

Abstract
The Campylobacter jejuni pgl locus encodes an N-linked protein glycosylation machinery that can be functionally transferred into Escherichia coli. In this system, we analyzed the elements in the C. jejuni N-glycoprotein AcrA required for accepting an N-glycan. We found that the eukaryotic primary consensus sequence for N-glycosylation is N terminally extended to D/E-Y-N-X-S/T (Y, X≠P) for recognition by the bacterial oligosaccharyltransferase (OST) PglB. However, not all consensus sequences were N-glycosylated when they were either artificially introduced or when they were present in non-C. jejuni proteins. We were able to produce recombinant glycoproteins with engineered N-glycosylation sites and confirmed the requirement for a negatively charged side chain at position −2 in C. jejuni N-glycoproteins. N-glycosylation of AcrA by the eukaryotic OST in Saccharomyces cerevisiae occurred independent of the acidic residue at the −2 position. Thus, bacterial N-glycosylation site selection is more specific than the eukaryotic equivalent with respect to the polypeptide acceptor sequence.
Keywords: Campylobacter jejuni, consensus sequence, N-glycosylation, periplasm, oligosaccharyltransferase
Introduction
N-linked protein glycosylation is an essential and conserved process occurring in the endoplasmic reticulum (ER) of eukaryotic organisms. It is important for protein folding, oligomerization, quality control, sorting, and transport of secretory and membrane proteins (Helenius and Aebi, 2004). The oligosaccharyltransferase (OST) catalyzes the transfer of the oligosaccharide from the lipid donor dolichylpyrophosphate to the acceptor protein. In yeast, eight different membrane proteins have been identified that constitute the complex in vivo (Kelleher and Gilmore, 2006). STT3 is thought to represent the catalytic subunit of the OST (Yan and Lennarz, 2002; Nilsson et al, 2003). It is the most conserved subunit in the OST complex (Burda and Aebi, 1999).
Within recent years, it has been shown that the food-borne bacterial pathogen Campylobacter jejuni contains a general protein glycosylation system (Szymanski et al, 1999). The machinery required for glycosylation is encoded by 12 genes clustered in the so-called pgl locus (for protein glycosylation; Linton et al, 2005). The Pgl enzymes synthesize a heptasaccharide, GalNAc-α1,4-GalNAc-α1,4-(Glc-β1,3)-GalNAc-α1,4-GalNAc-α1,4-GalNAc-α1,3-Bac, where Bac is 2,4-diacetamido-2,4,6-trideoxy-D-Glc (Young et al, 2002) on a lipid carrier undecaprenylpyrophosphate (Feldman et al, 2005). The heptasaccharide is transferred to asparagine side chains present in a tripeptide consensus sequence of the type N-X-S/T (where X can be any amino acid except proline) by the bacterial OST, PglB (Nita-Lazar et al, 2005). Some biological functions of protein glycosylation in bacteria have been identified. Disruption of N-glycosylation in C. jejuni diminishes not only immunogenicity of several glycoproteins but also host cell adherence and invasion in vitro, and colonization of mice and chicks (Szymanski et al, 2002; Hendrixson and DiRita, 2004; Karlyshev et al, 2004).
The goal of this study was to elucidate the C. jejuni pgl system and to compare it to the eukaryotic system. It is known from eukaryotic N-glycosylation that biosynthesis, translocation and in particular folding of acceptor proteins strongly influence glycosylation (Pless and Lennarz, 1977; Chen and Helenius, 2000). Thus, we decided to address the substrate requirements for the bacterial N-glycosylation in vivo using native proteins in order to identify requirements that are not detained by short peptide acceptors used in vitro (Glover et al, 2005). From truncation and mutation experiments with the C. jejuni protein AcrA expressed in Escherichia coli strains bearing the pgl system together with analyses of native glycoproteins from C. jejuni, we found that bacterial N-glycoproteins require a negatively charged side chain at the −2 position to the glycosylated Asn for N-glycosylation to occur, resulting in the stringent acceptor sequence D/E-Y-N-X-S/T (Y, X≠P). This consensus pentapeptide was required but not sufficient for N-glycosylation in E. coli, whereas glycosylation site selection in yeast was independent of the negatively charged residue at the −2 position.
Results
What are the signals in the AcrA protein that trigger glycosylation?
In order to analyze the structural requirements of the acceptor protein for bacterial N-glycosylation, we performed studies on the C. jejuni N-glycoprotein, AcrA (Parkhill et al, 2000; Wacker et al, 2002), a periplasmic lipoprotein. The N-glycosylation sites of this 350 amino acid residue protein are identified (Nita-Lazar et al, 2005), and the crystal structure of a homologue of this protein, MexA from Pseudomonas aeroginosa, had recently been determined (Akama et al, 2004; Higgins et al, 2004). The structure of MexA shows three ordered domains, a β barrel hybrid domain, a lipoyl domain, and a coiled coil domain. As the MexA and AcrA proteins share 29% sequence identity and 50% similarity, and also belong to the periplasmic efflux protein family of proteins predicted to exhibit similar structures (Johnson and Church, 1999), we reasoned that both proteins feature a similar overall fold.
To find the minimal requirements in an acceptor protein for glycosylation, we aimed to identify the smallest domain of the AcrA protein that could be glycosylated. As was previously determined, the hypothetical coiled coil domain of AcrA contains three potential N-glycosylation sites, of which only the site at N123 is used (Nita-Lazar et al, 2005). We constructed a plasmid coding for the lipoyl and the glycosylation site containing coiled coil domains of AcrA (named ‘Lip', corresponding to residues D60-D210 of the mature AcrA polypeptide sequence), fused it N terminally to the signal sequence of OmpA (Choi and Lee, 2004) and C terminally to a hexa-His tag. The Lip domain sequence was chosen based on the domain margins of the lipoyl domain part in the crystal structure of MexA and a sequence alignment of MexA and AcrA (Figure 1). We further constructed seven truncated forms of Lip, stepwise deleting the residues flanking the natural glycosylation site at N123 without affecting the lipoyl domain sequence in Lip (Figure 2A). The peptide stretches of the different truncation variants containing the glycosylation site at N123 tested in this way are indicated in brackets, according to their numbering in the native AcrA polypeptide (Figure 2A). In some recombinant proteins, we also replaced K131 with Gln (K131Q) to reduce the extent of proteolytic cleavage by protease specifically acting between positions K131 and S132 (data not shown).
Figure 1.
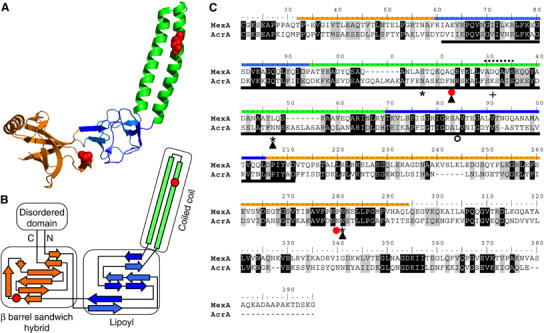
The structural homology of MexA and AcrA. (A) Crystal structure of the MexA protein (adapted from Higgins et al, 2004). Colors indicate the different domains. Red spheres show a space fill representation of the residues in MexA corresponding to the glycosylated N123 and N273 of AcrA based on the homology alignment (C). (B) Scheme of the secondary structure elements in MexA. Orange is the β barrel sandwich hybrid domain. It is connected to an unordered part of the protein containing the N and C termini. Blue: lipoyl domain. Green: coiled coil. Note the overall antiparallel organization of the polypeptide. Red circles indicate the residues in MexA, which align to N123 and N273 (C). (C) Polypeptide alignment of signal peptide processed MexA and AcrA amino acid sequences. The ruler is adjusted to the AcrA amino acid numbering. Colored lines indicate the sequences constituting the domains in MexA (see A, B). The dashed line designates the loop connecting the ascending and descending strands of the coiled coil in MexA. The black line below indicates the part of AcrA constituting the Lip sequence (see Figure 2); the red circles show the naturally glycosylated N123 and N273 of AcrA, the asterisk and the empty circle discriminate Asn residues in additionally introduced glycosylation sites that were active (N117, N145, N274) or inactive (N184, see Figure 3E). Filled triangles indicate glycosylation sites, which were glycosylated in the yeast expression system (see Figure 6). + shows the residue in the protease sensitive loop that was replaced by Gln (K131Q, see Figure 3). Black and gray shading shows identical and similar amino acid residues according to the BLOSUM62 substitution matrix. Note that the AcrA sequence corresponding to the coiled coil is longer than the homologous stretch of MexA.
Figure 2.
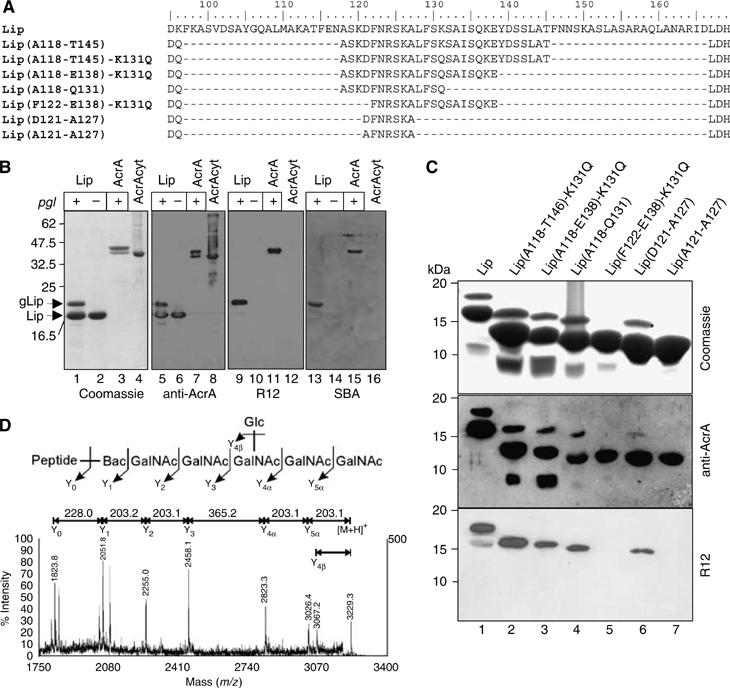
Glycosylation analysis of truncated forms of AcrA. (A) Sequence details of the hypothetical coiled coil domain (see Figure 1, the green domain) present in Lip and truncated forms thereof, which were tested as N-glycan acceptors in E. coli. The ruler indicates the amino acid numbering of the corresponding residues as in the AcrA sequence. Residues D95 and L167 belong to the predicted lipoyl domain and flank the sequence of the coiled coil that include the glycosylation at N123. The peptide stretches of the different Lip truncation variants containing the glycosylation site at N123 are indicated in brackets, according to the numbering in the AcrA sequence. According to this nomenclature, the full length, nontruncated Lip protein is Lip(K96-D166). Note that some residues were mutated due to cloning reasons or to render the protein more resistant to proteolytic cleavage (K131Q). (B) Characterization of the glycosylated lipoyl coiled coil domain of AcrA (Lip). SDS–PAGE analysis of four different Ni2+ affinity purified protein fractions by Coomassie Brilliant Blue staining (left panel), immunoblotting with anti-AcrA antiserum (middle left), R12 antiserum (middle right), or HRP-coupled SBA (right panel). Lip was expressed in presence (+, lanes 1, 5, 9, and 13) and absence (−, lanes 2, 6, 10, and 14) of a functional pgl locus, soluble AcrA only in its presence (+, lanes 3, 7, 11, and 15). Unglycosylated AcrA was purified from the cytoplasm (lanes 4, 8, 12, and 16; Nita-Lazar et al, 2005). Note that the glycosylated proteins are detected with the R12 antiserum and SBA. (C) Glycosylation analysis of truncated forms of Lip. Proteins were Ni2+ affinity purified from periplasmic extracts of Top10 cells expressing the corresponding proteins as indicated above the gel frame and analyzed by SDS–PAGE and immunoblotting. Top gel: Coomassie stained; middle gel: anti-AcrA; bottom: R12. (D) MALDI-MS/MS of m/z=3229.3 from the in-gel trypsinized protein band labeled with a star in panel C. The mass corresponds to the expected glycopeptide derived from the Lip(D121-A127) protein (GQTLFIIEQDQDFN123R). The inset shows the C. jejuni N-glycan attached to the peptide and the corresponding fragmentation pattern.
The Lip and the Lip truncation mutants were expressed in glycosylation proficient E. coli Top 10 cells bearing a plasmid containing the wild-type pgl locus from C. jejuni (pACYCpgl). As a negative control for glycosylation, we used cells carrying a plasmid with an inactive PglB (pACYCpglmut; Wacker et al, 2002). Recombinant proteins were purified from periplasmic extracts by Ni2+ affinity chromatography and analyzed by SDS–PAGE, and subsequent Coomassie staining or immunoblotting (for a typical purification see Supplementary Figure S1). Ni2+ affinity purified fractions from cells expressing Lip showed a major protein at a mass of 17 kDa and an additional protein band at a mass of 18.5 kDa when the wild-type pgl locus was present in the cells (Figure 2B, lane 1). Both of the proteins contained the same N terminus (D60VII…, data not shown), and they reacted with anti-AcrA antiserum (lanes 5 and 6) as did a full-length cytoplasmically expressed, nonglycosylated (lanes 4 and 8) or periplasmically expressed, glycosylated AcrA protein (lanes 3 and 7). As the 18.5 kDa protein was not present in extracts from glycosylation deficient cells (lanes 2 and 6), we hypothesized that this additional protein may be the singly glycosylated form of Lip. To confirm this, we tested the proteins reactivity towards R12 antiserum (lanes 9–12). This serum was raised against C. jejuni whole-cell extracts and has been shown to detect preferentially C. jejuni N-glycoproteins (Wacker et al, 2002). We also used the GalNAc-specific lectin soybean agglutinin (SBA), which recognizes the terminal GalNAc residues present on the C. jejuni N-glycans (Young et al, 2002; Linton et al, 2005). The 18.5 kDa band reacted with R12 antiserum as well as with SBA (lanes 9 and 13). Fragmentation analysis of tryptic peptides derived from the 18.5 kDa protein by MALDI-TOF/TOF confirmed the presence of an N-linked C. jejuni glycan attached to the peptide DFN123R (not shown).
The truncated forms of Lip expressed in glycosylation competent cells were analyzed in a similar manner. The decreasing size of the major protein species detected by SDS–PAGE in each elution fraction reflected the stepwise reduction of the proteins as expected (Figure 2C, lanes 2–6). With the exception of the Lip(F122-E138)-K131Q, all of the samples contained an additional slower mobility band. These proteins reacted with the anti-AcrA antiserum as well as with the R12 serum suggesting glycosylation. We performed MALDI-TOF/TOF analysis of in-gel trypsinized, R12-reactive protein derived from the Lip(D121-A127) expression. The MS/MS spectrum of m/z=3229.3 demonstrated fragmentation behavior indicative of the C. jejuni N-glycan structure (Figure 2D). A complete Y-ion series was observed down to the nonglycosylated peptide GQTLFIIEQDQDFNR (m/z=1824), which encompasses the Asn residue of the glycosylation site at N123 (underlined). These results demonstrated that, with the exception of Lip(F122-E138)-K131Q, all of the truncation variants were glycosylated. Comparing the primary sequence of the nonglycosylated Lip(F122-E138) protein and the glycosylated Lip(D121-A127) protein, we speculated that the residue D121 missing in the former construct is essential for glycosylation by PglB (see Figure 2A). To address the role of D121 in glycosylation, we mutated D121 to A in the Lip(D121-A127) protein and tested the resulting protein in the in vivo glycosylation assay (Figure 2C, lane 7). Lip(A121-A127) was not glycosylated showing that the Asp residue in the −2 position was essential for glycosylation.
The D121A mutation disrupts glycosylation at N123 in AcrA
To confirm the importance of the Asp at the −2 position for N-glycosylation, we analyzed the effect of different mutations in the full-length AcrA protein. For protein production and glycosylation, we used the strain CLM24 (Feldman et al, 2005) bearing the plasmid-borne pgl locus (Wacker et al, 2002) and a plasmid for the expression of soluble AcrA containing the PelB signal peptide for secretion and a C-terminal hexa-His tag (Nita-Lazar et al, 2005). The glycosylation status of the periplasmic AcrA was analyzed by SDS–PAGE and immunoblotting with anti-AcrA or R12 antisera. Cells containing the functional pgl locus produced three proteins reactive towards the anti-AcrA antiserum (Figure 3A, lane 2), which were respectively un-, mono- and diglycosylated forms of AcrA as shown by their reactivity towards the R12 antiserum (Figure 3A, R12, lane 2). Residual reactivity of the R12 antiserum towards nonglycosylated AcrA was also detected as it has been observed before (Wacker et al, 2002). The mutation N123Q inactivated the glycosylation site at N123 and resulted in only two protein bands in anti-AcrA immunoblots. The R12 antiserum detected monoglycosylated AcrA only (lane 3). The double mutant, AcrA-N123,273Q, produced a single band reactive towards anti-AcrA, but no signal was observed with R12 antiserum showing that glycosylation was absent despite the presence of a functional pgl machinery (lane 4). When a D121A mutation was made, only a single glycosylated AcrA protein was detected in glycosylation competent cells (lane 6). This result confirmed our finding that the Asp at the position −2 to the modified Asn represents a novel determinant for pgl system N-glycosylation. Replacement of D121A by a Glu residue rescued the glycosylation negative phenotype and resulted in un-, mono- and diglycosylated AcrA (lane 8). Hence, an amino acid with a negatively charged side chain at position −2 was sufficient for glycosylation at N123.
Figure 3.
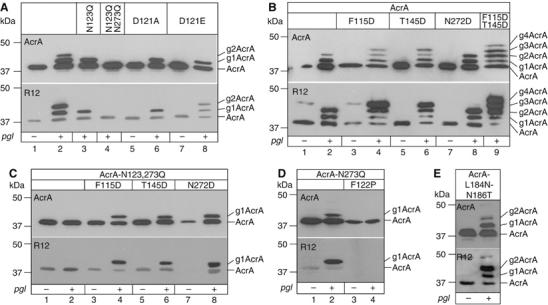
SDS–PAGE analysis of N-glycosylation site mutants of AcrA. All top panels are immunoblots probed with anti-AcrA antiserum, bottom panels represent identical samples detected with the C. jejuni N-glycan specific R12 antiserum. + and − indicate the presence of pACYCpgl or pACYCpglmut in the cells. All samples contained a plasmid expressing soluble AcrA with an N-terminal signal peptide. The different point mutations in the AcrA protein are indicated. Numbers on the left of the gel frame show the electrophoretic mobility of the molecular weight marker. The numbers of N-glycans in the different glycoforms of AcrA are indicated at the right of the gel frames. (A) Analysis of the naturally used glycosylation sites N123 and N273. (B) Analysis of artificially introduced glycosylation sites at positions N117, N147, and N274, in the presence of active natural glycosylation sites. (C) Analysis of artificially introduced glycosylation sites at positions N117, N147, and N274, in the absence of other glycosylation sites. (D) Pro in position −1 of the natural glycosylated N123 abolishes glycosylation. (E) A sequon that was mutated into the putative lipoyl domain of AcrA was not active.
Engineering additional glycosylation sites into AcrA
We tested the activation of all three cryptic N-X-S/T sequences within the AcrA polypeptide for glycosylation by introducing an Asp residue at the −2 position. Accordingly, we mutated the residue in the −2 position of sites N117, N147, and N274 (F115, T145, and N272) of wild-type AcrA to Asp. The mutations were placed either into the wild-type AcrA or into the glycosylation inactive AcrA-N123,273Q. Expression of AcrA-F115D in glycosylation competent E. coli cells led to the detection of four proteins with anti-AcrA antiserum (Figure 3B, lane 4). The slowest migrating protein triggered the strongest signal of all four bands with the R12 antiserum indicating the presence of multiple C. jejuni N-glycans. Based on the electrophoretic mobilities in SDS–PAGE, we concluded that the F115D mutation had resulted in an AcrA protein with three glycosylation sites. The AcrA-T145D also generated triply glycosylated AcrA (lane 6). When both mutations were introduced in the same AcrA protein (AcrA-F115D-T145D), five proteins reactive towards the anti-AcrA antiserum were detected (lane 9). The protein with the slowest mobility triggered a strong signal with the R12 antiserum despite the weak staining observed with anti-AcrA antiserum, and it appeared larger than AcrA-T145D or AcrA-F115D on SDS–PAGE. Taken together, these data indicated that the AcrA-F115D-T145D protein was glycosylated at all four sites. A different result was obtained with the third cryptic site of AcrA, N274. Expression of AcrA-N272D did not trigger the detection of an additional glycosylation. In fact, the glycosylation pattern was similar to the one obtained with wild-type AcrA (compare lanes 8 and 2). Due to our assay, we are not able to judge if the site N273 or N274 was glycosylated in the AcrA-N272D protein. It is likely that glycosylation of the native site N273 inhibited modification of N274 – or vice versa – due to spatial restrictions.
Next, we tested if the introduced N-glycosylation sites were used in absence of other sites within the same protein (Figure 3C). Therefore, all three point mutations were individually inserted into the AcrA-N123,273Q double mutant protein (lanes 1 and 2). Periplasmic extracts of glycosylation competent cells expressing either AcrA-N123,273Q-F115D (lane 4), AcrA-N123,273Q-T145D (lane 6), or AcrA-N123,273Q-N272D (lane 8) contained a singly glycosylated, anti-AcrA-reactive protein. We concluded that glycosylation reactions occurring on the same protein are self-sufficient. The absence of triply glycosylated AcrA-N272D represented an exception to this rule. Probably, two adjacent Asn residues participating in overlapping glycosylation sites cannot be modified within the same molecule.
Proline at position −1 inhibits N-glycosylation
A Pro residue between the Asn acceptor and the hydroxyamino acid of the N-X-S/T consensus sequon inactivates the consensus site (Bause et al, 1995; Nita-Lazar et al, 2005). We observed that a Pro residue between the acidic side chain residue at the −2 position and the Asn acceptor abolished glycosylation in E. coli as well (Figure 3D). Expression of AcrA-N273Q in glycosylating cells produced un- and monoglycosylated AcrA (lane 2), whereas the AcrA-N273Q-F122P protein was not glycosylated at all (lane 4).
The bacterial consensus sequence is required but not sufficient for glycosylation
We also identified a region in the AcrA protein where it was not possible to generate an active glycosylation site. The mutant AcrA-L184N-N186T was constructed such that it contained the glycosylation site (182DANVT186) in the sequence constituting the predicted lipoyl domain (see Figure 1). As shown in Figure 3E, there is no additional, glycosylated AcrA detectable when compared to the wild-type protein. Likewise, three periplasmic E. coli proteins that contained the consensus sequon in their native sequence could not be glycosylated in E. coli cells containing the pgl machinery (see Supplementary Figure S2). Thus, the consensus sequence alone is not sufficient for bacterial N-glycosylation.
In contrast, N-glycosylation of a heterologous protein was achieved with a form of the Cholera toxin B subunit (CtxB) bearing the N-terminal signal sequence of the OmpA protein and a C-terminal hexa-His tag. A bacterial N-glycosylation consensus sequence was created by introducing the point mutation W88D, generating the sequence 88DNNKT92. When expressed in the presence of the functional pgl locus, CtxB-W88D produced an additional band (Figure 4A). This slower migrating protein was glycosylated as shown by its reaction with R12 antiserum. Although N-glycosylation was inefficient, these results demonstrated site-directed glycosylation of a heterologous protein in E. coli.
Figure 4.
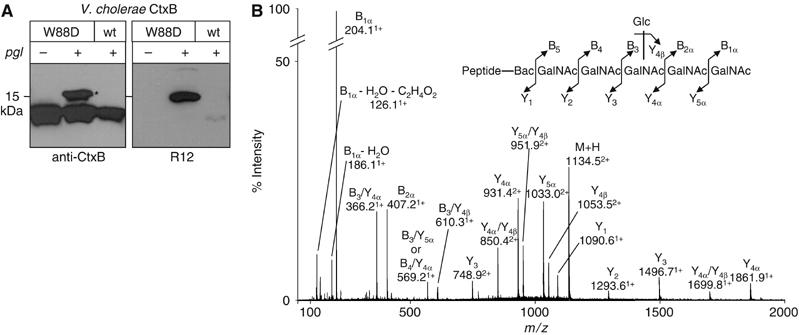
Bacterial N-Glycosylation of a non-C. jejuni protein. (A) Periplasmic extracts derived from cells expressing wild-type CtxB and CtxB-W88D were analyzed by immunoblotting with anti-CtxB antiserum (left panel) and R12 (right panel). Expression was performed in SCM7 E. coli cells containing either the glycosylation competent (+) or incompetent (−) pgl machinery on a plasmid. (B) Ni2+ purified, in-gel trypsinized protein corresponding to the glycosylated CtxB-W88D (marked with an asterisk in (A)) was analyzed by NanoESI-MS/MS. The fragmentation spectrum of the doubly charged pseudomolecular ion at m/z=1134.5, corresponding to the tryptic CtxB glycopeptide bearing the C. jejuni N-glycan is shown. The inset illustrates the C. jejuni N-glycan attached to the expected peptide, with sequential losses of HexNAc (203 Da) and Hex (162 Da) residues confirming the expected structure.
C. jejuni glycoprotein sites contain Asp or Glu residues in the −2 position
Six N-glycosylation sites from C. jejuni were identified previously by glycopeptide analyses of C. jejuni glycoproteins by mass spectroscopy (Young et al, 2002). Additional glycopeptide sequences were now obtained from glycoproteins in membrane extracts of C. jejuni cell cultures. Glycoproteins from inner membrane extracts were purified by SBA affinity chromatography and SDS–PAGE (Supplementary Figure S3) followed by in-gel tryptic digestion and NanoLC-MS/MS. The N-glycosylation sites were identified by the N-glycan signature found attached to their predicted peptide masses. The majority of the newly identified proteins were annotated as membrane proteins in the Campylobacter genome database, but their functions are not known (see Supplementary Table SI). The glycosylation sequons determined here plus the previously reported ones and other glycoprotein sites located by mutation experiments are listed in Table I. Only Asp and Glu were present at the −2 position (Figure 5). There were equal numbers of sequons with Thr or Ser at +2 and more containing Asp than Glu at −2. Pro is not only absent from position +1 as in eukaryotic N-glycosylation sites (Gavel and von Heijne, 1990) but also from position −1. Asp showed a preference for hydrophobic residues at −1 (Supplementary Figure S4). The over-abundance of Lys in the glycopeptide set is probably due to the superior behavior of smaller tryptic peptides in the mass spectrometry.
Table 1.
Alignment of glycosylated sequons found in C. jejuni
| Cj gene | Annotationa | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cj0081 | cydA | K | L | A | K | E | N | N | D | T | I | A | M | A |
| Cj0114 | pppb | N | Q | N | I | E | N | N | F | T | S | E | I | Q |
| Cj0152c | pmp | V | K | E | K | D | F | N | I | S | L | D | K | D |
| F | F | K | D | E | Q | N | N | T | S | A | T | V | ||
| Cj0200c | pppb | A | Q | I | Y | D | N | N | K | T | L | L | I | D |
| Cj0289c | Peb3b | D | F | G | K | D | F | N | V | S | K | I | K | P |
| Cj0313 | pmp | F | N | I | N | D | L | N | L | S | S | F | A | S |
| Cj0367c | AcrAc | K | A | I | F | D | N | N | N | S | T | L | L | P |
| N | A | S | K | D | F | N | R | S | K | A | L | F | ||
| Cj0397c | hp | E | N | I | N | D | F | N | N | T | T | L | Q | N |
| Cj0399 | pmp | D | I | V | S | D | L | N | N | T | Q | K | G | E |
| Cj0414 | CmeCd | L | N | I | P | E | A | N | Y | S | I | D | N | K |
| S | W | E | K | E | T | N | S | S | I | T | K | N | ||
| Cj0530 | ppp | L | N | F | E | D | F | N | A | S | V | N | D | A |
| Cj0599 | ppp | Q | L | S | K | D | L | N | S | T | L | D | N | K |
| Q | S | K | L | D | N | N | I | T | I | D | E | K | ||
| K | A | E | L | E | A | N | I | T | N | Y | K | Q | ||
| Cj0610c | ppp | T | L | I | Q | D | A | N | I | S | F | I | D | N |
| Cj0648 | hp | L | K | A | Y | E | S | N | T | S | I | I | K | A |
| K | V | L | F | E | G | N | V | T | Y | I | G | S | ||
| Cj0734c | HisJc | S | K | N | K | E | S | N | A | S | V | E | L | K |
| Cj0958c | pmp | P | L | K | I | E | Q | N | I | T | Q | K | N | Q |
| N | L | F | V | D | E | N | G | S | Q | V | L | K | ||
| Cj0982c | ppp | A | V | P | K | D | S | N | I | T | S | V | E | D |
| Cj1053c | pmp | I | L | C | K | D | I | N | V | S | K | A | Y | F |
| Cj1126c | PglBe | A | M | M | K | D | Y | N | Q | S | N | V | D | L |
| Cj1621 | ppp | T | Y | S | L | D | L | N | K | T | C | V | L | K |
| Cj1670c | CgpAb | P | F | K | T | D | Q | N | I | T | L | V | A | P |
| V | T | I | P | E | K | N | S | S | K | Q | E | S | ||
| S | S | K | Q | E | S | N | S | T | A | N | V | E | ||
| VirB10f | D | Q | T | S | E | E | N | V | S | K | N | I | S | |
| K | K | E | E | D | N | N | I | T | K | L | A | K | ||
| aHp, hypothetical protein; pmp, putative membrane protein; ppp, putative periplasmic protein. | ||||||||||||||
| bYoung et al (2002). | ||||||||||||||
| cNita-Lazar et al (2005). | ||||||||||||||
| dM Pos, University of Zürich, personal communication. | ||||||||||||||
| eData not shown. | ||||||||||||||
| fLarsen et al (2004). | ||||||||||||||
| Letters in bold indicate amino acids in positions –2, 0, and +2 of the predicted C. jejuni N-glycosylation site. | ||||||||||||||
Figure 5.

Statistical analysis of active N-glycosylation sites found in native C.jejuni glycoproteins. Fractional occurrence of amino acids in the region from −6 to +6 of the glycosylated Asn residue (32 sequons). The figure was prepared from the data of Table I, by means of WebLogo (Crooks et al, 2004). A full color version of this figure is provided in Supplementary Figure S4.
N-Glycosylation of AcrA in Saccharomyces cerevisiae
In eukaryotic glycosylation sites, amino acids with an acidic side chain are disfavored at the −2 position (Petrescu et al, 2004). To check if the acidic residue also determines AcrA site usage in eukaryotes, we analyzed glycosylation of the AcrA protein by the eukaryotic OST from S. cerevisiae. An expression plasmid was constructed that contained the sequence coding for the soluble part of the AcrA protein fused to the α mating type factor signal sequence for targeting and transport to the ER. The recombinant gene was expressed under the control of the constitutive TEF promoter (Mumberg et al, 1995). Glycoproteins were isolated from cell lysates by lectin affinity purification using Concanavalin A (ConA). Purified samples were separated by SDS–PAGE and AcrA protein was detected by the specific antiserum (Figure 6). ConA-purified extracts derived from a yeast strain expressing the wild-type AcrA protein resulted in a ladder of three protein bands when probed with the AcrA antiserum. When the same sample was treated with EndoH prior to SDS–PAGE, the signal collapsed to a single band, which appeared at a smaller molecular mass than the fastest migrating band of the untreated sample. We concluded that AcrA was partially glycosylated in S. cerevisiae, with a maximum of three N-glycans present on the protein. To identify which of the five potential N-X-S/T glycosylation sequons was used, mutant forms of AcrA, which had one of the putative Asn acceptor residues changed to Leu, were expressed and analyzed. Mutagenesis of Asn residues N123, N147, and N274 to Leu reduced the number of glycoforms to two, whereas the N117L or N273L mutations resulted in a wild-type glycosylation pattern. We concluded that in yeast, N-glycosylation of AcrA occurred at positions N123, N147, and N274, whereas N123 and N273 were utilized by the prokaryotic OST.
Figure 6.
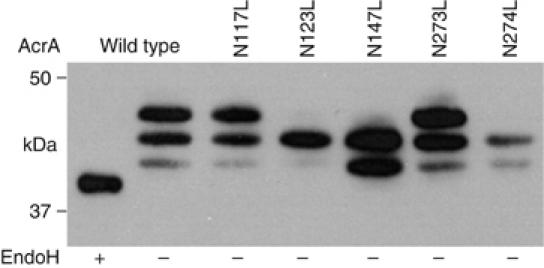
AcrA N-glycosylation site usage in yeast. AcrA was expressed in yeast cells with an N-terminal signal sequence for secretion. Shown are ConA lectin-purified fractions from cells expressing either wild-type AcrA or glycosylation site point mutants as indicated above the gel frame to analyze the site usage. EndoH treatment (+) was performed to show that all three bands obtained with wild-type AcrA contained one or more high-mannose N-glycans.
Discussion
We determined that the amino acid sequence D/E-X-N-Y-S/T, where X and Y can be any amino acid except Pro, is required for efficient glycosylation of an acceptor protein by the C. jejuni pgl machinery in living cells of E. coli or C. jejuni. In one instance, the introduction of a bacterial consensus site alone was shown to direct N-glycan attachment to a recombinant non-C. jejuni acceptor protein in vivo. The role of the acidic amino acid residue appeared to be specific for the recognition of N-glycosylation sites in bacteria but not in the eukaryote S. cerevisiae.
Similarity of the bacterial PglB and the eukaryotic central unit of the OST, STT3, has suggested a common evolutionary origin of the two general N-glycosylation systems. The enlarged data set now containing 32 active C. jejuni N-glycosylation sites allowed us to compare the acceptor protein sequences around N-glycosylation sites found in bacteria and eukaryotes. With the striking exception of the Asp/Glu at position −2, residues found around C. jejuni glycosylation sites are generally similar to those from bioinformatic (Ben-Dor et al, 2004; Petrescu et al, 2004) and experimental (Shakin-Eshleman et al, 1996; Mellquist et al, 1998) analyses of eukaryotic N-glycosylation sites. At positions −2 and −1, aromatic residues are favored and, remarkably, Asp or Glu disfavored in eukaryotes (Ben-Dor et al, 2004). The Campylobacter system also showed a high occurrence of hydrophobic residues at position −1 when Asp was at −2 (see Supplementary Figure S4), less of the acidics, and never Pro at position +1 as in eukaryotes (Gavel and von Heijne, 1990). Although the quantification of glycosylation efficiency by immunoblot analysis is inaccurate, our data from Coomassie staining (Figure 2C) show that the native, full-length AcrA protein is an optimal glycosylation substrate, whereas truncated versions of this protein or the nonglycosylated CtxB-W88D are suboptimal substrates for PglB, suggesting a role for the protein conformation in the reaction.
That the structure of the acceptor protein must be compatible and specific for glycosylation to occur was suggested by the complete absence of Pro from the −1 position. C. jejuni proteins and domains have co-evolved with the pgl machinery, and thus are tuned for accepting an N-glycan. This is not the case for native E. coli proteins. Although the primary sequence requirements are fulfilled in several secreted proteins, their potential glycosylation sites may be located within domains, which do not allow changes in local conformation necessary for N-glycosylation by PglB. This supports the hypothesis that unfolding or flexibility is required for protein domains to be compatible with glycosylation. We propose that the conformation of the protein domain bearing the acceptor site must be compatible with an OST-induced conformational change leading to the activated structure. Structural analysis of used and not used glycosylation sequons in the absence and presence of a functional OST will be needed to test this hypothesis. Within the framework of this model, it is evident that rigid, terminally folded protein domains have therefore a lower potential to serve as glycan acceptors than thermodynamically less stable folding intermediates.
Nevertheless, engineering of an acceptor site into the CtxB framework resulted in glycosylation of this protein, albeit with low efficiency (Figure 4A). Compatible acceptor structures may preferably reside in loops or flexible domains rather than within the core of the glycoprotein as suggested by the crystal structures of CtxB (Zhang et al, 1995) and OmpH1 (Muller et al, 2005). Whereas the former protein had to be modified for glycosylation to occur (CtxB-W88D, Figure 4), the OmpH1 (Cj0982c) protein is a native C. jejuni N-glycoprotein (see Table I and Supplementary Table SI). Both of these glycosylation sites are located in loops, suggesting that exposure and probably flexibility are two acceptor site characteristics.
Glycosylation of engineered sites by insertion of the bacterial consensus sequence illustrates that the pgl system of C. jejuni is a general protein modification system like its eukaryotic equivalent. The extended acceptor sequence suggests an increased stringency and specificity for the polypeptide acceptor, compared to the eukaryotic system. Indeed, our results obtained from the expression of the AcrA protein in yeast showed that the eukaryotic N-glycosylation system selected one glycosylation site more than the prokaryotic system. Site N123 was used by both glycosylation machineries, excluding the possibility that an Asp residue at the −2 position was not compatible with eukaryotic glycosylation. Of particular interest was the glycosylation of the sequence 271DNNNST276 in the bacterial and eukaryotic hosts. The Asp at position 271 determined the use of N273 in E. coli; however, in yeast, glycosylation occurred at position N274. Site N273 was also not used when N274 was mutated to Leu in the eukaryote (Figure 6), whereas the N274 site was activated in the bacterial system by introducing an Asp at position 272 (Figure 4). These results are best explained by a model proposing an active conformation of the acceptor peptide common to the bacterial and eukaryotic acceptor sites. Such a conformation might be induced by the binding of the OST to the substrate. However, the two systems differ in the recognition process of the acceptor site. In prokaryotes, an Asp/Glu residue at the −2 position is always required, whereas in the eukaryotic system, the site selection is less sequence specific, but might require a precise location of the acceptor sequon within a protein domain. This altered substrate specificity of the eukaryotic OST correlates with additional subunits in the eukaryotic system (Jones et al, 2005), suggesting defined roles of some of these subunits in the substrate recognition process (Wilson et al, 2005). The observation of an increased stringency and specificity for the protein acceptor in the bacterial system is in sharp contrast to the other substrate of the reaction, the lipid-linked oligosaccharide. In the C. jejuni system, a wide variety of bactoprenylpyrophosphate-linked oligosaccharides can serve as substrates (Feldman et al, 2005), whereas most eukaryotic systems prefer the Glc3Man9GlcNAc2 oligosaccharide (Burda and Aebi, 1998).
In eukaryotes, several monosaccharides in the glycan are thought to play signaling roles in coordinating the folding and quality control of proteins in the ER. The broad acceptor site specificity and the high stringency for a defined oligosaccharide structure makes the N-glycan a versatile, flexible molecular signal that can be attached to many different positions of secretory proteins (Helenius and Aebi, 2004). To increase the flexible use of such a polyvalent regulatory molecule eukaryotes have evolved a temporal separation of N-glycosylation and folding. During cotranslational translocation, secretory and membrane proteins are glycosylated when the nascent protein emerges from the translocon pore (Whitley et al, 1996). Thus, N-glycosylation occurs before or during domain folding in the ER (Kowarik et al, 2002), in a condition when the protein is still flexible allowing OST-induced conformational changes (Bulleid et al, 1992).
The establishment of recombinant protein glycosylation in E. coli is an important step towards the production of pharmacologically and commercially important N-glycoproteins in E. coli. In combination with the recent advances in engineering the N-glycan structure, the definition of the larger sequon opens the possibility of producing novel glycoproteins of biotechnological interest.
Materials and methods
Construction of plasmids
Template DNA for acrA gene derivatives was from the C. jejuni strain 81–176. acrA is also named cmeA. The E. coli expression vector backbone was the pBR322 based vector pEC415 (Schulz et al, 1998) for AcrA, Lip variants, and CtxB. The lip protein sequence (Lip, plasmid pMIK8) was amplified from pET24(AcrA) (Wacker et al, 2002). All point mutations were introduced with the QickChange Kit from Stratagene. Expression plasmids for Lip variants were constructed starting from pMIK8. The plasmid was modified and restricted to remove the sequence coding for the putative coiled coil domain. Pairs of 5′ phosphorylated, complementary oligonucleotides served as inserts to produce the Lip variant genes (see Figure 2A). The signal peptides for AcrA and AcrA point mutants were from PelB, for CtxB and Lip variants from OmpA. Insertion of a hexa-His tag in ctxB was carried out by QickChange mutagenesis. For the expression of AcrA in yeast (pSN9), acrA was amplified from pET24-AcrA by polymerase chain reaction (PCR) with oligonucleotides introducing the α mating factor signal sequence at the N terminus and a hexa-His tag to the C terminus. This fragment was cloned into the multiple cloning site of pRS426-TEF (Mumberg et al, 1995). Expression vectors for AcrA mutated in the glycosylation sites were constructed as follows: a segment of acrA containing the mutation to a glycosylation site was PCR amplified from the expression vectors pET-24-AcrA-Asn1, pET-24-AcrA-Asn2, pET-24-AcrA-Asn3, pET-24-AcrA-Asn4.1, and pET-24-AcrA-Asn4.2 (Nita-Lazar et al, 2005) and ligated into pSN9 by the use of appropriate restriction enzymes. The resulting plasmids—pSN11, pSN12, pSN13, pSN14 and pSN15—encode for AcrA with the point mutation N117L, N123L, N147L, N273L or N274L, respectively. All vectors were confirmed by restriction analysis and sequencing (Microsynth, Balgach, Switzerland) of the entire acrA cDNA. All of the used strains and plasmids are listed in Supplementary Table SII. Oligonucleotide sequences are available on request; details of the clonings are described in the Supplementary Materials and methods section.
Protein expression, purification, and analysis (E. coli)
E. coli CLM24, SCM7 (Alaimo et al, 2006) or Top10 cells bearing pACYCpgl or pACYCpglmut, and a plasmid coding for an acceptor protein were grown in volumes of 0.5–2 l to an OD of 0.5 at 37°C in LB medium containing ampicillin and chloramphenicol. For induction, arabinose was added to 0.02% (w/v) and cultures were grown for 2–3 h. Proteins in periplasmic extracts were purified by Ni2+ affinity chromatography and/or analyzed as described (Feldman et al, 2005). For the separation of glycosylated from nonglycosylated protein (see Supplementary Figure S1), eluates of the Ni2+ purification were directly loaded onto a 1 ml column packed with SBA agarose resin (Vector Laboratories Inc., Burlingame, CA, USA). After washing with PBS, bound protein was eluted from the SBA agarose column with PBS containing 0.5 M galactose. SDS–PAGE was performed according to Lämmli, immunodetection was performed with antisera anti-AcrA, R12 (Wacker et al, 2002), anti-OppA (Igarashi et al, 1997), anti-CtxB-HRP (ViroStat, Portland, USA), and anti-C terminal hexa-His tag (Invitrogen, Paisley, UK).
C. jejuni cultivation and protein preparation
For the isolation of C. jejuni N-glycoproteins, strain NCTC11168 was grown on Mueller–Hinton agar under microaerophilic conditions (10% CO2, 5% O2, 85% N2) at 37°C. Cells from two plates of overnight growth were resuspended in 5 ml of Mueller–Hinton broth and used to inoculate 1.5 l of this culture medium. Cultures were grown under microaerophilic conditions at 37°C for 24 h with shaking at 150 r.p.m. Bacterial cells from 3 l of culture media were harvested by centrifugation at 10 000 g for 15 min and immediately frozen at −75°C. Frozen cell pellets were thawed on ice in 10 mM HEPES buffer, pH 7.5 and broken by mechanical disruption in the presence of protease inhibitors. Extracts were clarified by centrifugation at 27 000 g for 30 min and cell debris discarded. Membrane protein fractions were obtained from clarified cell extracts by centrifugation at 100 000 g for 1 h. Proteins from the inner membrane were extracted by 1% (w/v) sarcosine treatment at 4°C for 15 min followed by centrifugation at 100 000 g for 1 h. Outer membrane proteins were extracted by solubilization in 1 M NDSB-201 (Calbiochem, San Diego, CA, USA)/1% (v/v) n-octyl polyoxyethylene in 20 mM Na2HPO4 pH 7.5 at 4°C for 15 min followed by centrifugation at 100 000 g for 1 h.
Purification and analysis of N-glycoproteins (C. jejuni)
Glycoproteins from the membrane extracts were isolated by affinity chromatography on SBA agarose (Sigma-Aldrich, St Louis, MO, USA). Extracts were dialyzed against PBS (100 mM NaCl, 50 mM Na2HPO4, pH 7.5) and passed through an SBA-agarose column previously equilibrated in PBS. The column was washed with two column volumes of PBS and bound glycoprotein was eluted with 0.1 M GalNAc in PBS. Glycoprotein-containing fractions were pooled, dialyzed against Milli-Q water, and freeze-dried. The glycoproteins were separated by SDS–PAGE on 12.5% (v/v) homogeneous polyacrylamide gels and stained with Coomassie Blue stain or silver stained (Rademaker et al, 1998). For subsequent lectin probing, the gels were electroblotted onto PVDF membrane and developed with SBA alkaline phosphatase conjugate (EY Laboratories, San Mateo, CA, USA) as previously described (Young et al, 2002).
Protein expression, purification, and analysis (S. cerevisiae)
Cells from strain SS328 (MATα ade2-101 his3Δ200 lys2-801 ura3-52; Vijayraghavan et al, 1989) harboring the AcrA expression plasmids were grown to a final OD600 of 1.0 and harvested by centrifugation. The cells were lysed (Y-PER, yeast protein extraction reagent, Pierce, Rockford, IL, USA) in the presence of a protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland) and cleared by low-speed centrifugation. In the case of mechanical lysis, the membranes were then solubilized at 4°C for 30 min in the presence of 1% Triton X-100. The glycoproteins were then enriched from the supernatant by incubating the supernatant with ConA-sepharose 4B (Sigma-Aldrich). The proteins were eluted by boiling the beads in the presence of Lämmli sample buffer. Protein expression and glycosylation were analyzed by SDS–PAGE followed by immunodetection using anti-AcrA antiserum. For deglycosylation, proteins were incubated with EndoH (New England Biolabs) for 2 h at 37°C in the appropriate buffer.
MS analysis
In-gel tryptic digestion and NanoLC-MS/MS using a Q-TOF Ultima hybrid quadrupole time-of-flight mass spectrometer (Waters, Milford, MA, USA) was carried out as previously described (Young et al, 2002). MALDI-TOF/TOF was carried out as described (Alaimo et al, 2006; see Supplementary Materials and methods). For NanoESI-MS/MS, dried tryptic peptides from in-gel digested proteins were resuspended in 10 μl 0.1% trifluroacetic acid and desalted with a C18 ZipTip (Millipore, Bedford, MA) with elution in 10 μl (50% methanol, 0.1% formic acid). NanoESI-MS/MS analysis was performed using a Q-TOF mass spectrometer (Micromass/Waters, Manchester, UK) fitted with a NanoMate HD source (Advion BioSciences, Ithaca, NY) in positive ion mode. MS/MS analyses were performed with collision energy of 30%.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Tables
Supplemetary Material and Methods
Acknowledgments
We are especially grateful to I Donzé for excellent technical assistance, to MF Feldman and K Ilg for critically reading the manuscript, and to W Findlay for preparing the WebLogos. We thank M Berkmen and J Beckwith for the AppA expression plasmid, K Igarashi for anti-OppA and anti-PotD antisera and the Functional Genomics Center Zurich (FGCZ) for providing technology. This work was supported by grants from the Gebert-Rüf Stiftung, the Swiss National Science Foundation (to MA), the Zürich Glycomics Initiative (GlycoInit), and the NRC Genomics and Health Initiative (to NMY).
References
- Akama H, Matsuura T, Kashiwagi S, Yoneyama H, Narita S, Tsukihara T, Nakagawa A, Nakae T (2004) Crystal structure of the membrane fusion protein, MexA, of the multidrug transporter in Pseudomonas aeruginosa. J Biol Chem 279: 25939–25942 [DOI] [PubMed] [Google Scholar]
- Alaimo C, Catrein I, Morf L, Marolda CL, Callewaert N, Valvano MA, Feldman MF, Aebi M (2006) Two distinct but interchangeable mechanisms for flipping of lipid-linked oligosaccharides. EMBO J 25: 967–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bause E, Breuer W, Peters S (1995) Investigation of the active site of oligosaccharyltransferase from pig liver using synthetic tripeptides as tools. Biochem J 312 (Part 3): 979–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Dor S, Esterman N, Rubin E, Sharon N (2004) Biases and complex patterns in the residues flanking protein N-glycosylation sites. Glycobiology 14: 95–101 [DOI] [PubMed] [Google Scholar]
- Bulleid NJ, Bassel-Duby RS, Freedman RB, Sambrook JF, Gething MJ (1992) Cell-free synthesis of enzymically active tissue-type plasminogen activator. Protein folding determines the extent of N-linked glycosylation. Biochem J 286 (Part 1): 275–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda P, Aebi M (1998) The ALG10 locus of Saccharomyces cerevisiae encodes the alpha-1,2 glucosyltransferase of the endoplasmic reticulum: the terminal glucose of the lipid-linked oligosaccharide is required for efficient N-linked glycosylation. Glycobiology 8: 455–462 [DOI] [PubMed] [Google Scholar]
- Burda P, Aebi M (1999) The dolichol pathway of N-linked glycosylation. Biochim Biophys Acta 1426: 239–257 [DOI] [PubMed] [Google Scholar]
- Chen W, Helenius A (2000) Role of ribosome and translocon complex during folding of influenza hemagglutinin in the endoplasmic reticulum of living cells. Mol Biol Cell 11: 765–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Lee SY (2004) Secretory and extracellular production of recombinant proteins using Escherichia coli. Appl Microbiol Biotechnol 64: 625–635 [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman MF, Wacker M, Hernandez M, Hitchen PG, Marolda CL, Kowarik M, Morris HR, Dell A, Valvano MA, Aebi M (2005) Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc Natl Acad Sci USA 102: 3016–3021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavel Y, von Heijne G (1990) Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng 3: 433–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover KJ, Weerapana E, Numao S, Imperiali B (2005) Chemoenzymatic synthesis of glycopeptides with PgIB, a bacterial oligosaccharyl transferase from Campylobacter jejuni. Chem Biol 12: 1311–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A, Aebi M (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem 73: 1019–1049 [DOI] [PubMed] [Google Scholar]
- Hendrixson DR, DiRita VJ (2004) Identification of Campylobacter jejuni genes involved in commensal colonization of the chick gastrointestinal tract. Mol Microbiol 52: 471–484 [DOI] [PubMed] [Google Scholar]
- Higgins MK, Bokma E, Koronakis E, Hughes C, Koronakis V (2004) Structure of the periplasmic component of a bacterial drug efflux pump. Proc Natl Acad Sci USA 101: 9994–9999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi K, Saisho T, Yuguchi M, Kashiwagi K (1997) Molecular mechanism of polyamine stimulation of the synthesis of oligopeptide-binding protein. J Biol Chem 272: 4058–4064 [DOI] [PubMed] [Google Scholar]
- Johnson JM, Church GM (1999) Alignment and structure prediction of divergent protein families: periplasmic and outer membrane proteins of bacterial efflux pumps. J Mol Biol 287: 695–715 [DOI] [PubMed] [Google Scholar]
- Jones J, Krag SS, Betenbaugh MJ (2005) Controlling N-linked glycan site occupancy. Biochim Biophys Acta 1726: 121–137 [DOI] [PubMed] [Google Scholar]
- Karlyshev AV, Everest P, Linton D, Cawthraw S, Newell DG, Wren BW (2004) The Campylobacter jejuni general glycosylation system is important for attachment to human epithelial cells and in the colonization of chicks. Microbiology 150: 1957–1964 [DOI] [PubMed] [Google Scholar]
- Kelleher DJ, Gilmore L (2006) An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16: 47R–62R [DOI] [PubMed] [Google Scholar]
- Kowarik M, Kung S, Martoglio B, Helenius A (2002) Protein folding during cotranslational translocation in the endoplasmic reticulum. Mol Cell 10: 769–778 [DOI] [PubMed] [Google Scholar]
- Larsen JC, Szymanski C, Guerry P (2004) N-linked protein glycosylation is required for full competence in Campylobacter jejuni 81–176. J Bacteriol 186: 6508–6514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton D, Dorrell N, Hitchen PG, Amber S, Karlyshev AV, Morris HR, Dell A, Valvano MA, Aebi M, Wren BW (2005) Functional analysis of the Campylobacter jejuni N-linked protein glycosylation pathway. Mol Microbiol 55: 1695–1703 [DOI] [PubMed] [Google Scholar]
- Mellquist JL, Kasturi L, Spitalnik SL, Shakin-Eshleman SH (1998) The amino acid following an Asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry 37: 6833–6837 [DOI] [PubMed] [Google Scholar]
- Muller A, Thomas GH, Horler R, Brannigan JA, Blagova E, Levdikov VM, Fogg MJ, Wilson KS, Wilkinson AJ (2005) An ATP-binding cassette-type cysteine transporter in Campylobacter jejuni inferred from the structure of an extracytoplasmic solute receptor protein. Mol Microbiol 57: 143–155 [DOI] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122 [DOI] [PubMed] [Google Scholar]
- Nilsson I, Kelleher DJ, Miao Y, Shao Y, Kreibich G, Gilmore R, von Heijne G, Johnson AE (2003) Photocross-linking of nascent chains to the STT3 subunit of the oligosaccharyltransferase complex. J Cell Biol 161: 715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nita-Lazar M, Wacker M, Schegg B, Amber S, Aebi M (2005) The N-X-S/T consensus sequence is required but not sufficient for bacterial N-linked protein glycosylation. Glycobiology 15: 361–367 [DOI] [PubMed] [Google Scholar]
- Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, Jagels K, Karlyshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Rajandream MA, Rutherford KM, van Vliet AH, Whitehead S, Barrell BG (2000) The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403: 665–668 [DOI] [PubMed] [Google Scholar]
- Petrescu AJ, Milac AL, Petrescu SM, Dwek RA, Wormald MR (2004) Statistical analysis of the protein environment of N-glycosylation sites: implications for occupancy, structure, and folding. Glycobiology 14: 103–114 [DOI] [PubMed] [Google Scholar]
- Pless DD, Lennarz WJ (1977) Enzymatic conversion of proteins to glycoproteins. Proc Natl Acad Sci USA 74: 134–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademaker GJ, Pergantis SA, Blok-Tip L, Langridge JI, Kleen A, Thomas-Oates JE (1998) Mass spectrometric determination of the sites of O-glycan attachment with low picomolar sensitivity. Anal Biochem 257: 149–160 [DOI] [PubMed] [Google Scholar]
- Schulz H, Hennecke H, Thony-Meyer L (1998) Prototype of a heme chaperone essential for cytochrome c maturation. Science 281: 1197–1200 [DOI] [PubMed] [Google Scholar]
- Shakin-Eshleman SH, Spitalnik SL, Kasturi L (1996) The amino acid at the X position of an Asn-X-Ser sequon is an important determinant of N-linked core-glycosylation efficiency. J Biol Chem 271: 6363–6366 [DOI] [PubMed] [Google Scholar]
- Szymanski CM, Burr DH, Guerry P (2002) Campylobacter protein glycosylation affects host cell interactions. Infect Immun 70: 2242–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski CM, Yao R, Ewing CP, Trust TJ, Guerry P (1999) Evidence for a system of general protein glycosylation in Campylobacter jejuni. Mol Microbiol 32: 1022–1030 [DOI] [PubMed] [Google Scholar]
- Vijayraghavan U, Company M, Abelson J (1989) Isolation and characterization of pre-mRNA splicing mutants of Saccharomyces cerevisiae. Genes Dev 3: 1206–1216 [DOI] [PubMed] [Google Scholar]
- Wacker M, Linton D, Hitchen PG, Nita-Lazar M, Haslam SM, North SJ, Panico M, Morris HR, Dell A, Wren BW, Aebi M (2002) N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 298: 1790–1793 [DOI] [PubMed] [Google Scholar]
- Whitley P, Nilsson IM, von Heijne G (1996) A nascent secretory protein may traverse the ribosome/endoplasmic reticulum translocase complex as an extended chain. J Biol Chem 271: 6241–6244 [DOI] [PubMed] [Google Scholar]
- Wilson CM, Kraft C, Duggan C, Ismail N, Crawshaw SG, High S (2005) Ribophorin I associates with a subset of membrane proteins after their integration at the sec61 translocon. J Biol Chem 280: 4195–4206 [DOI] [PubMed] [Google Scholar]
- Yan Q, Lennarz WJ (2002) Studies on the function of oligosaccharyl transferase subunits. Stt3p is directly involved in the glycosylation process. J Biol Chem 277: 47692–47700 [DOI] [PubMed] [Google Scholar]
- Young NM, Brisson JR, Kelly J, Watson DC, Tessier L, Lanthier PH, Jarrell HC, Cadotte N, St Michael F, Aberg E, Szymanski CM (2002) Structure of the N-linked glycan present on multiple glycoproteins in the Gram-negative bacterium, Campylobacter jejuni. J Biol Chem 277: 42530–42539 [DOI] [PubMed] [Google Scholar]
- Zhang RG, Westbrook ML, Westbrook EM, Scott DL, Otwinowski Z, Maulik PR, Reed RA, Shipley GG (1995) The 2.4 A crystal structure of cholera toxin B subunit pentamer: choleragenoid. J Mol Biol 251: 550–562 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Tables
Supplemetary Material and Methods