core 20S proteasome (Orlowski, 2001; Touitou et al, 2001; Liu et al, 2003). The translation initiation factors eIF3α and eIF4G, p53 and ornithine decarboxylase (ODC) have also been recently shown to be cleaved by the 20S proteasome both in vivo and in vitro (Baugh and Pilipenko, 2004; Asher et al, 2005a, 2005b). We show here that pure 20S proteasome can constitutively process p105 in vitro where the C-terminal segment is completely degraded, leaving behind p50 containing the RHR and GRR. In separate experiments with the C-terminal of p105 fused to GST, we show that the same C-terminal segment also degrades leaving behind GST-GRR. We used 25-fold molar excess of substrate relative to 20S proteasome in our reactions. Therefore, each proteasome molecule does carry out multiple rounds of catalysis like any efficient enzyme. We further produced evidence indicating that constitutive p105 processing may not require ubiquitination in vivo, as a nonfunctional E1 enzyme or the absence of ubiquitination essential lysines does not affect the level of processing. These results suggest that the 20S proteasome might be responsible for p105 processing in vivo. Although E1-defective ts mutant cell lines and arginine mutants are traditionally used to show possible direct involvement of the 20S proteasome in protein degradation, one cannot unambiguously conclude from these experiments that the 20S proteasome is the physiologically functional proteasome. For example, the E1 enzyme activity is not completely eliminated in ts20 mutant cells. Although a more robust proof of the in vivo function of the 20S proteasome is yet to come, it is perceivable that the 20S proteasome indeed functions in vivo independent of its regulatory subunits. A large cellular fraction of the 20S proteasome is known to be free of regulatory subunits (Orlowski and Wilk, 2003). There are other circumstantial evidences that support the notion that the 20S, and not the 26S proteasome, carries out p105 processing in vivo. For example, p105 can associate with both the α- and β-subunits of the 20S proteasome; this binding is further enhanced in the presence of the viral protein Tax, resulting in the enhanced processing of p105 (Rousset et al, 1996).
Cotranslational mode of p105 processing
Lin et al (1998, 2000) proposed a cotranslational mechanism of p50 generation, which suggested that p50 was not generated from a fully translated p105, but rather that the ribosome occasionally paused on or near mRNA sequences coding for residue 530, followed by the recruitment of the proteasome, leading to the generation of p50. Several arguments are inconsistent with the cotranslational only model. Firstly, nucleotide sequences around the proposed pausing site are unremarkable, in the sense that there are no rare codons present around this region of mRNA, which would warrant ribosome pausing during translation. Secondly, we show that although the C-terminally truncated p105 can generate p50, there is another smaller p50-related product that is generated from the shorter precursors. We show that the precursors must extend beyond the ARD to avoid the generation of the smaller p50-related product. From the size of this product, it appears that this abnormal cleavage is occurring near residue 365. We have previously shown that the NLS of p50 is masked by the C-terminal ARD of p105 (Moorthy and Ghosh, 2003). Therefore, it is possible that the intramolecular interaction between the ARD and p50 prevents abnormal cleavage and generation of the cryptic p50-related product. This again strengthens the notion that at least the synthesis of the ARD is necessary for the faithful processing of p105 into p50. The strongest argument favoring the cotranslational model came from lack of stringent precursor–product relationship between p105 and p50 in pulse–chase experiments. We show here clearly that p50 is generated from p105 during the chase period. Our conclusion is consistent with earlier reports (Fan and Maniatis, 1991). However, as in these previous cases, we also observe that a significant fraction of p105 remains unprocessed even after a prolonged period of chase. Processing appears to be efficient in the early times of chase. It is known that unprocessed p105 carries out important cellular functions by forming stable inhibitory complexes with a large number of proteins including the members of NF-κB family, Tpl2, c-FLIP, LYL1, ZUD5 (Ferrier et al, 1999; Beinke et al, 2003; Li et al, 2003; Zhang et al, 2004). We propose that the competition between the proteasome machinery and other p105-interacting proteins for p105 is a critical determinant of the processing event. p105 that is bound by Tpl2, FLIP, LYL1, ZUD5, etc. cannot undergo processing and the fate of p105 is determined perhaps within a short period after its synthesis. Consistent with this model, Harhaj et al (1996) showed that newly synthesized p105 is processed more rapidly compared to its accumulated form that is associated with NF-κB subunits.
Post-translational model of p105 processing
It has long been thought that the 20S proteasome can degrade unfolded proteins but spares stable domains from degradation. We observe that the known folded domains, the RHR of p50, GST, and GFP, are relatively resistant to degradation by the 20S proteasome in vitro. In p105, the positive degradation signal for the proteasome is present downstream of the GRR sequence. Our experiments clearly demonstrate that endoproteolysis of p105 at the region between residues 430 and 465 leads to p50 generation. We show that in the absence of the GRR, no processing occurs, instead p105 is completely degraded. Thus, the GRR seems to serve as an effective stop signal. How exactly the GRR blocks degradation of p50 is still not known. As shown in Figure 5A, by perhaps inherently being a poor substrate for the proteasome, the GRR may protect the rest of p50 from degradation. This protection mechanism might be similar to the one utilized by other high-stability proteins, such as those containing long glutamine stretches. It is known that poly-Glu peptides are not degraded by the proteasome (Venkatraman et al, 2004).
Based on our results, we can propose a plausible model for p105 processing (Figure 6). p105 can open the, otherwise closed, gate of the 20S proteasome and the segment following the GRR can access the central catalytic core. The exact mechanism of this gate opening is not known. One possible gate opening mechanism is that the highly flexible, relatively simple sequence present at the junction of GRR/processing region may not require complete gate opening for entry into the catalytic center. Once cleavage is initiated within this region, the proteasome can further degrade the C-terminus easily by unfolding the ARD. Free IκBα has been shown to be unstable and not completely well-folded (Croy et al, 2004). It is possible that the C-terminal IκB-like region of p105 may also share similar traits and can be unfolded relatively easily by the proteasome. We propose that a stringent sequence requirement of the GRR/processing region junction is not necessary. The processing region is long, ranging from residue 430 to 530, and any segment within this region can serve as an initiator site when linked directly to the GRR. Therefore, internal deletions within this region do not seem to negatively affect p105 processing. In addition, a cryptic site is generated when most of the ARD is deleted. This suggests that the C-terminal IκB-like region interacts with the N-terminal p50 RHR, masking the NLS. This presents the correct processing sites for proteasomal cleavage. In shorter precursors, without the ARD, additional cryptic sites are exposed, leading to the formation of other smaller products.
Figure 6.
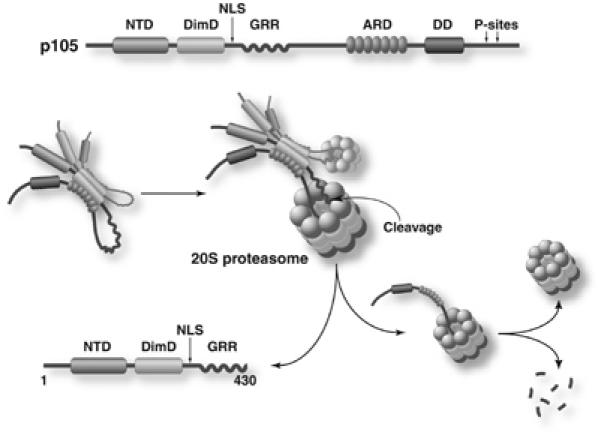
Model for p105 processing. In vitro studies have shown that p105 exists as a dimer. Folding back of the C-terminal of p105 masks the NLS and presents the correct processing region for cleavage by the 20S proteasome. Upon cleavage, the C-terminal is preferentially degraded, while the GRR protects the N-terminal from further degradation, thereby generating p50.
Materials and methods
E. coli expression and protein purification
The mouse p105(1–971), p105Δ(356–498), p105(1–495), and p105(245–495) were subcloned into the pET28 vector (Novagen) with an N-terminal His tag. The p105Δ(356–498) DNA template was a gift from Dr Li Lin (Johns Hopkins University). The p105(365–971), p105(365–800), and p105(435–971) constructs were subcloned into the pGEX-4T2 vector (Amersham Biosciences) with an N-terminal GST tag. The GST-GFP construct was created by subcloning GFP into the pGEX-4T2 vector. p105(365–971) was then subcloned into the GST-GFP vector to obtain GST- p105(365–971)-GFP.
His-tagged p105(1–971) was purified by a first step Nickel affinity chromatography (Probond Resin, Invitrogen) under native conditions, followed by size-exclusion chromatography on a Superdex 200 column in 20 mM Tris pH 7.5, 0.25 M NaCl, and 1 mM DTT. In the second method, the Nickel column eluate was denatured in 7 M urea and run on a Superdex 200 gel filtration column (Amersham Biosciences) under denaturing conditions. p105 and contaminant p50-like product eluted in separate fractions. The p105 fractions were pooled and refolded by dialysis in buffer containing 20 mM Tris pH 7.5, 0.5 M NaCl, 1 mM DTT, 10% glycerol, and 0.1% Triton X-100. The refolded protein was then loaded on a Superdex 200 gel filtration column under native conditions. The other His-tagged p105 proteins were purified by Nickel affinity chromatography followed by gel filtration chromatography. The GST-tagged proteins were purified by a two-step process: Glutathione Sepharose 4B column (Amersham Biosciences) followed by gel filtration chromatography. Purification of p50RHR(39–363) was described earlier (Ghosh et al, 1995). It should be noted that purified p50RHR migrates as a doublet on SDS–PAGE gel.
Antibodies
The p50NLS(sc-114), p50(sc-7178), GST(sc-138), p53(sc-99), and all secondary HRP-conjugated antibodies were purchased from Santa Cruz. GFP(A-6455) and HA(HA 11) antibodies were from Invitrogen and Covance, respectively. CTp105 antibody was a gift from Dr Nancy Rice.
20S proteasome degradation assay
Mammalian 20S proteasome was purchased from Boston Biochem Inc. (Cambridge, MA) and was also provided by Dr Rechsteiner and Dr Pratt, University of Utah. For the degradation reactions, the proteins and the proteasome were mixed in a molar ratio of 25:1 in a buffer containing 20 mM Tris pH 7.0, 250 mM NaCl, 10 mM MgCl2, and 1 mM DTT at 37°C. Samples were removed at various time points and the reaction was stopped by adding SDS–PAGE loading dye and boiling. Protein bands were separated by SDS–PAGE and visualized by Western blotting. Proteasome inhibitors MG132 (1 mM) and epoxomicin (10 μM) (Calbiochem) were used to ensure the specificity of the reactions. Similar to the recombinant p50RHR, p50, generated in vitro by the proteasome, migrated as a doublet on SDS–PAGE gel.
Proteasome activity assay
The Proteasome activity assay kit was from Calbiochem. The cuvette assay was followed, with some modifications. The total volume of reaction was reduced to 500 μl, and the amount of substrate was reduced two-fold. The fluorescent signal (excitation 380 nm, emission 460 nm) was monitored for 20 min.
Protein identification by mass spectrometry
Protein analysis was performed at the Scripps Center for Mass Spectrometry. In solution trypsin digestion of human 20S proteasome combined with LC-MS/MS was performed. Protein identification was made by comparison of a peptide map to NCBI's nonredundant database.
Mammalian cell culture and transient protein expression
The mouse ts20 cell lines were maintained in DMEM (CellGro) supplemented with 10% fetal bovine serum (Omega) at 35°C (permissive temperature). The cells were grown at the permissive temperature for 24 h and then moved to the restrictive temperature (39°C). Lysates were prepared after 24 h.
HEK293T cells were maintained in DMEM supplemented with 10% fetal bovine serum. Series of mouse p105 deletion mutants were generated by PCR and cloned into pEYFP-C1 vector (Clontech) to express N-terminal YFP fusion proteins in mammalian cells. To generate YFP-p105-GFP double fusion proteins, various YFP-p105 fusion sequences were amplified by PCR and subcloned into pEGFP-N1 vector (Clontech). p105Δ(356–498) sequence was cloned into two vectors, pEYFP-C1 to express YFP-tagged protein, and pRCβactin to express HA-tagged mutant. HEK293T cells were transfected using Lipofectamin Plus reagent (Invitrogen). Cell lysates were prepared 24–36 h after transfection or as indicated.
Pulse–chase studies
HEK293T cells were transfected as described above. Growth media were removed 24 h after transfection and cells were washed twice with labeling media (RPMI 1640 free of methionine and cystine supplemented with 10% dialyzed FCS plus glutamine), followed by pulse-radiolabeling with 0.1 mCi/ml 35S-Met for 30 min. Pulse media were replaced with chase media (labeling media plus 500 μg/ml cystine-HCl, 100 μg/ml methionine) after 30 min. Pulsed-labeled cells were lysed in lysis buffer (100 mM NaCl, 20 mM Tris pH 8.0, 1% Triton X-100) in the presence of protease and phosphatase inhibitors. A 1 mg portion of protein extract was used for each immunoprecipitation using Flag antibody. The immunoprecipitated proteins were separated by SDS–PAGE and analyzed by fluorography.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Dr S Joseph, Dr A Hoffmann, and members of the Ghosh laboratory for critical discussion of the manuscript; Dr M Rechsteiner and Dr G Pratt for providing 20S proteasome and helpful discussion; Dr GN DeMartino, Dr S Wilk, Dr L Deng, and Dr R Luo for providing 20S proteasome; Dr Ozer and Dr JM Roberts for the ts20 cell line; B Andrews and Dr P Jennings for purified GFP; Dr N Rice and M Ernst for CTp105 and p50NLS(1157) antibodies; Dr A Hoffman for several antibodies and reagents; J Ngo for helping with Figure 6. AKM is supported by a postdoctoral fellowship from Universitywide AIDS Research Fellowship. OVS is supported by GAANN predoctoral fellowship and Molecular Biophysics Training Grant. We would also like to acknowledge NIH/AI for funding to GG (CA71718 AI064326 and GM071862).
Competing interests statement: The authors declare that they have no competing financial interests.
References
- Asher G, Bercovich Z, Tsvetkov P, Shaul Y, Kahana C (2005a) 20S proteasomal degradation of ornithine decarboxylase is regulated by NQO1. Mol Cell 17: 645–655 [DOI] [PubMed] [Google Scholar]
- Asher G, Tsvetkov P, Kahana C, Shaul Y (2005b) A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev 19: 316–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh JM, Pilipenko EV (2004) 20S proteasome differentially alters translation of different mRNAs via the cleavage of eIF4F and eIF3. Mol Cell 16: 575–586 [DOI] [PubMed] [Google Scholar]
- Beinke S, Deka J, Lang V, Belich MP, Walker PA, Howell S, Smerdon SJ, Gamblin SJ, Ley SC (2003) NF-kappaB1 p105 negatively regulates TPL-2 MEK kinase activity. Mol Cell Biol 23: 4739–4752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank V, Kourilsky P, Israel A (1991) Cytoplasmic retention, DNA binding and processing of the NF-kappa B p50 precursor are controlled by a small region in its C-terminus. EMBO J 10: 4159–4167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, von Kessler DP, Park W, Wang B, Ma Y, Beachy PA (1999) Nuclear trafficking of Cubitus interruptus in the transcriptional regulation of Hedgehog target gene expression. Cell 98: 305–316 [DOI] [PubMed] [Google Scholar]
- Chowdary DR, Dermody JJ, Jha KK, Ozer HL (1994) Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol Cell Biol 14: 1997–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A, Finley D, Varshavsky A (1984) Ubiquitin dependence of selective protein degradation demonstrated in the mammalian cell cycle mutant ts85. Cell 37: 57–66 [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Gonen H, Bercovich B, Cohen S, Fajerman I, Israel A, Mercurio F, Kahana C, Schwartz AL, Iwai K, Orian A (2001) Mechanisms of ubiquitin-mediated, limited processing of the NF-kappaB1 precursor protein p105. Biochimie 83: 341–349 [DOI] [PubMed] [Google Scholar]
- Coux O, Goldberg AL (1998) Enzymes catalyzing ubiquitination and proteolytic processing of the p105 precursor of nuclear factor kappaB1. J Biol Chem 273: 8820–8828 [DOI] [PubMed] [Google Scholar]
- Croy CH, Bergqvist S, Huxford T, Ghosh G, Komives EA (2004) Biophysical characterization of the free IkappaBalpha ankyrin repeat domain in solution. Protein Sci 13: 1767–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan CM, Maniatis T (1991) Generation of p50 subunit of NF-kappa B by processing of p105 through an ATP-dependent pathway. Nature 354: 395–398 [DOI] [PubMed] [Google Scholar]
- Ferrier R, Nougarede R, Doucet S, Kahn-Perles B, Imbert J, Mathieu-Mahul D (1999) Physical interaction of the bHLH LYL1 protein and NF-kappaB1 p105. Oncogene 18: 995–1005 [DOI] [PubMed] [Google Scholar]
- Finley D, Ciechanover A, Varshavsky A (1984) Thermolability of ubiquitin-activating enzyme from the mammalian cell cycle mutant ts85. Cell 37: 43–55 [DOI] [PubMed] [Google Scholar]
- Fu D, Kobayashi M, Lin L (2004) A p105-based inhibitor broadly represses NF-kappa B activities. J Biol Chem 279: 12819–12826 [DOI] [PubMed] [Google Scholar]
- Ghosh G, van Duyne G, Ghosh S, Sigler PB (1995) Structure of NF-kappa B p50 homodimer bound to a kappa B site. Nature 373: 303–310 [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB (1998) NF-kappaB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 16: 225–260 [DOI] [PubMed] [Google Scholar]
- Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D (2000) A gated channel into the proteasome core particle. Nat Struct Biol 7: 1062–1067 [DOI] [PubMed] [Google Scholar]
- Harhaj EW, Maggirwar SB, Sun SC (1996) Inhibition of p105 processing by NF-kappaB proteins in transiently transfected cells. Oncogene 12: 2385–2392 [PubMed] [Google Scholar]
- Hoppe T, Matuschewski K, Rape M, Schlenker S, Ulrich HD, Jentsch S (2000) Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell 102: 577–586 [DOI] [PubMed] [Google Scholar]
- Inoue J, Kerr LD, Kakizuka A, Verma IM (1992) I kappa B gamma, a 70 kd protein identical to the C-terminal half of p110 NF-kappa B: a new member of the I kappa B family. Cell 68: 1109–1120 [DOI] [PubMed] [Google Scholar]
- Lang V, Janzen J, Fischer GZ, Soneji Y, Beinke S, Salmeron A, Allen H, Hay RT, Ben-Neriah Y, Ley SC (2003) betaTrCP-mediated proteolysis of NF-kappaB1 p105 requires phosphorylation of p105 serines 927 and 932. Mol Cell Biol 23: 402–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lonard DM, Jung SY, Malovannaya A, Feng Q, Qin J, Tsai SY, Tsai MJ, O'Malley BW (2006) The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell 124: 381–392 [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang J, Chen D, Shu HB (2003) Casper/c-FLIP is physically and functionally associated with NF-kappaB1 p105. Biochem Biophys Res Commun 309: 980–985 [DOI] [PubMed] [Google Scholar]
- Lin L, DeMartino GN, Greene WC (1998) Cotranslational biogenesis of NF-kappaB p50 by the 26S proteasome. Cell 92: 819–828 [DOI] [PubMed] [Google Scholar]
- Lin L, DeMartino GN, Greene WC (2000) Cotranslational dimerization of the Rel homology domain of NF-kappaB1 generates p50–p105 heterodimers and is required for effective p50 production. EMBO J 19: 4712–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Ghosh S (1996) A glycine-rich region in NF-kappaB p105 functions as a processing signal for the generation of the p50 subunit. Mol Cell Biol 16: 2248–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou HC, Nolan GP, Ghosh S, Fujita T, Baltimore D (1992) The NF-kappa B p50 precursor, p105, contains an internal I kappa B-like inhibitor that preferentially inhibits p50. EMBO J 11: 3003–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CW, Corboy MJ, DeMartino GN, Thomas PJ (2003) Endoproteolytic activity of the proteasome. Science 299: 408–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorthy AK, Ghosh G (2003) p105.Ikappa Bgamma and prototypical Ikappa Bs use a similar mechanism to bind but a different mechanism to regulate the subcellular localization of NF-kappa B. J Biol Chem 278: 556–566 [DOI] [PubMed] [Google Scholar]
- Orian A, Schwartz AL, Israel A, Whiteside S, Kahana C, Ciechanover A (1999) Structural motifs involved in ubiquitin-mediated processing of the NF-kappaB precursor p105: roles of the glycine-rich region and a downstream ubiquitination domain. Mol Cell Biol 19: 3664–3673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski M (2001) Selective activation of the 20 S proteasome (multicatalytic proteinase complex) by histone h3. Biochemistry 40: 15318–15326 [DOI] [PubMed] [Google Scholar]
- Orlowski M, Wilk S (2000) Catalytic activities of the 20 S proteasome, a multicatalytic proteinase complex. Arch Biochem Biophys 383: 1–16 [DOI] [PubMed] [Google Scholar]
- Orlowski M, Wilk S (2003) Ubiquitin-independent proteolytic functions of the proteasome. Arch Biochem Biophys 415: 1–5 [DOI] [PubMed] [Google Scholar]
- Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A (2004) An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol 11: 830–837 [DOI] [PubMed] [Google Scholar]
- Rousset R, Desbois C, Bantignies F, Jalinot P (1996) Effects on NF-kappa B1/p105 processing of the interaction between the HTLV-1 transactivator Tax and the proteasome. Nature 381: 328–331 [DOI] [PubMed] [Google Scholar]
- Salvat C, Acquaviva C, Scheffner M, Robbins I, Piechaczyk M, Jariel-Encontre I (2000) Molecular characterization of the thermosensitive E1 ubiquitin-activating enzyme cell mutant A31N-ts20. Requirements upon different levels of E1 for the ubiquitination/degradation of the various protein substrates in vivo. Eur J Biochem 267: 3712–3722 [DOI] [PubMed] [Google Scholar]
- Sears C, Olesen J, Rubin D, Finley D, Maniatis T (1998) NF-kappa B p105 processing via the ubiquitin–proteasome pathway. J Biol Chem 273: 1409–1419 [DOI] [PubMed] [Google Scholar]
- Touitou R, Richardson J, Bose S, Nakanishi M, Rivett J, Allday MJ (2001) A degradation signal located in the C-terminus of p21WAF1/CIP1 is a binding site for the C8 alpha-subunit of the 20S proteasome. EMBO J 20: 2367–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL (2004) Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell 14: 95–104 [DOI] [PubMed] [Google Scholar]
- Verma R, Deshaies RJ (2000) A proteasome howdunit: the case of the missing signal. Cell 101: 341–344 [DOI] [PubMed] [Google Scholar]
- Voges D, Zwickl P, Baumeister W (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem 68: 1015–1068 [DOI] [PubMed] [Google Scholar]
- Zhang J, Xu LG, Han KJ, Shu HB (2004) Identification of a ZU5 and death domain-containing inhibitor of NF-kappaB. J Biol Chem 279: 17819–17825 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information