

Abstract
Misfolded proteins of the endoplasmic reticulum (ER) are targeted to the cytoplasm for proteasomal degradation. Key components of this process are ER membrane-bound ubiquitin ligases. These ligases associate with the cytoplasmic AAA-ATPase Cdc48p/p97, which is thought to support the release of malfolded proteins from the ER. Here, we characterize a yeast protein complex containing the ubiquitin ligase Hrd1p and the ER membrane proteins Hrd3p and Der1p. Hrd3p binds malfolded proteins in the ER lumen enabling their delivery to downstream components. Therefore, we propose that Hrd3p acts as a substrate recruitment factor for the Hrd1p ligase complex. Hrd3p function is also required for the association of Cdc48p with Hrd1p. Moreover, our data demonstrate that recruitment of Cdc48p depends on substrate processing by the Hrd1p ligase complex. Thus, the Hrd1p ligase complex unites substrate selection in the ER lumen and polyubiquitination in the cytoplasm and links these processes to the release of ER proteins via the Cdc48p complex.
Keywords: Cdc48/p97, ERAD, Hrd1, Hrd3, protein degradation
Introduction
The cytosolic ubiquitin–proteasome system facilitates the breakdown of endoplasmic reticulum (ER) proteins that fail to fold properly or are metabolically regulated (Hampton, 2002; Meusser et al, 2005; Romisch, 2005). The key enzymes in this process are ubiquitin ligases, which recognize the substrate protein and mediate coupling of ubiquitin moieties. Selection of ER-lumenal substrates and their polyubiquitination in the cytoplasm are spatially separated by the ER membrane. As a consequence, a transport step is required which dislocates the substrate molecules from the ER into the cytoplasm prior to their degradation.
In the yeast Saccharomyces cerevisiae, Hrd1p/Der3p is one of two ubiquitin ligases that were found to accomplish ubiquitination of misfolded ER proteins (Bays et al, 2001a; Deak and Wolf, 2001). In addition, this ligase operates in the metabolic regulation of Hydroxymethylglutaryl-CoA reductase (HMGR), the rate-limiting enzyme in sterol synthesis (Hampton et al, 1996). Hrd1p is composed of two distinct domains: an N-terminal domain with six transmembrane segments and a C-terminal domain that harbors the catalytic active really interesting new gene (RING)-H2 motif (Bays et al, 2001a). In mammals, two homologues of Hrd1p were identified, HRD1 and gp78. Human HRD1 is topologically equivalent to Hrd1p, and functions in the basal regulation of HMGR and in the degradation of CD3-δ (Kikkert et al, 2004). gp78, originally identified as autocrine motility factor receptor (AMFR), contains five predicted transmembrane domains, a cytosolic RING-domain, followed by a CUE-motif and a C-terminal domain (Fang et al, 2001; Zhong et al, 2004).
The AAA-ATPase Cdc48p, in mammals known as p97 or valosine-containing protein (VCP), in complex with Ufd1p and Npl4p is required for the ubiquitin-dependent proteasomal degradation of cytosolic (Dai et al, 1998; Fu et al, 2003) as well as ER-resident proteins (Bays et al, 2001b; Ye et al, 2001; Braun et al, 2002; Jarosch et al, 2002; Rabinovich et al, 2002). The specific function of Cdc48p in protein dislocation from the ER is still a matter of intense research. The Cdc48p–Ufd1p–Npl4p complex might provide a driving force for pulling proteins out of the ER due to its AAA-ATPase activity (Rouiller et al, 2000; Zhang et al, 2000; Ye et al, 2003). Alternatively, the Cdc48p complex may be required for the release of already exported substrates from the cytosolic surface of the ER (Rape et al, 2001).
How is the function of Cdc48p linked to dislocation and ubiquitination of aberrant ER proteins? Recent studies have demonstrated that human HRD1 as well as gp78 associate with p97 and the ER-membrane protein Derlin-1 (Zhong et al, 2004; Ye et al, 2005). Derlin-1 is a mammalian homologue of yeast Der1p and is involved in the US11-induced turnover of MHC class I molecules (Lilley and Ploegh, 2004; Ye et al, 2004). Association of Derlin-1 with p97 is independent of HRD1, suggesting that at least two distinct protein complexes containing p97 function at different steps of protein dislocation. The interaction of gp78 and HRD1 with p97 does not depend on their activity. gp78 contains a stretch of 49 amino acid at its C-terminus that mediates p97 binding (Zhong et al, 2004; Ye et al, 2005). Conversely, in yeast, the ER-membrane protein Ubx2p was found to confer interaction of the Cdc48p complex and the ubiquitin ligases Hrd1p and Doa10p (Neuber et al, 2005; Schuberth and Buchberger, 2005).
Yeast Hrd1p stoichiometrically associates with Hrd3p, an ER-resident glycoprotein that is composed of a large N-terminal ER-lumenal domain, a single transmembrane span, and a short C-terminal cytosolic region (Gardner et al, 2000). The ER-lumenal domain shows significant similarity to SEL-1 (the Caenorhabditis elegans suppressor and/or enhancer of lineage-12) (Ponting, 2000). Both proteins contain multiple copies of a 40-amino-acid sequence stretch, which were classified as subtypes of tricopeptide repeats. The N-terminal part of the ER-lumenal domain seems to be essential for its function in degradation of misfolded proteins, whereas the C-terminal part interacts with the ER-lumenal loops of Hrd1p (Gardner et al, 2000). The mammalian Hrd3p homologue SEL1L associates with HRD1 and p97 (Lilley and Ploegh, 2005). The interaction of the SEL1L–HRD1 complex and the AAA-ATPase seems to be established by Derlin-1 and its homologue Derlin-2.
Membrane-bound misfolded proteins might be easily accessible to Hrd1p-Hrd3p. Contrariwise, ER-lumenal substrates have to be escorted to this ligase complex. The small, membrane protein Der1p may play a role in this process. Der1p consists of four transmembrane segments and a short cytosolic C-terminal domain (Hitt and Wolf, 2004). Der1p is involved in the degradation of soluble model substrates, such as mutant carboxypeptidase Y (CPY*) (Knop et al, 1996), but is dispensable when CPY* is fused to a membrane anchor (Taxis et al, 2003). Derlin-1 interacts with p97/VCP and was found in association with US11 as well as glycosylated and deglycosylated degradation intermediates of MHC class I molecules (Lilley and Ploegh, 2004; Ye et al, 2004). Thus, it was proposed that Derlin-1 forms a channel for protein dislocation.
In this work, we have focused on a systematic investigation of protein interactions and functions of Hrd1p in living cells. We demonstrate the association of Hrd1p and Hrd3p with Der1p, and characterize this heterogenic complex. Hrd1p and Hrd3p appear to form the functional core of the complex, whereas Der1p is only weakly associated and probably represents an auxiliary component required for the breakdown of a subset of proteins. Moreover, we were able to separate ER-lumenal and cytosolic functions of this ligase complex. In the ER lumen, Hrd3p and Der1p recruit substrates to Hrd1p independently of each other. However, binding of substrates to either Der1p or Hrd3p is not sufficient to initiate the degradation of a soluble ER protein. In the cytoplasm, the ligase complex binds to the Cdc48p complex via Hrd1p. Importantly, we show that the ER-lumenal domain of Hrd3p, and the ubiquitination activity mediated by Hrd1p and Ubc7p is essential for the recruitment of the Cdc48p complex to the Hrd1p ligase complex. From this study, we suggest that Hrd3p establishes the junction between the recruitment of substrate proteins in the ER lumen, and their ubiquitination by Hrd1p at the cytoplasmic face of the ER membrane.
Results
The Hrd1p ligase complex associates with Cdc48p
We set out to determine the molecular environment of the ubiquitin ligase Hrd1p, making use of nondenaturing immunoprecipitation from solubilized membrane preparations from the yeast S. cerevisiae. To this end, we constructed yeast strains that express HA- or myc-epitope-tagged versions of Hrd1p, Hrd3p, Der1p, and Npl4p (see Figure 8 for a schematic topology of these proteins) from the respective chromosomal loci under the control of their endogenous promoters. Cells expressing those proteins displayed degradation kinetics of CPY* comparable to wild type (data not shown) and the tags did not affect protein expression (Supplementary Figure 1). Hrd3p was co-precipitated from solubilized microsomes specifically and efficiently with Hrd1p-HA (Figure 1A, lane 5). Conversely, Hrd1p was detected when HA-Hrd3p was pulled down (lane 6). Although the ligase function of Hrd1p depends on the E2s Ubc1p, Ubc6p, and Ubc7p (Friedlander et al, 2000; Bays et al, 2001a), none of these E2s nor the Ubc7p membrane anchor Cue1p (Biederer et al, 1997) were found to be associated with Hrd1p or Hrd3p (lanes 4–6, and data not shown). In addition, we were not able to detect an interaction between Hrd1p and Sec61p, the core component of the translocation complex, which was discussed to have a function in protein dislocation (Tsai et al, 2002) (lanes 4–6). In contrast, Hrd1p and Hrd3p were precipitated with Der1p-myc (Figure 1B) and Der1p was pulled down with Hrd1p-HA or myc-Hrd3p (data not shown).
Figure 8.
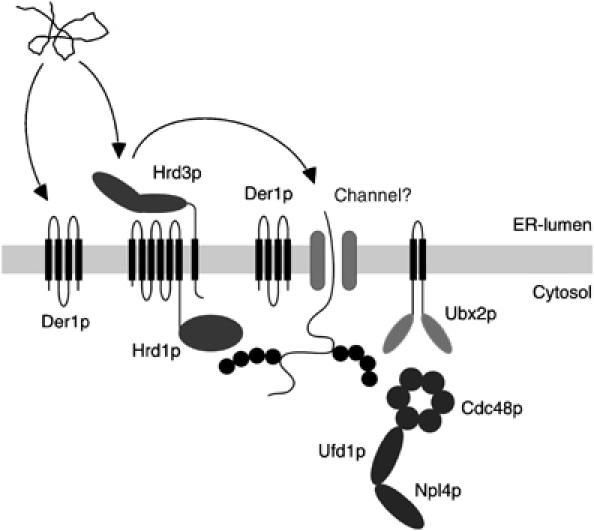
The Hrd1p ligase complex provides a linchpin between substrate selection in the ER and the binding of Cdc48p in the cytoplasm. Misfolded ER proteins bind to the ER-luminal domain of Hrd3p and possibly to Der1p. Subsequently, these substrates are dislocated into the cytoplasm. It is still a matter of debate how these proteins pass the ER membrane. Der1p as well as membrane-bound ubiquitin ligases such as Hrd1p or Doa10p may participate in this process. During or after dislocation, the substrates are ubiquitinated by Hrd1p. In the cytoplasm, binding of Cdc48p–Npl4p–Ufd1p supports the dislocation of ER proteins and may also target them to the proteasome. Recruitment of the Cdc48p complex to Hrd1p depends on the membrane protein Ubx2p/Sel1p as well as the catalytic activity of the ubiquitin ligases.
Figure 1.
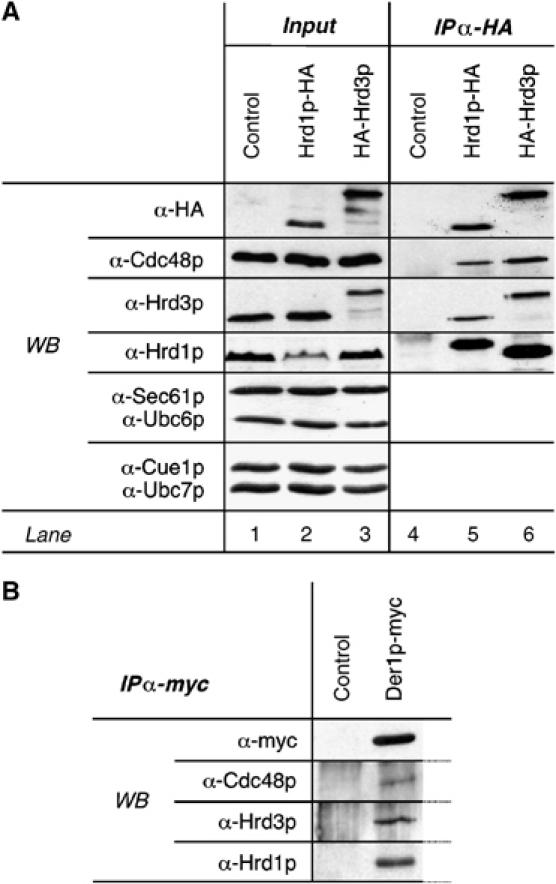
Hrd1p, Hrd3p, and Der1p form a heterogenic complex and associate with cytosolic Cdc48p. (A) Microsomes were isolated from logarithmically growing cells expressing Hrd1p-HA or HA-Hrd3p (see Materials and methods). Subsequently, membranes were solubilized with NP40 using nondenaturing conditions and HA-tagged proteins were immunoprecipitated. Precipitates were analyzed by SDS–PAGE and immunoblotting using specific antibodies. The Hrd1p antiserum reacts only poorly with the C-terminally HA-tagged Hrd1p protein (see also Supplementary Figure 1). The signal in lane 2 is an unspecific crossreaction of the Hrd1p antiserum. (B) Der1p-myc was immunoprecipitated from isolated microsomes as in (A) and the precipitate analyzed via immunoblotting.
To confirm the interactions of Hrd1p, Hrd3p, and Der1p, we performed a gel filtration experiment from solubilized microsome preparations (data not shown). Hrd1p and Hrd3p comigrated on a Sepharose size-fractionation column. The protein peak of Der1p overlapped with the protein peaks of Hrd1p and Hrd3p but was broader. Taken together, these data show that both the E3 ligase Hrd1p, and its partner protein Hrd3p are associated with Der1p, and presumably, these proteins form a heterogenic complex. In the following, we refer to the complex of Hrd1p, Hrd3p, and Der1p as the ‘Hrd1p ligase complex'.
The function of the Cdc48p–Ufd1p–Npl4p complex in dislocation or mobilization of proteins from the ER membrane depends on its interaction with ubiquitin ligases in the ER membrane. In line with this, Cdc48p was specifically co-precipitated with Hrd1p-HA, HA-Hrd3p, and Der1p-myc (Figures 1A and B). Vice versa, Hrd1p, Hrd3p, and Der1p were copurified with myc-tagged Npl4p, a cofactor of Cdc48p involved in proteasomal protein degradation (Supplementary Figure 2A). From these results, we conclude that the Hrd1p ligase complex interacts with the Cdc48p–Ufd1p–Npl4p complex.
Formation of the Hrd1p ligase complex is independent of its catalytic activity
In a next step, we analyzed the composition of the Hrd1p ligase complex in more detail. Deletion of DER1 did not affect the association of Hrd1p and Hrd3p (Figure 2A, compare lanes 2 and 4, and Figure 2B, lanes 2 and 3). Hrd3p still bound to a truncated version of Hrd1p lacking the cytoplasmic C-terminal domain (Figure 2A, lane 6, and Figure 2B, lane 5; see Supplementary Figure 3A for degradation kinetics of Hrd1p-Δcyt). In addition, deletion of UBC7 or the RING-motif within Hrd1p did not affect the association of Hrd3p (Figure 2A, lanes 5 and 9, and Figure 2B, lane 6; see Supplementary Figure 3B for degradation kinetics of Hrd1p-ΔRING). Der1p was also detected in precipitates from a Hrd1p-Δcyt strain (Figure 2A, lane 7). Thus, the catalytic activity of Hrd1p is not essential for the formation of the Hrd1p ligase complex. The amount of Hrd3p associated with Hrd1p was slightly increased when the RING-motif or the complete RING-domain of Hrd1p was deleted, although the total input levels of the proteins remained unchanged (Figure 2A, compare lanes 2 and 6, 7 and 9). This might hint to a dynamic nature of the Hrd1p–Hrd3p complex.
Figure 2.
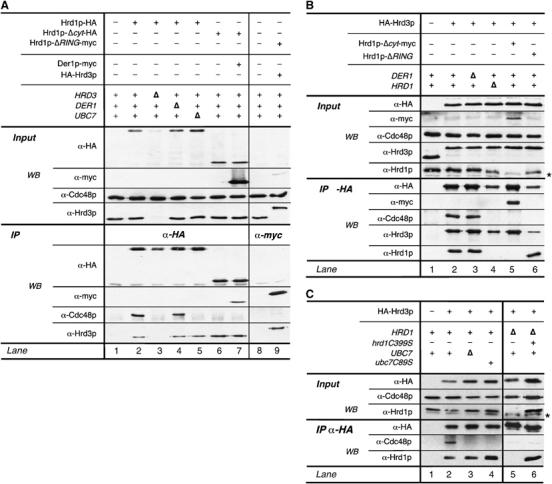
Hrd3p and the catalytic activity of Hrd1p are essential for the recruitment of Cdc48p. (A) Hrd1p-HA and epitope-tagged mutant forms of Hrd1p lacking either the cytoplasmic C-terminus (Hrd1p-Δcyt) or the RING-domain (Hrd1p-ΔRING; for description of the mutants, see Materials and methods section) were immunoprecipitated from solubilized membrane preparations of the indicated yeast strains as described in Figure 1. Precipitated material was subsequently analyzed by immunoblotting. (B, C) HA-Hrd3p was immunoprecipitated from the indicated yeast strains. Note that the Hrd1p polyclonal antibody was raised against a region not present in Hrd1p-Δcyt and hence this serum does not react with Hrd1p-Δcyt. (*) Refers to unspecific cross-reacting material. (▵) Indicates deletion of the respective gene.
The association of Cdc48p with the Hrd1p ligase complex depends on Hrd3p and the activity of Hrd1p and Ubc7p
From the experiments presented in Figure 1, we could not distinguish whether Cdc48p binds only to one component of the Hrd1p ligase complex, or whether all proteins contribute to the recruitment of the Cdc48p complex. We found that Der1p is not required for the binding of Cdc48p to Hrd1p-HA (Figure 2A, lane 4). In contrast, Cdc48p was not co-precipitated with Hrd1p-HA when HRD3 was deleted (lane 3). The reduced amounts of interacting Cdc48p might be explained by the diminished levels of Hrd1p in HRD3-deleted cells (Plemper et al, 1999; Gardner et al, 2000; Figure 2A, lane 2 and 3 input). Hrd3p did not bind Cdc48p in the absence of Hrd1p (Figure 2B, lane 4). These findings suggest that both, Hrd1p and Hrd3p, participate in the efficient binding of Cdc48p to the Hrd1p ligase complex. This view was further strengthened by immunoprecipitations of an myc-tagged version of Npl4p from extracts derived from HRD1- or HRD3-deleted cells (Supplementary Figure 2B).
When we removed the soluble C-terminal domain of Hrd1p, the interaction with Cdc48p was lost (Figure 2A, lane 6). This region contains the catalytically active RING-motif. Deletion of only the RING-motif also abolished the Cdc48p interaction (lane 9). Moreover, Hrd1p failed to associate with Cdc48p in cells lacking Ubc7p (lane 5). Consistently, HA-Hrd3p did not bind to Cdc48p when either the RING-motif of Hrd1p or UBC7 was knocked-out (Figure 2B, lanes 6 and 7; Figure 2C, lane 3). These results strongly suggest that Cdc48p is only recruited to catalytically active Hrd1p that is associated with Hrd3p. To further corroborate these findings, we used inactive point mutations of Hrd1p and Ubc7p in pull-down experiments with HA-Hrd3p (Figure 2C). Hrd1p(C399S) and Ubc7p(C89S) lack active-site cysteine residues in the respective proteins and abolish degradation of CPY* (Supplementary Figure 3C). Hrd1p(C399S) co-precipitated with HA-Hrd3p, but association of Cdc48p was lost (Figure 2C, compare lanes 2 and 8). Likewise, catalytically inactive Ubc7p(C89S) is not capable to trigger binding of Cdc48p to Hrd1p and Hrd3p (Figure 2C, compare lanes 2 and 4). These data confirm our assumption that ubiquitination mediated by Hrd1p and Ubc7p is an essential prerequisite for the binding of Cdc48p to the Hrd1p ligase complex. Summing up, the interaction of the Hrd1p complex and Cdc48p requires Hrd3p and the ubiquitinating activity of Hrd1p in concert with its E2 Ubc7p.
Der1p binds to Hrd1p, Hrd3p, and Cdc48p
Next, we were interested in the association of Der1p with Hrd1p/Hrd3p. We found small but significant amounts of Hrd1p as well as Hrd3p in immunoprecipitates of Der1p-myc (Figure 3). Interestingly, Der1p appears to bind Hrd1p and Hrd3p independently from the other Hrd proteins (lanes 3 and 4). Cdc48p was also co-purified with Der1p-myc even from cells lacking Hrd1p or Hrd3p. Since neither Hrd1p nor Hrd3p interacts with Cdc48p in the absence of its partner protein, we propose that Der1p itself displays a weak but significant binding activity towards Cdc48p.
Figure 3.
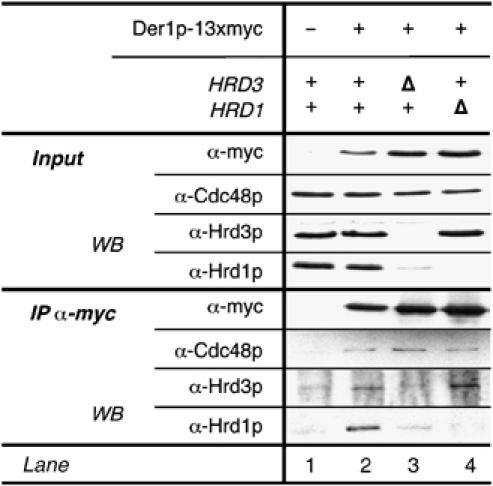
Der1p binds to the Hrd1p complex and interacts with Cdc48p. An myc-tagged version of Der1p as immunoprecipitated from isolated membrane preparations of the indicated yeast strains under nondenaturing conditions and co-precipitating proteins were identified via immunoblotting.
Hrd3p and Der1p autonomously bind substrate proteins
Earlier studies propose that Hrd3p constitutes a substrate recruitment factor for the ubiquitin ligase Hrd1p (Plemper et al, 1999; Gardner et al, 2000). Therefore, we set out to investigate the binding activity of Hrd3p to CPY*, a mutant form of carboxypeptidase Y (CPY) that is retarded in the ER and degraded in a Hrd1p/Hrd3p-dependent manner. In contrast to wild-type CPY, CPY* was specifically co-precipitated with myc-Hrd3p (Figure 4A, compare lanes 2 and 4). Deletion of DER1, which disrupts the degradation of CPY* (Knop et al, 1996; Hitt and Wolf, 2004), further increased the amount of Hrd3p-bound CPY* (lane 3).
Figure 4.
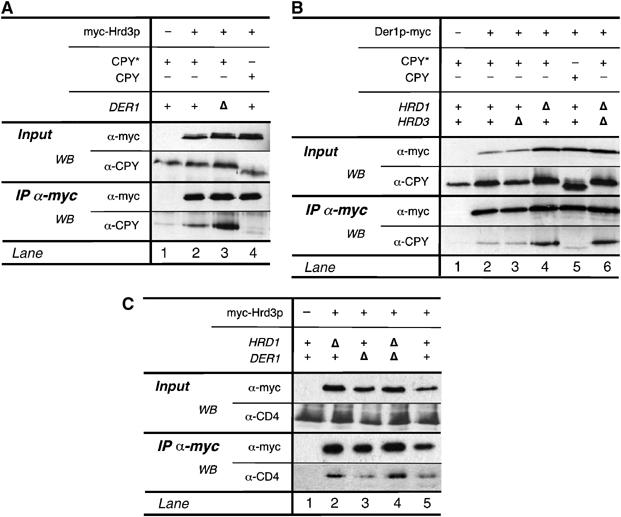
Hrd3p and Der1p autonomously bind the misfolded protein CPY*. myc-Hrd3p (A) and Der1p-myc (B) were precipitated from the indicated yeast strains expressing CPY* or wild-type CPY. Co-precipitated material was analyzed by Western blotting. (C) CD4 interacts with Hrd3p. myc-Hrd3p was immunopurified from yeast cells heterologously expressing CD4. The relevant genotype of the yeast strains used is given.
Der1p-myc did not bind significant levels of CPY* (Figure 4B, compare lanes 2 and 5) and the amount of CPY* bound to Der1p did not change in HRD3-deleted cells (Figure 4B, lane 3). In contrast, cells lacking Hrd1p displayed increased levels of CPY* that was associated with Der1p, indicating that this ubiquitin ligase functions downstream of Der1p (Figure 4B, compare lanes 2 and 4). If Der1p functions downstream of Hrd3p but upstream of Hrd1p, deletion of both HRD3 and HRD1 should diminish binding of CPY*. However, the amount of Der1p-associated CPY* was comparable in Δhrd1 and Δhrd1Δhrd3 cells, respectively (Figure 4B, compare lanes 4 and 6). This result suggests that Der1p is able to bind substrate molecules independently of Hrd3p, at least in cells lacking the ubiquitin ligase Hrd1p.
Next, we investigated whether Hrd3p also binds to a membrane-bound ER degradation substrate. To this end, we expressed a codon-adjusted version of human CD4 protein in yeast cells (Meusser and Sommer, 2004). Degradation of CD4 depends on Hrd1p and Hrd3p but is independent from Der1p (Meusser and Sommer, 2004; Supplementary Figure 3). CD4 efficiently co-precipitated with myc-Hrd3p (Figure 4C, lane 5). The binding of CD4 to Hrd3p was not affected by deletion of DER1 (lane 3). In contrast, the amount of bound CD4 was increased in Δhrd1 strains. This confirms that Hrd1p functions downstream of Hrd3p and that Der1p is dispensable for the binding of this membrane protein to the Hrd1p–Hrd3p complex (lanes 2 and 4). We view these studies regarding CD4 with attention because we failed to co-precipitate CD4 specifically with Hrd1p-HA or Hrd1p-Δcyt-HA (data not shown).
A small amount of CPY* precipitated with Hrd1p-HA (Figure 5, lane 2). Deletion of DER1 and HRD3 did not change the levels of Hrd1-HA-bound CPY* (lane 7), although the cellular amount of the protein was increased. Truncating the C-terminus of Hrd1p (Hrd1p-Δcyt) significantly increased the quantity of bound CPY* (compare lanes 2 and 3). Here, deletion of HRD3 slightly decreased levels of CPY* bound to Hrd1p-Δcyt, whereas no change was visible in cells deficient for Der1p (lanes 4 and 5). These results suggest that Der1p and Hrd3p function in parallel with regard to substrate recruitment to Hrd1p. In cells lacking both proteins, significantly less CPY* was found in association with Hrd1p-Δcyt when compared to the single knockout cells (compare lanes 3, 4, 5, and 8). These findings indicate that both Der1p and Hrd3p are required for substrate binding to Hrd1p. Both proteins appear to function autonomously upstream of Hrd1p. However, binding of the substrate to Hrd3p or Der1p alone is not sufficient for its breakdown because cells lacking either protein display severe degradation defects.
Figure 5.
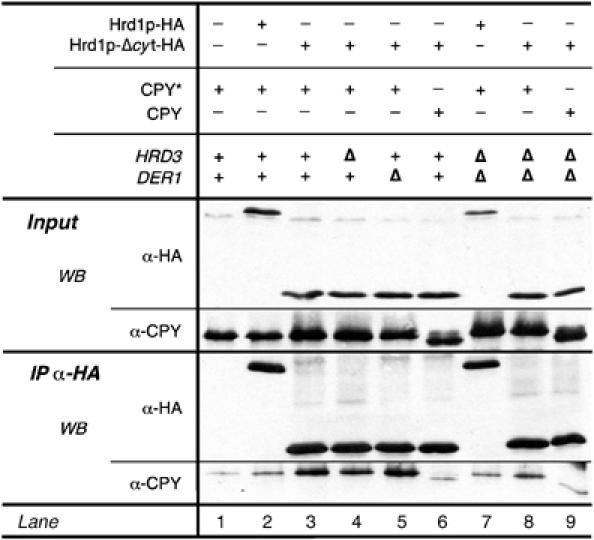
Hrd3p and Der1p are capable of recruiting CPY* to the Hrd1p ligase. Hrd1p-HA and Hrd1p-Δcyt-HA were pulled down from solubilized membrane fractions isolated from the indicated yeast strains. Bound CPY* or CPY was detected by immunoblotting using a specific antibody.
Hrd3p links substrate recruitment to ubiquitination and Cdc48p binding to the Hrd1p ligase complex
We have shown that Hrd3p is essential for the recruitment of Cdc48p to the Hrd1p ligase complex. In addition, Hrd3p is also required for the binding of substrate molecules. To analyze further the function of this protein, we expressed C-terminally truncated versions of Hrd3p from its chromosomal locus (Figure 6A). Mutant Hrd3p-Δ801 lacked the C-terminal cytoplasmic part, whereas mutant Δ769 lacked additionally the transmembrane domain. In the Δ664 mutant, a region of 100 amino acids upstream of the transmembrane domain was also deleted. The function of these Hrd3p truncations was tested by monitoring CPY* degradation in pulse-chase experiments (Figure 6B and C). In cells expressing Hrd3p-Δ801 or the Δ769 mutant, CPY* was degraded with wild-type kinetics. In contrast, CPY* degradation was severely affected in the Hrd3p-Δ664 strain. Hrd1p and Cdc48p still interacted with Hrd3p-Δ801 and Δ769 (Figure 7, lanes 2, 3, and 4), but not with the Δ664 construct (lane 5). The migration behavior of Hrd3p-Δ664 on SDS–polyacrylamide gels indicated a different glycosylation pattern when compared to wild-type or the functional Δ801 and Δ769 versions, which in turn pointed out at least partial mislocalization of this protein to a different secretory compartment. Indeed, a significant portion of the Hrd3p-Δ664 cofractionated with Golgi and vacuolar marker proteins in sucrose-gradient centrifugation experiments, whereas wild-type Hrd3p as well as the Δ801 and Δ769 mutants were exclusively found in the ER fractions (data not shown). These data imply that the cytoplasmic as well as the transmembrane region of Hrd3p is dispensable for the function of the Hrd1p ligase complex because Hrd3p mutants devoid of those regions were fully functional and were still able to confer recruitment of Cdc48p. This additionally supports our finding that Hrd3p does not bind directly to Cdc48p but rather mediates Cdc48p association with the Hrd1p ligase complex by another mechanism.
Figure 6.
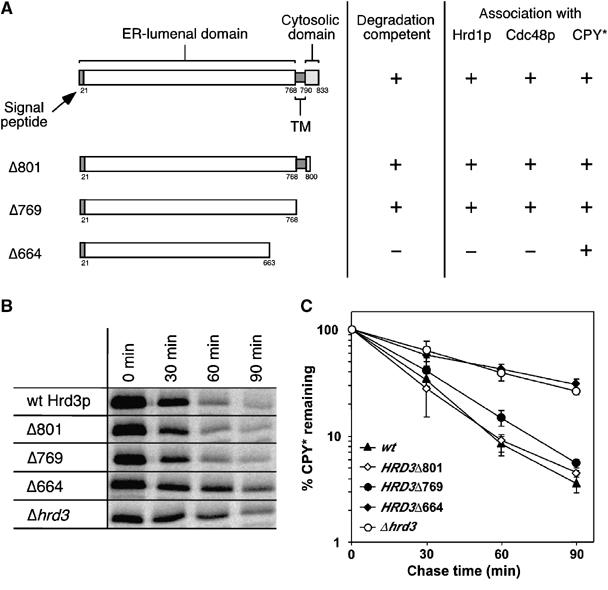
The C-terminal part of the ER-lumenal domain of Hrd3p is essential to trigger efficient degradation of CPY*. (A) Schematic domain structure of Hrd3p with representation of C-terminal truncation variants and a summary of the results. (B) A C-terminal region within the ER-lumenal domain of Hrd3p is essential for efficient CPY* degradation. Strains expressing wild-type Hrd3p and the various truncation mutants were essayed for CPY* degradation by pulse-chase analysis. Displayed is a representative experiment. (C) Quantitative evaluation of three pulse-chase experiments with error bars indicating the standard deviation of mean values.
Figure 7.
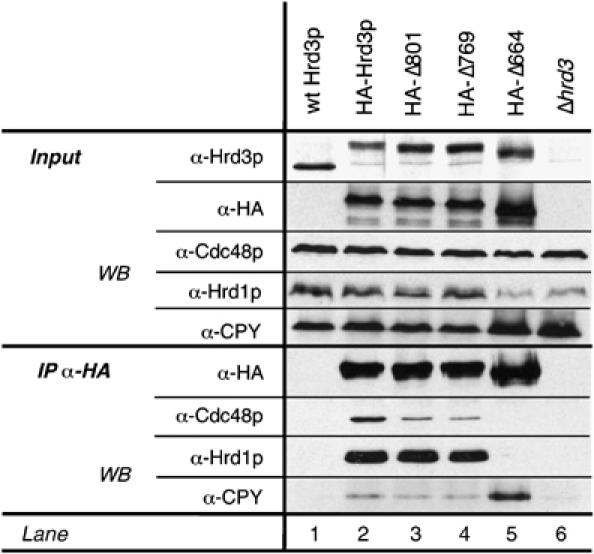
Hrd3p connects ER-lumenal substrate recognition and cytosolic Cdc48p binding. HA-Hrd3p and HA-tagged truncations mutants (Hrd3p-Δ801, -Δ769, and -Δ664) were pull-down from solubilized microsomal preparations. Precipitates were analyzed for Hrd3p-bound Cdc48, Hrd1p, and CPY* by immunoblot using specific antibodies.
Full-length Hrd3p and the Δ801 and Δ769 mutants bound weakly to CPY* (Figure 7, lanes 2–4). The amount of Hrd3p-bound CPY* was strongly increased in the Δ664 mutant (lane 5). This may be explained by the inability of this mutant to deliver substrate molecules to downstream components such as Hrd1p. Thus, Hrd3p appears to have at least two distinct functions that can be separated: the N-terminal part of the ER-lumenal domain binds substrate proteins, whereas a C-terminal portion of the protein interacts with Hrd1p.
Discussion
A key to efficient degradation of aberrant ER proteins is the tight linkage of substrate selection within the ER lumen, the attachment of ubiquitin at the cytosolic face of the ER, and the subsequent delivery of the substrates to the proteasome. Here, we provide evidence that the yeast Hrd1p ligase complex meets all these requirements: it binds substrates in the ER lumen, catalyzes their ubiquitination, and recruits the cytoplasmic Cdc48p complex, which is required for the delivery of ER proteins to the proteasome. Hrd1p is the central component of this ubiquitin ligase complex that contains at least two additional proteins, Hrd3p and Der1p. Interaction of Hrd1p and Hrd3p does not depend on the presence of Der1p and seems to involve a short ER lumenal region of Hrd3p located close to the transmembrane segment. The membrane anchor and the cytosolic tail of Hrd3p are dispensable for Hrd1p binding and the function of the ligase complex. Der1p binds to both Hrd1p and Hrd3p independently of the other Hrd proteins and possibly constitutes an auxiliary factor.
Our data imply that the Hrd3p lumenal domain provides crucial functions to the ligase complex. As long as this domain is associated with Hrd1p, proteolysis proceeds normally. However, when the ER-lumenal part of Hrd3p is disconnected from Hrd1p, turnover of ER proteins is disturbed. In this case, Hrd3p associates with increased amounts of malfolded CPY*, probably because it can no longer deliver substrate molecules to downstream components such as Hrd1p. The Hrd1p ligase complex also binds increased amounts of malfolded CPY* when ubiquitination on the cytosolic surface is abrogated. Under these conditions, substrate binding partly depends on Hrd3p, whereas deletion of DER1 has no detectable effect. Since Hrd1p binds only low amounts of malfolded substrates in the absence of Hrd3p and Der1p, we propose that Hrd3p functions as a substrate receptor of the Hrd1p ligase complex (Figure 8). Hrd3p may either directly interact with ER proteins destined for proteolysis, or provide a scaffold for factors of the ER quality control that are loaded with misfolded proteins.
Overexpression of HRD1 can at least partly suppress the degradation deficient phenotype of HRD3-deleted cells (Plemper et al, 1999; Gardner et al, 2000). This finding indicates that Hrd1p, at least when present in increased amounts, may associate with additional substrate-recruiting factors and thereby overcome the loss of Hrd3p. Indeed, deletion of HRD3 and DER1 did not completely abolish the binding of CPY* to Hrd1p, although the amount of bound substrate was largely diminished. A N-terminally truncated form of Hrd3p still binds to Hrd1p and maintains it at wild-type levels, although the function of the Hrd1p ligase complex is abolished (Gardner et al (2000) and data not shown). This demonstrates that under normal expression conditions, Hrd3p is a crucial factor for the function of the Hrd1p ligase complex.
At the cytosolic face of the ER membrane, Hrd1p mediates the ubiquitination of substrate proteins and binds to Cdc48p. In yeast, Cdc48p recruitment to membrane-bound ubiquitin ligases depends on Ubx2p/Sel1p (Neuber et al, 2005; Schuberth and Buchberger, 2005). Our data now imply that the ubiquitination of substrates by Hrd1p is an additional prerequisite for the binding of Cdc48p. Hrd1p does not associate with Cdc48p in mutants defective in the RING-domain or deleted for the Hrd1p-specific E2 UBC7. The active-site mutant Hrd1p also binds significantly less Ubx2p than the wild-type protein (data not shown). Consistently, the interaction of Ubx2p with the Doa10p ubiquitin ligase was also largely diminished when the RING-finger of Doa10p was mutated (Neuber et al, 2005). Furthermore, Hrd1p fails to associate with Cdc48p in the absence of the substrate recruitment factor Hrd3p. It is unlikely that Hrd3p directly binds to Cdc48p because Hrd3p mutants lacking the cytoplasmic and the transmembrane segment are still able to support Cdc48p association with Hrd1p. These observations strongly suggest that ubiquitinated substrate proteins establish the binding of the Cdc48p complex to Hrd1p (Figure 8). Contrariwise, the interaction of the mammalian membrane-bound ubiquitin ligases HRD1 and gp78 with p97 does not depend on their catalytic activity (Zhong et al, 2004; Ye et al, 2005). As shown for gp78, some ligases may contain distinct domains that confer p97 binding, but these are absent in yeast Hrd1p or Doa10p. In addition, gp78 binds polyubiquitinated proteins via its cytoplasmic CUE domain. The architecture of gp78 may thus display a different mode of coupling retrotranslocation with ubiquitination and degradation.
The function of Der1p in the ligase complex is less clear. To some extent, Der1p seems to contribute to the recruitment of some substrates, since reduced binding of CPY* to the ligase complex is only observed in the absence of both Hrd3p and Der1p. However, Cdc48p recruitment to Hrd1p is not disturbed in Δder1 cells, while HRD3 deletion abolishes Cdc48p binding. This is in line with the observation that Der1p mutants affect turnover of only a subset of Hrd1p substrates (Hitt and Wolf, 2004; Supplementary Figure 3). Accordingly, the binding of heterologously expressed CD4 to Hrd3p is not affected in DER1-deleted cells. It has recently been proposed that Derlin-1, a mammalian homolog of Der1p, functions as an export channel for malfolded ER proteins (Lilley and Ploegh, 2004; Ye et al, 2004). Our data neither do support nor rule out this possibility. The observation that Der1p itself displays weak interaction with Cdc48p indicates that it fulfills functions on both sides of the membrane.
Since the Hrd1p complex recruits misfolded proteins in the ER lumen and ubiquitinates them at the cytoplasmic face of the ER membrane, it is expected to reside in spatial proximity to the dislocation channel. Earlier studies hypothesized that the Sec61p import channel also transports proteins back into the cytosol. In our experimental setup, we failed to detect an association of Sec61p with the Hrd1p ligase complex. Hrd1p contains six transmembrane segments and it is tempting to speculate that the Hrd1p ligase complex itself forms a channel within the lipid bilayer, which facilitates the export of proteins from the ER. Such a function was earlier proposed for the transmembrane ubiquitin ligases Doa10p (Swanson et al, 2001) and gp78 (Zhong et al, 2004). Der1p may serve as an auxiliary component of this channel that mediates the initial step in feeding a soluble substrate into an export pore. In such a scenario, Hrd1p serves as a linchpin that recruits all necessary components required for substrate selection, dislocation, ubiquitination, and substrate mobilization from the ER membrane.
Materials and methods
Materials and reagents
Monoclonal 12CH5 anti-hemagglutinin (HA) and 9E10 monoclonal anti-myc antibodies were covalently coupled to Protein-G sepharose beads (Amersham Corporation). DH Wolf provided a polyclonal antiserum specific for a region within the cytoplasmic C-terminal tail of Hrd1p. Antibodies specific for Cue1p, Ubc7p, Ubc6p, and Sec61p were described previously (Sommer and Jentsch, 1993; Biederer et al, 1997). Antibodies directed against CD4 (Santa Cruz) and CPY (Molecular Probes) are commercially available. Polyclonal antibodies specific for Cdc48p and Hrd3p were raised in rabbit and affinity-purified.
Yeast strains and plasmids
Yeast strains used in this study are listed in Supplementary Table 1. Standard protocols were followed for preparation of yeast media, yeast sporulation, and tetrad dissection (Ausubel, 1993–2004). The lithium acetate method as described was used to transform yeast cells (Gauss et al, 2005). All yeast strains were haploid descendents of the wild-type strain DF5 and had the following genotype: trp1-1(am), his3-Δ200, ura3-52, lys2-801, leu2-3,-112. Yeast strains expressing CPY* were derived from YTX140 (Biederer et al, 1996) that additionally contained the prc1-1 allele. Specific methods based on polymerase–chain reactions were used to introduce C-terminally (Longtine et al, 1998; Knop et al, 1999) and internally epitope-tagged (Gauss et al, 2005) versions of Hrd1p, Hrd3p, Der1p, and Npl4p. These epitope-tagged forms replaced the endogenous copy of the respective genes at their natural chromosomal loci and were also expressed from the native promoter. A similar strategy was applied to construct yeast strains expressing chromosomally encoded Hrd1p-Δcyt (lacking aa 243–551), Hrd1p-ΔRING (lacking aa 347–401), and C-terminally truncated versions of Hrd3p (Hrd3p-Δ801, -Δ769, and -Δ664). The function of the epitope-tagged proteins was controlled by monitoring degradation of CPY*. Gene deletions were performed as described (Longtine et al, 1998; Gueldener et al, 2002). Plasmid YCP/der3C399S encoding mutant Hrd1p with the active-site RING-finger cysteine 399 replaced by serine has been described (Bordallo and Wolf, 1999). The active site cysteine 89 of Ubc7p was replaced by serine via site-directed mutagenesis and the resulting construct was integrated into its original locus, thereby replacing endogenous UBC7. CD4 was heterologously expressed in yeast from plasmid pBM108 as described (Meusser and Sommer, 2004).
Co-immunoprecipitation
Yeast cells from a 50 ml overnight culture were diluted in 100 ml to OD600=0.25 and grown to OD600=1. Cells were harvested by centrifugation at 2000 g for 2 min, washed once with 40 ml water supplemented with 1 mM PMSF, and resuspended in 400 μl IP15-LB (50 mM Tris–HCl, 200 mM KOAc, 1 mM EDTA, pH 7.5)+1 mM PMSF. The suspension was filled up to 2 ml with glass beads. Cells were disrupted by mixing on a Vortex at maximum speed for 2 min. Then, 3 ml IP15-LB were added, and the supernatant was clarified by centrifugation at 800 g at 4°C for 5 min. Microsomes were collected by centrifugation at 91 000 g at 4°C for 20 min, and solubilized in 500 μl IP15 (IP15-LB, 10% glycerol)+1% NP40 with agitation at 4°C for 2 h. After centrifugation at 20 000 g at 4°C for 15 min, 500 μl IP15 and 25 μl of a 50% suspension of protein-G beads covalently loaded with anti-HA or anti-myc antibodies in IP15 buffer were added to the solubilized microsomes. Beads were incubated with agitation at 4°C overnight. After washing the beads three times with 1 ml IP15+0.5% NP40, bound proteins were eluted with 50 μl 1 × SB. Aliquots of 5 μl were analyzed by SDS–PAGE (Laemmli, 1970) and immunoblotting. Usually, 10% of the input material was loaded on the gels for comparison.
Pulse-chase experiments and immunoprecipitation of CPY*
Pulse-chase experiments were carried out essentially as described by Biederer et al (1996). Exponentially grown cells were labeled at 30°C in synthetic dropout (SD) complete medium with 80 μCi/10 OD600 cells of a mixture of [35S]methionine and cysteine (in vivo cell labeling mixture, Amersham Corporation) for 12 min. Subsequently, cells were chased in fresh SD complete medium supplemented with 3.3 mM (NH4)3SO4, 0.013% methionine, 0.01% cysteine at 30°C. At the indicated time, aliquots were removed. The cells were lysed in 100 μl cold lysis buffer (50 mM Tris–HCl pH 7.5, 1% SDS, 1 mM PMSF) using glass beads. After dilution with 900 μl IP-dilution buffer (50 mM Tris–HCl pH 7.5, 1.1% Trition, 165 mM NaCl, 5.5 mM EDTA, 1 mM PMSF), the samples were cleared by centrifugation and subjected to immunoprecipitation with anti-CPY antibodies. The precipitated material was treated with 8 U/μl N-Glycosidase-F (Roche) in 10 μl IP-buffer, 0.02% SDS, 1% β-mercaptoethanol at 37°C for 1 h and analyzed via SDS–PAGE autoradiography.
Supplementary Material
Supplementary Figure 1
{kind=link}
Supplementary Figure 2
{kind=link}
Supplementary Figure 3
{kind=link}
Supplementary Figure Legends
Supplementary Table 1
Acknowledgments
We thank Corinna Volkwein and Angelika Wittstruck for excellent technical assistance and Alistair Garratt, Christian Hirsch, and the members of the Sommer group for critical comments on the manuscript. We also thank Dieter H Wolf for providing plasmids and reagents. Part of this work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) and the EU Network of Excellence RUBICON. RG thanks the Boehringer Ingelheim Fonds for support.
References
- Ausubel FM (eds) (1993–2004) Current Protocols in Molecular Biology. New York: John Wiley & Sons, Inc. [Google Scholar]
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY (2001a) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol 3: 24–29 [DOI] [PubMed] [Google Scholar]
- Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY (2001b) HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell 12: 4114–4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Volkwein C, Sommer T (1996) Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin–proteasome pathway. EMBO J 15: 2069–2076 [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Volkwein C, Sommer T (1997) Role of Cue1p in ubiquitination and degradation at the ER surface. Science 278: 1806–1809 [DOI] [PubMed] [Google Scholar]
- Bordallo J, Wolf DH (1999) A RING-H2 finger motif is essential for the function of Der3/Hrd1 in endoplasmic reticulum associated protein degradation in the yeast Saccharomyces cerevisiae. FEBS Lett 448: 244–248 [DOI] [PubMed] [Google Scholar]
- Braun S, Matuschewski K, Rape M, Thoms S, Jentsch S (2002) Role of the ubiquitin-selective CDC48(UFD1/NPL4)chaperone (segregase) in ERAD of OLE1 and other substrates. EMBO J 21: 615–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai RM, Chen E, Longo DL, Gorbea CM, Li CC (1998) Involvement of valosin-containing protein, an ATPase Co-purified with IkappaBalpha and 26 S proteasome, in ubiquitin–proteasome-mediated degradation of IkappaBalpha. J Biol Chem 273: 3562–3573 [DOI] [PubMed] [Google Scholar]
- Deak PM, Wolf DH (2001) Membrane topology and function of Der3/Hrd1p as a ubiquitin–protein ligase (E3) involved in endoplasmic reticulum degradation. J Biol Chem 276: 10663–10669 [DOI] [PubMed] [Google Scholar]
- Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM (2001) The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci USA 98: 14422–14427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T (2000) A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol 2: 379–384 [DOI] [PubMed] [Google Scholar]
- Fu X, Ng C, Feng D, Liang C (2003) Cdc48p is required for the cell cycle commitment point at Start via degradation of the G1-CDK inhibitor Far1p. J Cell Biol 163: 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY (2000) Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol 151: 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauss R, Trautwein M, Sommer T, Spang A (2005) New modules for the repeated internal and N-terminal epitope tagging of genes in Saccharomyces cerevisiae. Yeast 22: 1–12 [DOI] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY (2002) ER-associated degradation in protein quality control and cellular regulation. Curr Opin Cell Biol 14: 476–482 [DOI] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, Rine J (1996) Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell 7: 2029–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitt R, Wolf DH (2004) Der1p, a protein required for degradation of malfolded soluble proteins of the endoplasmic reticulum: topology and Der1-like proteins. FEMS Yeast Res 4: 721–729 [DOI] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T (2002) Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol 4: 134–139 [DOI] [PubMed] [Google Scholar]
- Kikkert M, Doolman R, Dai M, Avner R, Hassink G, van Voorden S, Thanedar S, Roitelman J, Chau V, Wiertz E (2004) Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J Biol Chem 279: 3525–3534 [DOI] [PubMed] [Google Scholar]
- Knop M, Finger A, Braun T, Hellmuth K, Wolf DH (1996) Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J 15: 753–763 [PMC free article] [PubMed] [Google Scholar]
- Knop M, Siegers K, Pereira G, Zachariae W, Winsor B, Nasmyth K, Schiebel E (1999) Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15: 963–972 [DOI] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL (2004) A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429: 834–840 [DOI] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL (2005) Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc Natl Acad Sci USA 102: 14296–14301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Meusser B, Hirsch C, Jarosch E, Sommer T (2005) ERAD: the long road to destruction. Nat Cell Biol 7: 766–772 [DOI] [PubMed] [Google Scholar]
- Meusser B, Sommer T (2004) Vpu-mediated degradation of CD4 reconstituted in yeast reveals mechanistic differences to cellular ER-associated protein degradation. Mol Cell 14: 247–258 [DOI] [PubMed] [Google Scholar]
- Neuber O, Jarosch E, Volkwein C, Walter J, Sommer T (2005) Ubx2 links the Cdc48 complex to ER-associated protein degradation. Nat Cell Biol 7: 993–998 [DOI] [PubMed] [Google Scholar]
- Plemper RK, Bordallo J, Deak PM, Taxis C, Hitt R, Wolf DH (1999) Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J Cell Sci 112 (Part 22): 4123–4134 [DOI] [PubMed] [Google Scholar]
- Ponting CP (2000) Proteins of the endoplasmic-reticulum-associated degradation pathway: domain detection and function prediction. Biochem J 351 (Part 2): 527–535 [PMC free article] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S (2002) AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol 22: 626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S (2001) Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin-selective chaperone. Cell 107: 667–677 [DOI] [PubMed] [Google Scholar]
- Romisch K (2005) Endoplasmic reticulum-associated degradation. Annu Rev Cell Dev Biol 21: 435–456 [DOI] [PubMed] [Google Scholar]
- Rouiller I, Butel VM, Latterich M, Milligan RA, Wilson-Kubalek EM (2000) A major conformational change in p97 AAA ATPase upon ATP binding. Mol Cell 6: 1485–1490 [DOI] [PubMed] [Google Scholar]
- Schuberth C, Buchberger A (2005) Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat Cell Biol 7: 999–1006 [DOI] [PubMed] [Google Scholar]
- Sommer T, Jentsch S (1993) A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature 365: 176–179 [DOI] [PubMed] [Google Scholar]
- Swanson R, Locher M, Hochstrasser M (2001) A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev 15: 2660–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxis C, Hitt R, Park SH, Deak PM, Kostova Z, Wolf DH (2003) Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J Biol Chem 278: 35903–35913 [DOI] [PubMed] [Google Scholar]
- Tsai B, Ye Y, Rapoport TA (2002) Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol 3: 246–255 [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414: 652–656 [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, Rapoport TA (2003) Function of the p97–Ufd1–Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J Cell Biol 162: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA (2005) Inaugural Article: Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci USA 102: 14132–14138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA (2004) A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429: 841–847 [DOI] [PubMed] [Google Scholar]
- Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, Gorman MA, Kondo H, Dokurno P, Lally J, Leonard G, Meyer H, van Heel M, Freemont PS (2000) Structure of the AAA ATPase p97. Mol Cell 6: 1473–1484 [DOI] [PubMed] [Google Scholar]
- Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S (2004) AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem 279: 45676–45684 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure Legends
Supplementary Table 1