

Abstract
The AAA+ATPase p97/VCP, helped by adaptor proteins, exerts its essential role in cellular events such as endoplasmic reticulum-associated protein degradation or the reassembly of Golgi, ER and the nuclear envelope after mitosis. Here, we report the three-dimensional cryo-electron microscopy structures at ∼20 Å resolution in two nucleotide states of the endogenous hexameric p97 in complex with a recombinant p47 trimer, one of the major p97 adaptor proteins involved in membrane fusion. Depending on the nucleotide state, we observe the p47 trimer to be in two distinct arrangements on top of the p97 hexamer. By combining the EM data with NMR and other biophysical measurements, we propose a model of ATP-dependent p97(N) domain motions that lead to a rearrangement of p47 domains, which could result in the disassembly of target protein complexes.
Keywords: AAA ATPase, conformational change, Cryo-EM, p47, p97
Introduction
p97 belongs to the diverse AAA+ (ATPase Associated with various cellular Activities) family of proteins involved in a variety of apparently unrelated cellular processes. AAA+ proteins are characterised by the presence of one or two conserved AAA domains (∼230–250 residues, D1 and D2 domains), which contain the Walker A and B motifs. AAA+ proteins are generally oligomeric (typically hexamers) and are believed to have common functions in the unfolding or disassembly of macromolecules or multiprotein complexes (Ogura and Wilkinson, 2001). Several p97 cofactors have been identified that link p97 with a variety of cellular activities (reviewed in Dreveny et al, 2004b). For example, p97 binds to the Ufd1–Npl4 adaptor complex mediating events during endoplasmic reticulum (ER)-associated degradation (ERAD, Bays et al, 2001; Ye et al, 2001; Braun et al, 2002; Jarosch et al, 2002), nuclear envelope formation (Hetzer et al, 2001) and spindle disassembly after mitosis (Cao et al, 2003).
p97 also functions in membrane fusion utilising the p47 adaptor protein to mediate the reformation of Golgi membranes and reassembly of the nuclear envelope at the end of mitosis (reviewed in Meyer, 2005; Uchiyama and Kondo, 2005). The p97–p47 complex is proposed to disassemble t-t SNARE (target-target soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complexes (Acharya et al, 1995), in a role reminiscient of that of the homologous AAA+ ATPase NSF (N-ethylmaleimide-sensitive factor) and its adaptor α-SNAP (soluble NSF attachment protein) in disassembling v-t SNAREs (vesicle-target-SNARE). During Golgi regrowth, it is proposed that p47 mediates the binding of p97 to the t-SNARE syntaxin 5 post fusion (Kondo et al, 1997; Rabouille et al, 1998). An additional protein, VCIP135 (valosin-containing protein, VCP/p97–p47 complex-interacting protein, p135), binds to the p97–p47-syntaxin 5 complex and dissociates it using ATP hydrolysis events in p97, preparing syntaxin 5 for further rounds of membrane fusion (Uchiyama et al, 2002). Recently, NSF–α-SNAP complexes and the p97–p47–VCIP135 complexes were shown to act sequentially in cell cycle-dependent reformation of the ER network (Kano et al, 2005). These events also involve the t-SNARE syntaxin 18 (Kano et al, 2005). Interestingly, p47 was shown to bind to ubiquitin via its N-terminal UBA domain (Meyer et al, 2002; Yuan et al, 2004b) and this binding is essential for efficient p97–p47 mediated reassembly of Golgi cisternae (Meyer et al, 2002). In yeast, p47 orthologues interact with ubiquitylated proteins in vivo and are described to be involved in ubiquitin mediated proteolytic events, probably delivering substrates to the 26S proteasome (Hartmann-Petersen et al, 2004; Schuberth et al, 2004).
p97 comprises three domains, the N-terminal cofactor binding domain and two AAA ATPase domains (D1 and D2). Three-dimensional (3D) reconstructions of p97 in different nucleotide states obtained by cryo-electron microscopy (cryo-EM) and small angle X-ray scattering have shown that the N-domains are mostly flexible and together with the D1 and D2 domains can undergo significant conformational change during ATP binding and hydrolysis (Zhang et al, 2000; Rouiller et al, 2002; Beuron et al, 2003; Davies et al, 2005). Crystal structures of p97 at low resolution (3.5–4.7 Å) in different nucleotide states reveal a consistent picture of a hexameric arrangement of subunits with N-domains interacting with D1 in the D1 hexamer plane and with only minor conformational changes (DeLaBarre and Brunger, 2003, 2005; Huyton et al, 2003). The adaptor protein p47 contains three domains connected by long flexible regions. The solution structures for all three p47 domains (UBA, SEP and UBX) and dynamics of p47 fragments have been reported (Yuan et al, 2001, 2004b; Soukenik et al, 2004). A 2.9 Å crystal structure of p97(N–D1) hexamer bound to p47(UBX) domain revealed that p47 interacts with the p97(N) domain via a conserved loop within the UBX domain (S3/S4) that inserts into a hydrophobic pocket between the two p97(N) subdomains (Dreveny et al, 2004a). Despite this body of data, no detailed information exists regarding the structural arrangements within the full-length p97–p47 complex, other than low-resolution 2D EM projections (Rouiller et al, 2000). Six peripheral densities were attributed to p47, which is not consistent with biochemical studies suggesting a p47 trimer binding to a p97 hexamer (Kondo et al, 1997).
One of the major cellular roles for p97 is to provide mechanical force via ATPase activity to disassemble or unfold target proteins or protein complexes, a force which is mostly thought to be mediated through various adaptor proteins in vivo, although direct substrate binding has also been reported (Ye et al, 2003). We have combined cryo-EM, high-field NMR and other biophysical methods to capture and analyse the nucleotide-dependent conformational states of the physiological p97–p47 complex. A comparison between these states points to possible conformational changes that are necessary for p97 action on target substrates specific to the p97–p47 pathway and thus provide new insights into p97 function. Interestingly, we found that the p97–p47 complex resembles the NSF 20S complex structurally despite the differences between these ATPases and corresponding adaptor proteins (Brunger and DeLaBarre, 2003; Furst et al, 2003).
Results and discussion
p47 trimerisation and stoichiometry of the p97–p47 complex
In this study, we have carried out experiments, which allowed a qualitative appreciation of the oligomeric status of freshly purified full-length p47 using short timescale NMR experiments. Our earlier NMR studies on the p47(SEP–UBX) fragment revealed a weak propensity for oligomerisation via the SEP domain (Yuan et al, 2004b). Detailed analysis of 15N relaxation data indicated that the likely oligomeric state is either a dimer or trimer (Yuan et al, 2004b). SEP domain NMR signals are not observable in 1H-15N HSQC spectra of full-length p47, which can be attributed to their fast relaxation properties, revealed by 1D relaxation measurements and the fact that they become visible upon perdeuteration (Supplementary Figure 1A). The extent of broadening is similar to our earlier studies (Yuan et al, 2004b) and therefore consistent with the involvement of the SEP region in formation of either a dimeric or trimeric p47 species. Support for a trimeric p47 has been provided by gel-filtration and light scattering data (Kondo et al, 1997) as well as crosslinking experiments (Yuan et al, 2004b). Furthermore, analytical ultracentrifugation data presented in this study provides additional evidence for full-length p47 existing as a trimer (Supplementary Figure 1B).
As the individual UBA, SEP and UBX domains possess relaxation properties consistent with monomeric species (overall correlation times ∼4–6 ns) (Yuan et al, 2001, 2004b; Soukenik et al, 2004), we were able to investigate elements of the trimerisation interface by comparing chemical shifts between spectra of trimeric p47 and the separate, monomeric domains (Figure 1A) (Soukenik et al, 2004; Yuan et al, 2004b). Several well-resolved shifts occur in the SEP domain and delineate a contiguous patch at one edge of the β-sheet that could form part of the trimerisation boundary (Figure 1B). No perturbations are observed in the UBA or UBX domains and spectral overlap precludes the detailed comparison of the linker regions. These data combined with rigid-body docking using the program SymmDock (Schneidman-Duhovny et al, 2005) provide the means to construct a working model for the trimeric arrangement of SEP (Figure 1B). Interestingly, the N- and C-termini extend in opposite directions but are proximal to the trimer interface, this supports the premise that neighbouring linker regions are also important determinants for trimer formation.
Figure 1.
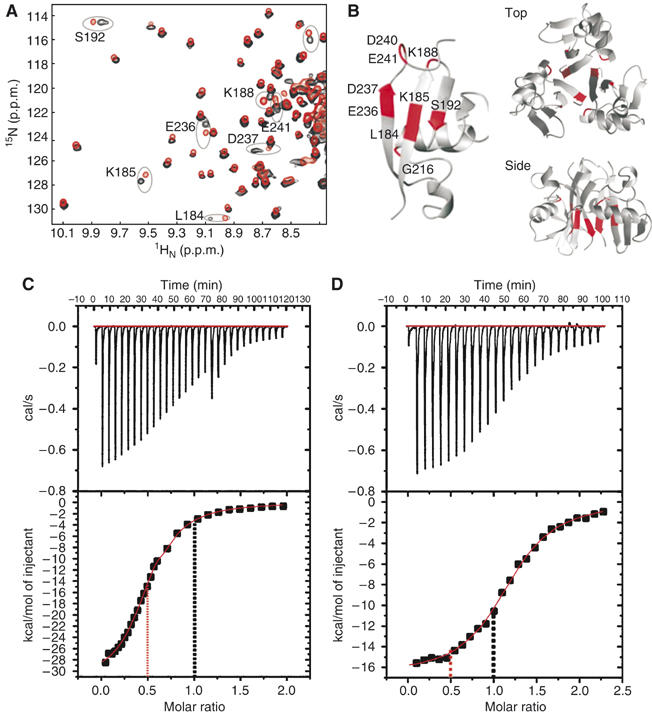
(A) Overlay of 1H-15N HSQC NMR spectra for 2H, 15N-labelled p47 (black) and a mixture of the individual 15N-labelled UBA, SEP and UBX domains from p47 (red). Key shifted resonances are labelled. (B) Ribbon representation of the p47(SEP) domain with chemical shift perturbations illustrating the likely trimer interface mapped in red (left) and a model for the trimer obtained by rigid-body docking (right) (top and side views). (C) Isothermal titration calorimetry of full-length p97–p47 and (D) p97–p47(SEP–UBX) domains showing a 1:0.5 and 1:1 stoichiometry respectively. The 1:0.5 and 1:1 ratios are indicated by a red and black dotted line in both plots, respectively.
Much controversy exists regarding the binding stoichiometry of p47 to p97 with data supporting both 3 and 6 p47 molecules per p97 hexamer (Kondo et al, 1997; Rouiller et al, 2000). In resolving this ongoing debate, we took extra care in producing, assessing and testing the activity of our p97–p47 complexes (Supplementary Figure 2A and B). To establish the stoichiometry of our p97–p47 complexes, we used isothermal titration calorimetry (Figures 1C and D). A ratio of 0.5 p47 monomer to 1 p97 protomer was observed with a Kd of 0.5 μM confirming that three p47 bind to a hexameric p97 (Figure 1C). In contrast, the stoichiometry of a shorter p47 fragment, comprising the SEP–UBX domains, was found to be of 1 p47 fragment bound to 1 p97 protomer (Figure 1D).
Furthermore, en face 2D EM projection averages of the respective complexes show that p97–p47(SEP–UBX) has significant additional six-fold density at the periphery of the p97 hexamer whereas the full-length complex contains central density and no additional peripheral density (data not shown). Scanning transmission electron microscopy mass measurements also confirm that the particles are no larger than a p97 hexamer in complex with 3 p47 (data not shown) consistent with the calorimetric data and analysis of the complex by native gel electrophoresis (Figure 1C and Supplementary Figure 2A).
Taken together our data firmly establish that three p47 molecules bind to a single p97 hexamer and indicate that p47 trimerisation mediated by the SEP domain and parts of the N-terminal linker are important determinants for this stoichiometry.
Cryo-EM reconstructions of the p97–p47 complex
We have obtained cryomicrographs of p97–p47 complex in the presence of ADP or AMPPNP (Figure 2 and Supplementary Table I). Manual picking of single particles was straightforward because of the good contrast of the particles (Figure 2A), and the resulting class averages (Figure 2C) presented a range of different views that were used for the angular reconstitution. 3D reconstructions were computed independently from separate initial models. The distribution of Euler angles for the AMPPNP and ADP data sets are shown in Figure 2B. Class averages obtained in the final iteration of the multireference alignments (MRA)/multivariate statistical analysis (MSA) and angle assignment correspond well to matching projections of the 3D model (Figure 2C). The parallel-striated appearance of p97 side views (Beuron et al, 2003; Figure 5A) is replaced in all data sets by a flat appearance of the bottom striation and additional density found at the top of the upper striation with small satellite densities connecting to this additional density (Figure 2C, arrows).
Figure 2.

Cryo-EM and class averages of the p97–p47 complex. (A) Field of a cryomicrograph of p97–p47 incubated with AMPPNP showing a range of different views with protein density shown in black. (B) Distribution of viewing angles of the class sums used for calculating the 3D models of the p97–p47 complex in the AMPPNP and ADP conformations. The γ-angles are clustered between (−60°, +60°) because of the use of the three-fold symmetry. (C) For each nucleotide condition, the top row contains representative p97–p47 class averages (protein is shown in white) and the bottom row the corresponding reprojection of the 3D map in the direction of the orientation assigned to the average. For the complex in the presence of AMPPNP, three classes are circled that represent typical top, side and tilted views (left to right). The arrows point to small satellite densities that are connected to the rest of the particle.
Figure 5.
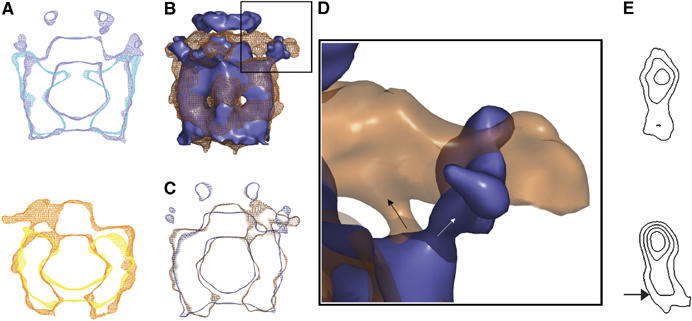
Pairwise comparison of the p97–p47 reconstructions. (A) Comparison of p97 (Beuron et al, 2003) and p97–p47 maps in the presence of AMPPNP (top, p97 alone in cyan; p97–p47 in blue) and ADP (bottom, p97 alone in yellow; p97–p47 in orange). Superimposed central vertical density sections are presented. In both cases, additional density is located centrally above the D1 ring. (B) Side view surface representation of the superimposed AMPPNP (slate) and ADP (mesh orange) maps. (C) Corresponding central vertical sections (same colour coding as in B). (D) Blown up surface representation of the area delineated in (B) left. The density (arrows) connecting the outer region of the D1 ring with peripheral density in the presence of AMPPNP or with the ‘propeller' in the presence of ADP emerges from the same point but extends in opposite directions. (E) Projected density of p97(D1–D2) domains in the p97–p47 maps (top, AMPPNP; bottom ADP) represented as iso-density contours.
The low-pass filtered, three-fold symmetrised 3D reconstructions are shown as surface representations in Figure 3 and Supplementary movies. The complex adopts a typical barrel-shaped arrangement formed by the stacking of D1 and D2 hexameric rings. In both maps, the top and bottom rings have an overall diameter of ∼110 Å and a combined height of 95 Å, which corresponds well to the diameter of p97 and the height of stacked D1 and D2 rings in crystal structures (DeLaBarre and Brunger, 2003, 2005; Huyton et al, 2003). Significant common density is found above the D1 plane at the top of the particle and consists of a large flat ‘cap' or ‘plug' sitting on the three-fold symmetry axis, from which other densities project. The height of the maps increases to ∼135 Å when including the ‘plug' and extends to ∼150 Å when enclosing the satellite densities (‘antennae') at the top of the AMPPNP map. The presence and connectivity of these antennae to the ‘plug' in the AMPPNP map is clearly visible on the class averages as well as in the corresponding reprojection (Figure 2C) and density sections through the map (Supplementary Figure 3). The same is observed for the 3D reconstruction of p97–p47 in the absence of nucleotide (data not shown).
Figure 3.
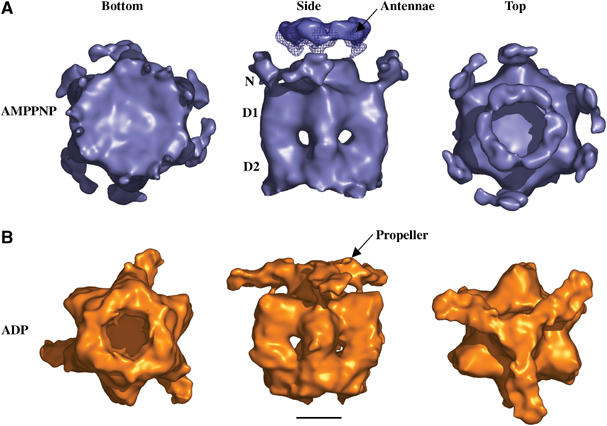
3D reconstructions of p97–p47 single particles imaged by cryo-EM. Surface representations of the three reconstructions low-pass filtered using a 20 Å Gaussian mask are shown (A) in the presence of AMPPNP and (B) in the presence of ADP. Three views are shown: bottom (looking from D2 to D1), side (perpendicular to the symmetry axis) and top (looking from D1 to D2). The position of the N, D1 and D2 domains of p97 are indicated. The ‘propeller' and ‘antennae' emerging from the central plug are indicated and represent densities attributed to p47. The mesh superimposed in Figure 3A (side view) shows the connection between the ‘antennae' and central ‘plug' in the non band-pass filtered AMPPNP surface map. The scale bar represents 50 Å.
The p97–p47 reconstructions also display a number of unique features. In the presence of AMPPNP, the D2 ring has no central pore (Figure 3A, bottom view) and small densities protrude outwards from the outer tip of the D1 ring (Figure 3A, side view and vertical density sections in Supplementary Figure 3). Connected ‘antennae' densities emerge from three points on the central ‘plug' and are contiguous giving rise to a ring-like appearance. In the presence of ADP, the D2 ring has a pore of ∼20 Å in diameter located at the end of a funnel shaped conduit (Figure 3B, bottom view). The particle is wider at the D1–D2 interface and three ‘arms' emerge from the central ‘plug' forming a three-fold symmetric ‘propeller-like' structure that links back onto the top of three D1 domains.
Similarities between the cryo-EM reconstructions of the p97–p47 and NSF–-SNAP–SNARE complexes
Recently, the 3D structure of NSF (D1 ATP hydrolysis mutant) in complex with α-SNAP–SNARE (20S complex) was determined by cryo-EM and single-particle methods in the presence of a mixture of ADP and ATP (EM3D accession code 1059; Furst et al, 2003). NSF is a close homologue of p97 and dissociates v-t SNARE complexes, thereby reactivating them to mediate further rounds of membrane fusion. Interestingly, the 3D reconstruction of the NSF 20S complex (filtered to 20 Å resolution) is similar to p97–p47 both in size and overall shape (Figure 4). The central ‘plugs' are comparable as are the positions of protrusions emanating form D1 attributed to N-domains (see below). The volume of the NSF 20S reconstruction accounts mainly for NSF due to the six-fold averaging used in the reconstruction of the complex (Furst et al, 2003). Owing to the conformational heterogeneity, the NSF 20S reconstruction does not contain all the density for the α-SNAP–SNARE proteins with the ‘plug' density representing only a fraction of the whole α-SNAP–SNARE complex (Furst et al, 2003). These similarities are interesting given the absence of any sequence and structural homology between p47 and α-SNAP, the corresponding adaptor for NSF. Furthermore, the residues implicated in α-SNAP binding by NSF(N) are located on the opposite side of the p47 binding site on p97(N) (Dreveny et al, 2004a). Despite the differences including ATPase domain arrangements between NSF and p97 (Brunger and DeLaBarre, 2003; Furst et al, 2003), both in complex with their respective adaptor protein (adaptor protein and parts of SNARE complex for NSF) show an overall similarity at this resolution. Especially, the similar location of the adaptor proteins on top of the hexamers is intriguing (Figure 4B and C), although further higher resolution structures would be necessary to substantiate any possible mechanistic analogies.
Figure 4.
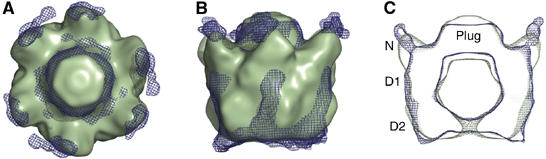
Superposition of the EM reconstructions of p97–p47 and the NSF 20S complex. The NSF 20S complex (Furst et al, 2003) was band pass filtered to a resolution of 20 Å. (A) Top view of the superposition of the NSF 20S complex (green) and the p97–p47 AMPPNP map (blue mesh; lacking the antennae). (B) Side view of the superimposed maps. (C) Vertical central section of superimposed maps. The N, D1 and D2 domains of p97 are indicated. Both maps are characterised by a central plug on top of the D1 hexameric ring, which is attributed to α-SNAP–SNARE densities for the NSF 20S complex and corresponds to the SEP domains of the p47 adaptor in the p97–p47 complex.
Distinct nucleotide-dependent conformations of the p97–p47 complex
When comparing our previous p97 maps (Beuron et al, 2003) with the new p97–p47 maps obtained under identical nucleotide conditions, we find good overall agreement for p97 (Figure 5A). This comparison allows us to identify density for p47 as being on top of the D1 ring (‘plug' and ‘antennae'). Apart from these clear extra densities, there are a number of other distinct differences. In the AMPPNP conformations (Figure 5A, top), the D1 and D2 rings have similar diameters with D1 stacking on top of D2. In p97–p47 the D2 ring is closed, whereas it is open in p97 alone. In the ADP conformations (Figure 5A, bottom), the D1 and D2 rings also show good agreement although in contrast to AMPPNP, the D2 ring is open in p97–p47 but closed in p97 alone. These differences in p97 clearly reflect p47 binding and illustrate the limitations of interpreting p97 nucleotide-dependent conformational changes in the absence of bound adaptor proteins.
Superimposed surface rendered representations for the p97–p47 AMPPNP (blue) and ADP (orange; Figure 5B) as well as vertical sections of the same comparison (Figure 5C) show that the maps differ substantially in both shape and protruding densities above the D1 plane. Intriguingly, thin densities emerge from the same point in the D1 ring in both maps, though projecting in different directions (arrows in Figure 5D). In the ADP state, p97 adopts a barrel shape with the widest point between the D1 and D2 domains, although the D1 ring in the ADP state has a slightly wider diameter (∼120 Å). By comparison, the D1 and D2 domains in the AMPPNP state have similar diameters with the D1 domain stacking directly on top of D2 (Figure 5C). This results in an apparent counter-clockwise rotation in D2 of ∼10° (viewing down the trimer axis from top) in the ADP map compared to the AMPPNP map (Figure 5E).
Proposed p47 domain arrangements in the p97–p47 complex
In order to interpret further the domain arrangements in the p97–p47 reconstructions, we performed a number of labelling studies. Electron micrographs of negatively stained nanogold labelled p47 N-termini (preceding the UBA) clearly show the gold marker at the centre of the hexamer in both the AMPPNP and nucleotide-free samples (Supplementary Figure 4A). In contrast, we observe a different labelling pattern in the presence of ADP (Supplementary Figure 4A, middle) reflecting a dispersed distribution of the observed gold particles, consistent with flexible or untethered p47 N-termini (Supplementary Figure 1C). In agreement with the location of p47 domains above the p97 hexamer, we observe both syntaxin 5 and ubiquitin (known p97 target proteins) as binding to the top of the p97–p47 complex (Supplementary Figure 4B and C).
Using the mapping information and comparisons with previous p97 reconstructions, we have provisionally assigned p47 domains within our reconstructions. These provisional assignments were further guided by the following key pieces of information: (1) existing high-resolution structures of p97, p47 and the p97(N–D1)–p47(UBX) complex (DeLaBarre and Brunger, 2003, 2005; Huyton et al, 2003; Dreveny et al, 2004a; Yuan et al, 2004b); (2) three p47 molecules bind to one p97 hexamer; (3) p47 binds to p97(N) domains via two binding sites, the p47(UBX) domain (Dreveny et al, 2004a) and the linker region between SEP and UBX (residues ∼246–273) (Uchiyama et al, 2002; Bruderer et al, 2004), which is confirmed by chemical shift perturbation data measured from CRIPT-TROSY NMR spectra of the p97–p47 complex (Supplementary Figure 1D); (4) p47 has a propensity to form trimers in solution mediated by the p47(SEP) domain and flanking regions, for which we propose a possible model (Figure 1B). A number of chemical shifts for interfacial residues are unperturbed in the CRIPT-TROSY data on the p97–p47 complex suggesting that the trimer is most likely maintained within the complex (Supplementary Figure 1D), consistent with the observed stoichiometry of the full-length complex.
In the AMPPNP map, we attribute the ‘plug' density on top of the p97 hexamer to represent the SEP trimer (∼200 residues). We attribute the ring of ‘antennae' density connected to the ‘plug' to be p47(UBA) domains, which is consistent with gold labelling studies and the length of the UBA–SEP linker (∼125 residues). The six peripheral densities connected to the outside of D1 (Figure 3A) are arranged as pairs of spatially close but morphologically distinct densities. The connecting density between these peripheral densities and the main body are still observed after band-pass filtering to 20 Å, indicating that it is a well-defined part of the reconstruction even at this resolution. We interpret the ‘kidney-shaped' density (12 500 Å3; ∼10 kDa) within each pair as p97(N) domains and the second more elongated density (18 000 Å3; ∼15 kDa) as p97(N)–p47(UBX). The observed volumes are much smaller than expected (∼20 and 34 kDa, respectively). This could reflect the flexible nature of these domains. When contoured at a lower threshold, thin density fills the gap between the two N-domains (data not shown) supporting a model of p47 binding to adjacent N-domains in agreement with the 3 p47:6 p97 binding stoichiometry.
In contrast, the ADP map displays a significantly different arrangement of density on top of p97 with the appearance of a distinctive ‘propeller-like' structure equivalent to a volume of ∼150 000 Å3 (∼120 kDa) when contoured using a similar threshold as for the whole map in Figure 3. The ‘propeller' includes the p47(SEP) trimer central ‘plug' and the ‘blade' which is connected to the D1 domain via similar density to the one observed in the AMPPNP map (arrows in Figure 5D). This density is present in our 20 Å band-pass filtered maps suggesting that it is a well-defined part of the EM reconstruction. In contrast to the AMPPNP map, we do not observe clear density for p47(UBA) domains consistent with gold labelling studies. In the ADP conformation, the p47(UBA) domains are probably flexibly tethered to p47(SEP) domains.
We have used the above information to manually dock known p97 and p47 structures into the p97–p47 maps in order to propose a tentative model for the domain arrangements of p47 (Figure 6). The crystal structure of p97(N)–p47(UBX) was modelled into peripheral density in the AMPPNP map and ‘propeller' density in the ADP map, respectively, taking into account both the p97 N–D1 and p47 SEP–UBX linker lengths (∼20 and ∼30 residues, respectively). In the ADP map we have positioned three p97(N) domains in the middle of the ‘propeller blade' assuming the connecting density to D1 is the p97 N–D1 linker. This then places three p47(UBX) domains at the tip of each ‘propeller blade' in order to maintain the interaction observed in the p97(N–D1)–p47(UBX) crystal structure (Dreveny et al, 2004a). This assignment also places the p97(N)–p47(UBX) moiety at a similar height in the ADP and AMPPNP nucleotide states. The three unbound N-domains in the ADP map could be modelled to positions similar to p97 crystal structures, which leads to a model of three N-domains above the D1 plane (bound to p47) forming a ‘propeller-like' structure and three N-domains in the plane of D1. The structures of the D1 and D2 rings from the crystal structures of p97 were fitted manually into both the AMPPNP and ADP maps and both show good agreement with the EM density (Figure 6). Furthermore, both EM envelopes show additional densities that would accommodate the C-terminal extension of p97, which is not fully visible in crystal structures (residues ∼700–806, ∼12 kDa; Figure 6C and D, horizontal arrows).
Figure 6.
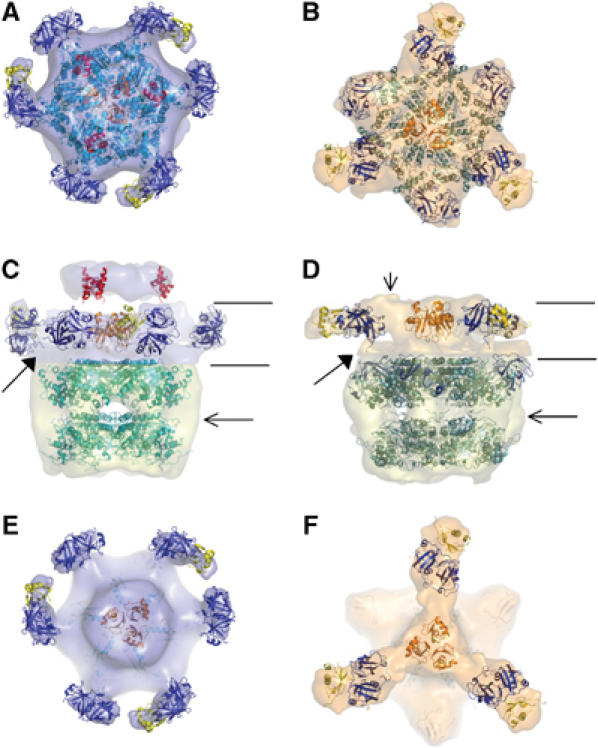
Provisional fitting of domains in the EM envelopes. p97(N) is represented in blue, p97(D1–D2) in cyan, p47(UBA) in red, p47(SEP) in orange and p47(UBX) in yellow. Left column (AMPPNP) and right column (ADP) with: top (A, B), side (C, D) and cutout top views above the D1 ring (E, F) indicated by the lines in C and D, are shown with transparent EM envelopes. Two contour levels were used in the side view surface representations. A low threshold was used for the moiety attributed to p47 to account for the flexibility. For both maps the volumes corresponding to p97(D1–D2–C) domains, shown in pale yellow, are similar (530 000 Å3; 440 kDa; ∼600 residues). For both maps, the p97(D1) and p97(D2) hexamers were fitted as found in the p97 crystal structures (Huyton et al, 2003; DeLaBarre and Brunger, 2005). The p97(N)–p47 density at the top of both maps was contoured at a threshold leading to a volume of 305 000 Å3 (260 kDa). The filled arrows point to the density where the p97(N–D1) linker has been docked in both nucleotide states. The horizontal arrows in C and D indicate the possible location of the p97 C-terminal extension not resolved in the crystal structures. The vertical arrow in D right, points to density in the EM map that could account for structured p47(SEP–UBX) linker or additional residues N-terminal of SEP.
A speculative model for ATP-dependent p97 driven domain rearrangements in p47
Nucleotide-dependent conformational changes of the p97–p47 complex would provide mechanistic insights into how p97 utilises its ATPase activity to act on bound adaptors and target substrates. Owing to the limited resolution of our reconstructions, we cannot propose a detailed mechanism of action for the p97–p47 complex. However, we do observe distinct conformational changes of the complex following incubation with ADP, namely significant conformational change in the D2 domain in the ADP state and a substantial reorganisation of the densities attributed to p47 on top of the D1 ring. We speculate that at some stage during ATP hydrolysis, as captured in the AMPPNP reconstruction, p97(N) domains form contacts with p47(UBX) domains at the periphery of the hexamer. ATP hydrolysis in D2 results in conformational changes that induce a rearrangement of p97(N)–p47(UBX) domains and the p47(SEP–UBX) linker to form part of the ‘propeller' structure. Finally, the SEP–UBX linker relocation could affect the position and properties of the SEP trimer. This could reorder the UBA–SEP linker to promote conformational changes in target proteins (Figure 7).
Figure 7.

Schematic representation of the proposed p47 domain rearrangements within the p97–p47 complex. p97(D1–D2) protomers are represented in light blue, with N-domains in dark blue. The p47 domains are shown as ellipses with N-terminal UBA domain coloured red, SEP orange and UBX yellow. Left shows the domain arrangements as interpreted in the AMPPNP p97–p47 EM reconstruction. The p97(N) domains sit above the D1 hexamer with three out of the six in direct contact with p47(UBX). The p47 SEP–UBX linker (grey) makes contact with the adjacent p97(N) before connecting to p47(SEP). A p47 SEP trimer sits centrally on top of the p97 hexamer with UBA domains shown vertically above connected by the UBA–SEP linker (grey). Right shows the domain arrangements as interpreted in the ADP p97–p47 EM reconstruction. The p97(N)–p47(UBX) domain complex has rotated ∼100° in the plane to form the extremities of the propeller structure (yellow arrow, left) breaking contact between the SEP–UBX linker and the adjacent p97(N) domain. p47(SEP) domain and p47 UBA–SEP linker rearrangements (grey arrow, left) result in the p47(UBA) domain experiencing a lateral displacement (red arrow, left). This nucleotide-dependent conformational change would result in mechanical force being applied to target proteins bound to p47 via UBA domains and may represent part of the p97–p47 disassembly activity.
It has been noted previously that p97(N) domain motion and flexibility is important for p97 function (Zhang et al, 2000; Rouiller et al, 2002; Beuron et al, 2003). Conserved glycines (Gly208, Gly209) within the p97(N–D1) linker region could form pivoting points that allow the N–D1 linker to rotate and relocate (Smith et al, 2004). Furthermore, N–D1 linker residues are involved in D1 nucleotide binding (Zhang et al, 2000; DeLaBarre and Brunger, 2003, 2005; Huyton et al, 2003), thereby connecting nucleotide binding and hydrolysis to potential N-D1 linker and N-domain relocations. The D1–D2 linker also contains conserved glycines and probably plays a similar role in transmitting nucleotide binding/hydrolysis events in D2 to conformational changes in D1 and N-domains.
In conclusion, we propose that changes occurring during nucleotide binding and hydrolysis in the D2 domain of p97 are transmitted and applied to the p97(N) and p47 adaptor domains. This long-range transmission of conformational change has also been suggested for p97 alone (Huyton et al, 2003; DeLaBarre and Brunger, 2003; Davies et al, 2005). We suggest a possible interplay between the two p47 linkers mediated by an SEP trimer rearrangement and p97(N) domain relocation, which would exert conformational changes on proposed p97 target proteins such as SNARE complexes or ubiquitylated substrates bound to the p47(UBA) domains. To address how these conformational changes disassemble protein complexes or act on substrates, further structures of p97 adaptor complexes bound to defined target proteins will be required. We also cannot exclude that p47 or its orthologues could have additional adaptor functions where nucleotide hydrolysis in p97 is not required.
Materials and methods
p97–p47 complex preparation
Endogenous full-length p97 (89 kDa) was purified as described previously (Kondo et al, 1997). This procedure involved high salt treatment in order to reduce the amount of prebound cytosolic p47 to a non-detectable level. Recombinant N-terminal (His6)p47 (41 kDa) was expressed and purified from Escherichia coli lysates. p97 was incubated with four times molar excess amount of (His6)p47 on ice for 30 min in 20 mM Tris–HCl, 150 mM KCl, 1 mM DTT, pH 7.4 and p97–(His6)p47 complexes were separated from free (His6)p47 using gel-filtration as described previously (Kondo et al, 1997).
Isothermal titration calorimetry (ITC)
ITC experiments were performed using a VP-ITC instrument (MicroCal Inc., USA). Recombinant protein samples were dialysed into 50 mM Tris–HCl, 150 mM KCl, 5 mM MgCl2, 5% glycerol, pH 7.4 (buffer A). Titrations were performed by injecting consecutive aliquots of p47 (129.5 μM) or p47(SEP–UBX) fragment (137 μM; p47 residues 171–370) into the ITC cell containing p97 (13.1 μM) at 25°C. ITC data were corrected for heats of dilution of the protein solution. Binding stoichiometry, enthalpy, entropy and binding constants were determined by fitting the corrected data to a one site binding model. The ITC data were fit using Origin 5.0 (MicroCal Inc., USA).
NMR studies
Full-length (His6)p47 and p97(His6) were expressed, purified and concentrated to mM concentrations. p47 is fully degraded into individual domains within 2–3 days at ambient temperature. This was confirmed by gel-filtration, SDS–PAGE and recording of NMR spectra. 15N-labelled full-length p47 and 2H, 15N-double labelled full-length p47 were produced in Silantes rich media (15N and 2H 15N respectively). The protein was purified from clarified cell lysate to homogeneity in two steps using a Ni2+-chelating column followed by gel-filtration. Purified proteins were buffer exchanged to 20 mM Tris–HCl at pH 7.4 and concentrated to mM concentrations. For the p97–p47 complexes with AMPPNP or ADP, the final concentrations of all components in the NMR samples were p47=0.1 mM, p97=0.35 mM and AMPPNP/ADP=3 mM. NMR spectra were recorded at 298 K on either a 500 or an 800 MHz Bruker spectrometer. For 1H–15N CRIPT-TROSY experiments, polarisation transfer times of 1.5, 2 and 2.5 ms were used to ensure optimal performance of the pulse sequence and the three spectra were added together using NMRPipe. All spectra were processed with NMRPipe and analysed with NMRView. Assignment of the p47 trimer was accomplished using the assignment available for fragments p47(1–174) and p47(171–370). In cases where identical chemical shifts were not observed for the fragments and full-length p47, the nearest available resonance was chosen and 3D 1H-15N-NOESY spectra were used for confirmation (Yuan et al, 2004a). Backbone assignments could be made for 100% of the residues from the three structured domains within full-length p47. Assignments could also be made for the majority of the two inter-domain linker regions 46–178 and 248–281 within the fragments but overlap precluded a full assignment of linker regions within full-length p47 (Yuan et al, 2004a).
The model for the p47 trimer was calculated using the rigid-body docking algorithm SymmDock with a user-defined trimerisation interface and C3 point group symmetry. The trimerisation interface was identified from chemical shift perturbations results (i.e. L184, K185, K188, S192, E236, D237, D240 and E241) (Schneidman-Duhovny et al, 2005). The top 10 solutions based on their geometric shape complementarity score show the same relative domain arrangement. The model burying the largest surface area was chosen for presentation.
Cryo-EM
5 μl drops of freshly diluted endogenous p97–(His6)p47 (∼70 μg/ml, equivalent to ∼740 nM of p97 in 20 mM Tris–HCl, 150 mM KCl, 1 mM DTT, pH 7.4) was applied to washed glow-discharged copper grids supporting a lacey carbon film (Agar Scientific, UK). The samples with added nucleotide (5 mM ADP or AMPPNP) were incubated for 30 min on ice. The drop was blotted and quench-frozen in liquid ethane. The grid was then transferred into a Gatan 626 cryo-holder and inserted into the microscope (CM200, FEI). The complex was imaged in vitreous ice without supporting carbon film and was found to adopt different orientations. Micrographs were recorded at an accelerating voltage of 200 kV at a nominal magnification of × 38 000 or × 50 000 on Kodak SO163 film under minimal electron dose illumination (∼20e−/Å2).
Image processing and 3D reconstruction
Micrographs were digitised with a Leafscan 45 scanner (Leaf Systems Inc.) to 2.6 Å per pixel. The defoci varied from 1.1 to 2.5 μm. The contrast transfer function (CTF) effects of the microscope were corrected by reversing phases and no amplitude correction was applied. Image processing was performed using the IMAGIC-5 software (van Heel et al, 1996). The centred (non-rotationally aligned) images were subjected to MSA (van Heel and Frank, 1981) giving rise to a first set of reference-free averages and the eigenimages were inspected for information about the rotational symmetry. A first set of references is extracted after this first cycle of classification for the initial round of MRA. Iterations of MRA and classification followed until good stable classes were obtained.
Independent starting models were built by back-projecting putative top, side and tilted views, which are given arbitrary Euler angles. The relative orientations of all the classes were subsequently defined by angular reconstitution using the model as an initial approximation of the structure (van Heel, 1987). Neither X-ray structures nor existing cryo-EM 3D maps were used as starting models. The 3D models were three-fold symmetrised before generating the anchor sets. Use of the three-fold symmetry has also been made in the Euler angles searches. The initial raw 3D reconstructions were then refined by several iterations of MRA using reprojections of the resulting model as references, MSA and Euler angle assignments followed by exact filter back-projections.
Between two and four initial 3D models were tried for each data set until a good agreement between the classes and the reprojections is achieved after one or two iterations of MRA, MSA and angle assignment. The results are summarised in Supplementary Table I. Fewer classes were used for the ADP data set probably due to some heterogeneity in the particle population. The numbers of particles in the final reconstructions are similar to previously published data on p97 (Rouiller et al, 2002). The Euler angle distributions for the ADP and AMPPNP data sets (Figure 2B) show that the orientations of the classes show good coverage of the asymmetric unit with a slight preference for side and tilted views. The three data sets processed independently led to similar overall features for the reconstructions (Figure 3). The resolution of the AMPPNP (and ADP) map was 24 (27), 20 (22) or 22 (25) Å according to the 0.5 FSC correlation criteria, the 3σ criteria or the 1-bit criteria respectively (Harauz and van Heel, 1986; Orlova et al, 1997; van Heel and Schatz, 2005). For surface rendering, the contour level was chosen to enclose a volume that would contain 110% of the expected mass (720 kDa; Figure 3), based on a density of 0.84 Da/Å3 (1.33 g/cm). Electron density maps have been submitted to the EMDB database under accession numbers EMD-1191 and EMD-1192.
Model fitting
The crystallographic models (1S3S: p97(N–D1)–p47(UBX), 1VAZ: p47(SEP), 1V92: p47(UBA), 1YPW: p97 full-length ADP/AMPPNP, 1YQI: p97 full-length ADP/ADP) were fitted manually into the p97–p47 cryo-EM reconstructions using the program ‘O' (Jones et al, 1991). Coordinate sets for p97(N), p97(D1), p97(D2), p47(UBA), p47(SEP) and p97(N)–p47(UBX) complex were used in the fitting procedures. Linker lengths between p97(N–D1), p47(UBA-SEP) and p47(SEP–UBX) were used as guides in the modelling process. All figures were prepared using the program Pymol (http://www.pymol.org). In Figure 6, two contour levels were used for p97(D1–D2) and p97(N)–p47 separately to take into account the flexibility of the p97(N)–p47 domains.
Supplementary Material
Supplementary data
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Movies
Supplementary Table 1
Acknowledgments
We thank the CBEM for excellent facilities. We are grateful to E Orlova and P Da Fonseca for help with the image processing and to S Islam for his help with the figures and movies. The Brookhaven National Laboratory STEM facility is a National Institutes of Health Supported Resource Centre, NIH 5 P41 EB2181, with additional support provided by Department of Energy and Office of Biological and Environmental Research. We also thank P Simpson for helpful discussions, G Kelly and T Frenkiel of the 800 MHz NMR service at NIMR. We thank the Wellcome Trust and the BBSRC for generous support.
References
- Acharya U, Jacobs R, Peters JM, Watson N, Farquhar MG, Malhotra V (1995) The formation of Golgi stacks from vesiculated Golgi membranes requires two distinct fusion events. Cell 82: 895–904 [DOI] [PubMed] [Google Scholar]
- Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY (2001) HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell 12: 4114–4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuron F, Flynn TC, Ma J, Kondo H, Zhang X, Freemont PS (2003) Motions and negative cooperativity between p97 domains revealed by cryo-electron microscopy and quantised elastic deformational model. J Mol Biol 327: 619–629 [DOI] [PubMed] [Google Scholar]
- Braun S, Matuschewski K, Rape M, Thoms S, Jentsch S (2002) Role of the ubiquitin-selective CDC48(UFD1/NPL4)chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J 21: 615–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruderer RM, Brasseur C, Meyer HH (2004) The AAA ATPase p97/VCP interacts with its alternative co-factors, Ufd1–Npl4 and p47, through a common bipartite binding mechanism. J Biol Chem 279: 49609–49616 [DOI] [PubMed] [Google Scholar]
- Brunger AT, DeLaBarre B (2003) NSF and p97/VCP: similar at first, different at last. FEBS Lett 555: 126–133 [DOI] [PubMed] [Google Scholar]
- Cao K, Nakajima R, Meyer HH, Zheng Y (2003) The AAA-ATPase Cdc48/p97 regulates spindle disassembly at the end of mitosis. Cell 115: 355–367 [DOI] [PubMed] [Google Scholar]
- Davies JM, Tsuruta H, May AP, Weis WI (2005) Conformational changes of p97 during nucleotide hydrolysis determined by small-angle X-Ray scattering. Structure 13: 183–195 [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, Brunger AT (2003) Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol 10: 856–863 [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, Brunger AT (2005) Nucleotide dependent motion and mechanism of action of p97/VCP. J Mol Biol 347: 437–452 [DOI] [PubMed] [Google Scholar]
- Dreveny I, Kondo H, Uchiyama K, Shaw A, Zhang X, Freemont PS (2004a) Structural basis of the interaction between the AAA ATPase p97/VCP and its adaptor protein p47. EMBO J 23: 1030–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreveny I, Pye VE, Beuron F, Briggs LC, Isaacson RL, Matthews SJ, McKeown C, Yuan X, Zhang X, Freemont PS (2004b) p97 and close encounters of every kind: a brief review. Biochem Soc Trans 32: 715–720 [DOI] [PubMed] [Google Scholar]
- Furst J, Sutton RB, Chen J, Brunger AT, Grigorieff N (2003) Electron cryomicroscopy structure of N-ethyl maleimide sensitive factor at 11 A resolution. EMBO J 22: 4365–4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauz G, van Heel M (1986) Exact filters for general geometry three dimensional reconstruction. Optik 73: 146–156 [Google Scholar]
- Hartmann-Petersen R, Wallace M, Hofmann K, Koch G, Johnsen AH, Hendil KB, Gordon C (2004) The Ubx2 and Ubx3 cofactors direct Cdc48 activity to proteolytic and nonproteolytic ubiquitin-dependent processes. Curr Biol 14: 824–828 [DOI] [PubMed] [Google Scholar]
- Hetzer M, Meyer HH, Walther TC, Bilbao-Cortes D, Warren G, Mattaj IW (2001) Distinct AAA-ATPase p97 complexes function in discrete steps of nuclear assembly. Nat Cell Biol 3: 1086–1091 [DOI] [PubMed] [Google Scholar]
- Huyton T, Pye VE, Briggs LC, Flynn TC, Beuron F, Kondo H, Ma J, Zhang X, Freemont PS (2003) The crystal structure of murine p97/VCP at 3.6A. J Struct Biol 144: 337–348 [DOI] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T (2002) Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol 4: 134–139 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kano F, Kondo H, Yamamoto A, Kaneko Y, Uchiyama K, Hosokawa N, Nagata K, Murata M (2005) NSF/SNAPs and p97/p47/VCIP135 are sequentially required for cell cycle-dependent reformation of the ER network. Genes Cells 10: 989–999 [DOI] [PubMed] [Google Scholar]
- Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G (1997) p47 is a cofactor for p97-mediated membrane fusion. Nature 388: 75–78 [DOI] [PubMed] [Google Scholar]
- Meyer HH (2005) Golgi reassembly after mitosis: the AAA family meets the ubiquitin family. Biochim Biophys Acta 1744: 108–119 [DOI] [PubMed] [Google Scholar]
- Meyer HH, Wang Y, Warren G (2002) Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1–Npl4. EMBO J 21: 5645–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ (2001) AAA+ superfamily ATPases: common structure–diverse function. Genes Cells 6: 575–597 [DOI] [PubMed] [Google Scholar]
- Orlova EV, Dube P, Harris JR, Beckmann E, Zemlin F, Markl J, van Heel M (1997) Structure of Keyhole Limpet Hemocyanin Type 1 (KLH1) at 15 Å resolution by electron cryomicroscopy and angular reconstitution. J Mol Biol 271: 417–437 [DOI] [PubMed] [Google Scholar]
- Rabouille C, Kondo H, Newman R, Hui N, Freemont P, Warren G (1998) Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell 92: 603–610 [DOI] [PubMed] [Google Scholar]
- Rouiller I, Butel VM, Latterich M, Milligan RA, Wilson-Kubalek EM (2000) A major conformational change in p97 AAA ATPase upon ATP binding. Mol Cell 6: 1485–1490 [DOI] [PubMed] [Google Scholar]
- Rouiller I, DeLaBarre B, May AP, Weis WI, Brunger AT, Milligan RA, Wilson-Kubalek EM (2002) Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat Struct Biol 9: 950–957 [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ (2005) Geometry-based flexible and symmetric protein docking. Proteins 60: 224–231 [DOI] [PubMed] [Google Scholar]
- Schuberth C, Richly H, Rumpf S, Buchberger A (2004) Shp1 and Ubx2 are adaptors of Cdc48 involved in ubiquitin-dependent protein degradation. EMBO Rep 5: 818–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GR, Contreras-Moreira B, Zhang X, Bates PA (2004) A link between sequence conservation and domain motion within the AAA+ family. J Struct Biol 146: 189–204 [DOI] [PubMed] [Google Scholar]
- Soukenik M, Diehl A, Leidert M, Sievert V, Bussow K, Leitner D, Labudde D, Ball LJ, Lechner A, Nagler DK, Oschkinat H (2004) The SEP domain of p47 acts as a reversible competitive inhibitor of cathepsin L. FEBS Lett 576: 358–362 [DOI] [PubMed] [Google Scholar]
- Uchiyama K, Jokitalo E, Kano F, Murata M, Zhang X, Canas B, Newman R, Rabouille C, Pappin D, Freemont P, Kondo H (2002) VCIP135, a novel essential factor for p97/p47-mediated membrane fusion, is required for Golgi and ER assembly in vivo. J Cell Biol 159: 855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama K, Kondo H (2005) p97/p47-mediated biogenesis of Golgi and ER. J Biochem (Tokyo) 137: 115–119 [DOI] [PubMed] [Google Scholar]
- van Heel M (1987) Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction. Ultramicroscopy 21: 111–124 [DOI] [PubMed] [Google Scholar]
- van Heel M, Frank J (1981) Use of multivariate statistics in analysing the images of biological macromolecules. Ultramicroscopy 6: 187–194 [DOI] [PubMed] [Google Scholar]
- van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M (1996) A new generation of the IMAGIC image processing system. J Struct Biol 116: 17–24 [DOI] [PubMed] [Google Scholar]
- van Heel M, Schatz M (2005) Fourier shell correlation threshold. J Struct Biol 151: 250–262 [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414: 652–656 [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2003) Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol 162: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Shaw A, Zhang X, Kondo H, Lally J, Freemont PS, Matthews S (2001) Solution structure and interaction surface of the C-terminal domain from p47: a major p97cofactor involved in SNARE disassembly. J Mol Biol 311: 255–263 [DOI] [PubMed] [Google Scholar]
- Yuan X, Simpson P, Kondo H, McKeown C, Dreveny I, Zhang X, Freemont PS, Matthews S (2004a) Complete backbone resonance assignments of p47: the 41kDa adaptor protein of the AAA ATPase p97. J Biomol NMR 28: 309–310 [DOI] [PubMed] [Google Scholar]
- Yuan X, Simpson P, McKeown C, Kondo H, Uchiyama K, Wallis R, Dreveny I, Keetch C, Zhang X, Robinson C, Freemont P, Matthews S (2004b) Structure, dynamics and interactions of p47, a major adaptor of the AAA ATPase, p97. EMBO J 23: 1463–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, Gorman MA, Kondo H, Dokurno P, Lally J, Leonard G, Meyer H, van Heel M, Freemont PS (2000) Structure of the AAA ATPase p97. Mol Cell 6: 1473–1484 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Movies
Supplementary Table 1