

Abstract
There are eight thermosensitive TRP (transient receptor potential) channels in mammals, and there might be other TRP channels sensitive to temperature stimuli. Here, we demonstrate that TRPM2 can be activated by exposure to warm temperatures (>35°C) apparently via direct heat-evoked channel gating. β-NAD+- or ADP-ribose-evoked TRPM2 activity is robustly potentiated at elevated temperatures. We also show that, even though cyclic ADP-ribose (cADPR) does not activate TRPM2 at 25°C, co-application of heat and intracellular cADPR dramatically potentiates TRPM2 activity. Heat and cADPR evoke similar responses in rat insulinoma RIN-5F cells, which express TRPM2 endogenously. In pancreatic islets, TRPM2 is coexpressed with insulin, and mild heating of these cells evokes increases in both cytosolic Ca2+ and insulin release, which is KATP channel-independent and protein kinase A-mediated. Heat-evoked responses in both RIN-5F cells and pancreatic islets are significantly diminished by treatment with TRPM2-specific siRNA. These results identify TRPM2 as a potential molecular target for cADPR, and suggest that TRPM2 regulates Ca2+ entry into pancreatic β-cells at body temperature depending on the production of cADPR-related molecules, thereby regulating insulin secretion.
Keywords: cyclic ADP-ribose, insulin, temperature, TRPM2
Introduction
TRP (transient receptor potential) channels were first described in Drosophila, where photoreceptors carrying trp gene mutations exhibited an abnormal transient responsiveness to continuous light (Montell and Rubin, 1989). In mammals, TRP channels comprise six related protein families (TRPC, TRPV, TRPM, TRPA, TRPML, TRPP) (Minke and Cook, 2002; Clapham, 2003; Montell, 2005). In general, TRP channels are ubiquitously expressed, indicating that most cells have a number of TRP channel proteins. While physiological functions for most TRP channels remain unknown, this wide distribution indicates that biological functions and activation mechanisms for these channels are diverse. Perhaps, TRP channels are best recognized for their contributions to sensory transduction, responding to temperature, nociceptive stimuli, touch, osmolarity, pheromones and other stimuli from both within and outside the cell. In a sense, their role is much broader than classical sensory transduction.
Among the huge TRP super-family of ion channels, some have been proven to be involved in thermosensation (Benham et al, 2003; Jordt et al, 2003; Patapoutian et al, 2003; Tominaga and Caterina, 2004). Insight into the molecular nature of temperature-gated ion channels came with the cloning of the capsaicin receptor, TRPV1 (also known as VR1) and the recognition that this ion channel protein could be activated by elevated temperatures with a threshold near 43°C (Caterina et al, 1997; Tominaga et al, 1998; Caterina and Julius, 2001). Three other TRPV channels (TRPV2, TRPV3 and TRPV4) and two TRPM channels (TRPM4 and TRPM5) have been cloned and characterized as thermosensors (Caterina et al, 1999; Guler et al, 2002; Peier et al, 2002b; Smith et al, 2002; Watanabe et al, 2002; Xu et al, 2002; Talavera et al, 2005). The threshold temperatures for the activation of these channels range from relatively warm (TRPV3, TRPV4, TRPM4 and TRPM5) to extremely hot (TRPV2). In contrast to the six heat-sensitive TRPV channels, TRPM8 and TRPA1 have been found to be activated by cold stimuli (McKemy et al, 2002; Peier et al, 2002a; Story et al, 2003). Many of the mammalian thermosensitive TRP channels (thermo-TRPs) identified to date can alternatively be activated by chemical stimuli, such as capsaicin for TRPV1 (Caterina et al, 1997; McKemy et al, 2002; Peier et al, 2002a; Bandell et al, 2004; Jordt et al, 2004; Moqrich et al, 2005), and a shift of voltage dependence by temperature change has been reported to be a fundamental mechanism for thermal activation in some of them (Brauchi et al, 2004; Voets et al, 2004; Nilius et al, 2005).
We sought to determine whether additional TRP channels might exhibit thermosensitivity. Among those we focused on TRPM2 (previously named TRPC7 or LTRPC2), a channel known to be activated by nicotinamide adenine dinucleotide (β-NAD+), adenosine 5′-diphosphoribose (ADPR) or hydrogen peroxide (Perraud et al, 2001; Sano et al, 2001; Hara et al, 2002), because it is phylogenetically very close to the cold-gated channel, TRPM8 (Clapham, 2003). While TRPM2 is dominantly expressed in the brain, it is also detected in many other tissues, including bone marrow, spleen, heart, liver and lung. Native TRPM2 currents have been recorded from U937 monocyte cell line (Sano et al, 2001), neutrophil cell line (Heiner et al, 2003), Jurkat T cells (Gasser et al, 2006), microglia (Kraft et al, 2004) and CRI-G1 insulinoma cells (Inamura et al, 2003). We observed that TRPM2 responses were evoked by heat over 35°C and that activation by β-NAD+ or ADPR was greatly potentiated by heat. A related molecule, cyclic ADP-ribose (cADPR) catalyzed from β-NAD+ by ADP-ribosyl cyclase (CD38) is a well-known messenger molecule for Ca2+ signaling in a variety of cells (Guse, 2000; Lee, 2002; Berridge et al, 2003), and has been believed to be important with regard to potential roles in insulin secretion from pancreatic β-sells (Takasawa and Okamoto, 2002), although there is some debate about it. However, cADPR was not found to be an agonist of TRPM2 at room temperature (Perraud et al, 2001; Hara et al, 2002). We found that cADPR activates TRPM2 at body temperature similar to β-NAD+ and ADPR. TRPM2 expression was observed in rat insulinoma RIN-5F cells and rat pancreatic β-cells where heat-evoked responses, including insulin secretion, were observed through endogenous TRPM2 proteins. Thus, TRPM2 activated by cADPR may play an important role in the regulation of insulin secretion in pancreas at body temperature.
Results
Heat-evoked TRPM2 activation in HEK293 cells
We determined the change of cytosolic-free Ca2+ concentration ([Ca2+]i) of the cells expressing human TRPM2 upon cold stimulus with a temperature as low as 5°C using a Ca2+-indicator dye, fura-2, since TRPM2 is phylogenetically very close to the cold stimulus-activated channel, TRPM8. We used CHO-K1 cells for this experiment because some [Ca2+]i increase was observed in HEK293 cells but not in CHO-K1 cells without TRPM2 expression upon cold stimulus. We could not detect any [Ca2+]i changes upon cold stimulus (data not shown). Surprisingly, however, human TRPM2-expressing HEK293 cells (hTRPM2-HEK) but not vector-transfected cells showed significant [Ca2+]i increases upon heating with a threshold of about 40°C (Figures 1A and B), suggesting that TRPM2 is activated by warm temperatures. This [Ca2+]i increase was abolished in the absence of extracellular Ca2+ (Figure 1C), suggesting that Ca2+ influx through TRPM2 channels causes the [Ca2+]i increase.
Figure 1.
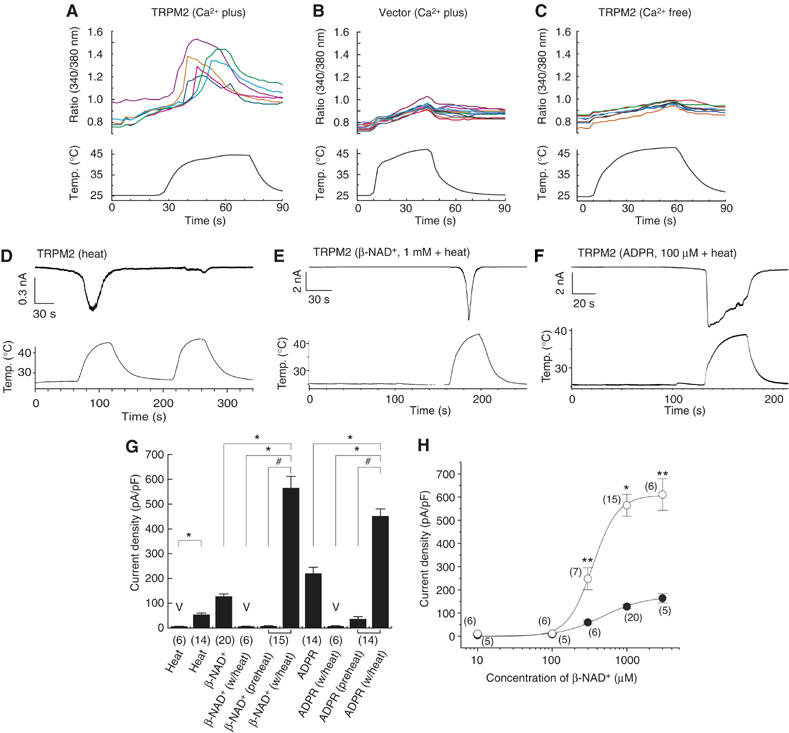
Heat-evoked responses in HEK293 cells expressing TRPM2. (A–C) Representative traces of [Ca2+]i change by heat in cells expressing TRPM2 (A), vector-transfected cells (B) or in the absence of extracellular Ca2+ (C). (D–F) Representative TRPM2-mediated whole-cell current responses by heat without (D) or with β-NAD+ (E) or ADPR (F). (G) Comparison of current densities of responses by the indicated stimuli in cells transfected with vector plasmid (V) or TRPM2 (±s.e.m.). ‘Preheat' indicates current responses just before heat stimulation. *P<0.01; #P<0.01. Numbers in parenthesis indicate cells tested. Vh: −60 mV. (H) Concentration-dependent profiles of β-NAD+ for TRPM2 activation with (open circle) or without (closed circle) heat (40°C). *P<0.01; **P<0.05. Numbers in parenthesis indicate cells tested. Vh: −60 mV.
Next, we measured TRPM2-mediated current responses in hTRPM2-HEK using the patch-clamp technique. We first confirmed TRPM2 activation by its reported stimuli, β-NAD+ and ADPR (Supplementary Figures 1A and B). At a holding potential of −60 mV, a gradual increase in inward current was observed several minutes after the establishment of the whole-cell configuration with a pipette solution containing β-NAD+ or ADPR at room temperature (25°C) (127.7±10.1 pA/pF for β-NAD+, n=20 and 217.1±23.0 pA/pF for ADPR, n=14), although β-NAD+-evoked responses might be induced by ADPR contaminated in the β-NAD+ as reported recently (Kolisek et al, 2005). Heat alone induced significant current responses in hTRPM2-HEK (54.5±6.0 pA/pF, n=14) but not in vector-transfected cells (6.4±0.7 pA/pF, n=6, P<0.01) (Figure 1D and Supplementary Figure 1C), indicating that TRPM2 can be activated by heat. We applied heat to cells already exposed to β-NAD+ (1 mM) or ADPR (100 μM). When temperature ramps were applied to cells showing small inward currents evoked by either β-NAD+ or ADPR, the current responses were immediately observed and robustly potentiated (Figures 1E and F). To facilitate the observation of a maximal TRPM2 response to heat, we applied heat ramps approximately 2–3 min after establishing the whole-cell configuration. Much larger current responses than those induced by heat, β-NAD+ or ADPR alone were detected under these circumstances (7.1±1.2 and 564.5±46.7 pA/pF before and after heat in β-NAD+-exposed cells, respectively, n=15, P<0.01; 36.4±9.5 and 452.2±28.7 pA/pF before and after heat in ADPR-exposed cells, respectively, n=14, P<0.01) (Figure 1G).
To examine how heat changes TRPM2 responsiveness, we measured TRPM2 currents in cells by serially applying a range of concentrations of β-NAD+ with or without heat. Substantial inward currents were observed upon heat stimulation at low concentrations of β-NAD+, which induced small currents by itself at 25°C. An increase in heat-evoked TRPM2 responses was detected in cells exposed to 300 μM β-NAD+ and even larger increases to 1 or 3 mM β-NAD+, indicating that the synergistic effects of the two stimuli depend on β-NAD+ concentration (Figure 1H). Whereas maximal current density during heating was 3.7 times bigger than that at 25°C, the EC50 for TRPM2 activation by β-NAD+ was not significantly changed by heat (349±16 and 477±66 μM with and without heat, respectively), suggesting that heat does not alter the kinetics of β-NAD+ binding to TRPM2.
TRPM2 activation by cyclic ADP-ribose and heat
We next examined whether a related molecule, cADPR, could interact with heat at TRPM2. When cADPR (100 μM) was included in the pipette, little current response was observed at 25°C. However, heat stimulation evoked large inward currents in these cells (Figures 2A and B). cADPR-activated TRPM2 currents showed desensitization upon repeated heat stimuli similar to that observed in β-NAD+-induced TRPM2 responses (Figure 2A). These results indicate that, at mammalian body temperature, TRPM2 functions as a molecular target of cADPR. Only one report (Kolisek et al, 2005), to date, describing the ability of cADPR to activate TRPM2 in heterologous expression systems might therefore be owing to the conduct of experiments at room temperature (Perraud et al, 2001; Hara et al, 2002). The similarities observed between β-NAD+- and cADPR-induced currents suggest that cADPR, like β-NAD+, binds to the Nudix motif of TRPM2 to exert its effects (Hara et al, 2002). Consistent with this hypothesis, a TRPM2 mutant lacking the Nudix motif (ΔNudix) failed to respond to the combination of heat and cADPR (6.8±1.1 pA/pF, n=5) (Figure 2B). The binding of cADPR to the Nudix motif of TRPM2 was further supported biochemically, by the finding that cADPR considerably inhibited binding of [32P]-β-NAD+ to TRPM2 (Figure 2C) and by the cADPR concentration dependence of TRPM2 currents evoked by cADPR with heat (Figure 2D).
Figure 2.
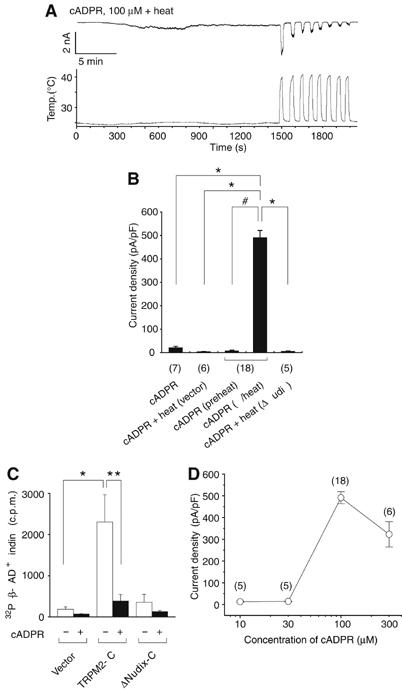
Heat and cADPR activate TRPM2 through binding to its Nudix motif in HEK293 cells. (A) Heat-evoked activation of TRPM2 by cADPR with repetitive heat stimuli. (B) Comparison of current densities of responses evoked by heat, cADPR (100 μM) or heat with cADPR in HEK293 cells transfected with vector plasmid, TRPM2 or TRPM2 mutant lacking Nudix motif (ΔNudix). ‘Preheat' indicates current responses just before heat stimulation. *P<0.01, two-tailed unpaired t-test. #P<0.01, two-tailed paired t-test. Numbers in parenthesis indicate cells tested. Vh: −60 mV. (C) β-NAD+ binding to the TRPM2 fusion proteins in the presence (+) or absence (−) of cADPR. *P<0.01; **P<0.05. (D) A dose-dependent profile of cADPR for the activation of TRPM2 at 40°C.
Body temperature gates TRPM2 directly
We then carried out more detailed analyses of heat-evoked TRPM2 current responses. The temperature-response profile with or without β-NAD+ or cADPR revealed that the temperature threshold was about 35°C with no significant difference in the presence or absence of β-NAD+ or cADPR (33.5±0.3°C, n=8; 35.0±0.8°C, n=10; 35.1±0.9°C, n=9 for heat alone, β-NAD+ plus heat or cADPR plus heat, respectively) (Figures 3A, Supplementary Figure 2A and data not shown). This finding differs from that reported for other thermo-TRPs exposed to simultaneous thermal and chemical stimulation; decreases and increases of the activation temperature threshold in the presence of other effective stimuli, protons and menthol were reported for TRPV1 and TRPM8, respectively (Tominaga et al, 1998; McKemy et al, 2002). To analyze the temperature dependence of TRPM2 activation more precisely, we made Arrhenius plots for the currents activated by heat with ADPR or cADPR at both negative and positive potentials, and found that the plots showed the similar temperature threshold for TRPM2 activation to that obtained in the temperature-response profiles (35.0±0.7°C, n=4; 33.6±1.5°C, n=5; 33.9±0.6°C, n=4 for cADPR plus heat, β-NAD+ plus heat or heat alone). Q10 values were 44.4±5.3 (n=11), 44.1±7.9 (n=12), 32.3±2.9 (n=4), 38.0±3.7 (n=5) and 15.6±2.0 (n=4) for ADPR-activated TRPM2 currents at +60 mV, ADPR-activated currents at −60 mV, cADPR-activated currents at −60 mV, β-NAD+-activated currents at −60 mV and currents activated by heat alone at −60 mV, respectively (Supplementary Figures 2B–D, and data not shown). Linear current–voltage (I–V) relationships were observed for heat-evoked currents both in the presence and absence of each ligand (Figure 3B, Supplementary Figure 2E and data not shown). As the observed reversal potentials were close to 0 mV (Erev=+1.1±2.3 mV, n=3 for heat; Erev=−1.9±1.2 mV, n=3 for heat plus cADPR), the heat-evoked responses most likely involve a nonselective cation channel. Heat-evoked TRPM2 currents exhibited a relatively higher permeability for divalent cations than monovalent cations (PCa/PCs=2.8±0.1, n=3; PMg/PCs=2.2±0.1, n=5; PNa/PCs=0.5±0.1, n=3) (Supplementary Figure 2F). This finding somehow contradicts previous reports in which Ca2+ was shown to be less permeant than Na+ in β-NAD+- or ADPR-induced currents (Perraud et al, 2001; Sano et al, 2001). The relative high permeability of heat-evoked TRPM2 responses to Ca2+ (PCa/PNa=5.83) might explain the robust [Ca2+]i increase upon heat stimulation (Figure 1A). These electrophysiological properties indicate that heat does activate TRPM2 and suggest that heat and ligands share some overlapping mechanism for TRPM2 activation. Temperature-induced change in cADPR-related enzyme activities might also be involved in the TRPM2 activation by heat with cADPR. HEK293 cells expressing mouse TRPM2 (mTRPM2-HEK) exhibited essentially similar current responses to those observed in hTRPM2-HEK (data not shown).
Figure 3.
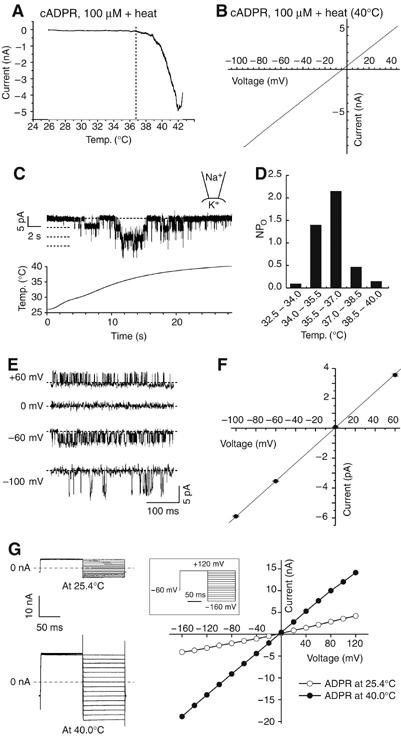
Electrophysiological properties of heat-gated current responses in HEK293 cells expressing TRPM2. (A) A representative temperature–response profile of heat-evoked TRPM2 currents in the presence of cADPR (100 μM). Vh: −60 mV. (B) Current–voltage relationship for a heat-evoked current in the presence of cADPR (100 μM) in hTRPM2-HEK. (C) A representative trace of heat-evoked single-channel responses at −60 mV in inside-out configuration. A lower panel shows bath temperature. Broken lines indicate 0, 1, 2 and 3 channel open levels. (D). NP0 values (calculated from data shown in C) plotted against bath temperatures. (E) Traces of heat-evoked TRPM2 single-channel currents in inside-out patches at the indicated holding potentials. Broken lines indicate the closed-channel level. (F) Current–voltage curve of mean single-channel amplitudes (±s.e.m.). (G) Representative current responses to the voltage step-pulses (−160 to +120 mV with 20 mV increment for 100 ms, inset) at 25.4 and 40.0°C (left), and current–voltage relationship (right).
How do heat-evoked TRPM2 currents behave at the single-channel level? To address this question, we performed single-channel recordings in inside-out membrane patches excised from hTRPM2-HEK. When we applied heat ramps to these patches, well-resolved single-channel currents were observed with a temperature threshold of about 34°C and maximal activities at about 36°C (Figure 3C). This finding demonstrates the existence of heat-gated ion channels within this patch whose activation does not depend upon soluble cytoplasmic components. Heat-evoked channel activity became relatively diminished when we further increased temperature over 37°C, suggesting that the optimal temperature for TRPM2 activation is near core body temperature. This phenomenon was more clearly recognized when NP0 values were plotted against temperatures (Figure 3D). The I–V relation at the single-channel level was almost identical to that established in the whole-cell configuration (Figures 3E and F). A slope conductance for Na+ as the sole charge carrier was 60.6 pS. These single-channel properties are like those described for β-NAD+- or ADPR-gated TRPM2 currents (Perraud et al, 2001; Sano et al, 2001; Hara et al, 2002). It has been reported that a shift of voltage dependence by temperature is the fundamental mechanism of temperature-evoked activation of TRPV1, TRPM8, TRPM4 or TRPM5 (Brauchi et al, 2004; Voets et al, 2004; Nilius et al, 2005). Therefore, we applied voltage step-pulses to heat-activated TRPM2 currents to examine whether a similar mechanism is involved in TRPM2 activation by warm stimulus. As shown in the Figure 3G, temperature elevation simply increased the slope without changing the linear I–V relationship, suggesting that temperature activation of TRPM2 involves a different mechanism from that reported for TRPV1, TRPM8, TRPM4 or TRPM5.
Expression of TRPM2 in pancreatic β-cells
To explore the potential physiological significance of heat-evoked activation of TRPM2, we first examined the expression of endogenous TRPM2. An anti-mouse TRPM2 antibody specifically recognized a protein with similar molecular weight (about 171 kDa) in lysates from hTRPM2-HEK or mTRPM2-HEK (Figure 4A). Specificity of the antibody was confirmed in the absorption experiment using the immunogenic peptide (Supplementary Figure 3A). We could not detect TRPM2 expression using this antibody in mouse DRG neurons, where TRPV1 is expressed (Supplementary Figures 3B and C). The fact that TRPM2 can be activated by cADPR with heat prompted us to examine TRPM2 expression in pancreatic β-cells where cADPR is known to be involved in insulin secretion (Takasawa et al, 1993). The antibody recognized TRPM2 in lysates from rat insulinoma RIN-5F cells (Figure 4A). We detected clear TRPM2-like immunoreactivity not only in hTRPM2-HEK or mTRPM2-HEK but also in RIN-5F cells (Figure 4B, and data not shown). TRPM2 was highly coexpressed with insulin, a marker for β-cells, but not with glucagon, a marker for α-cells in mouse pancreatic islets (Figure 4C), suggesting the important and specific function of TRPM2 in pancreatic β-cells.
Figure 4.
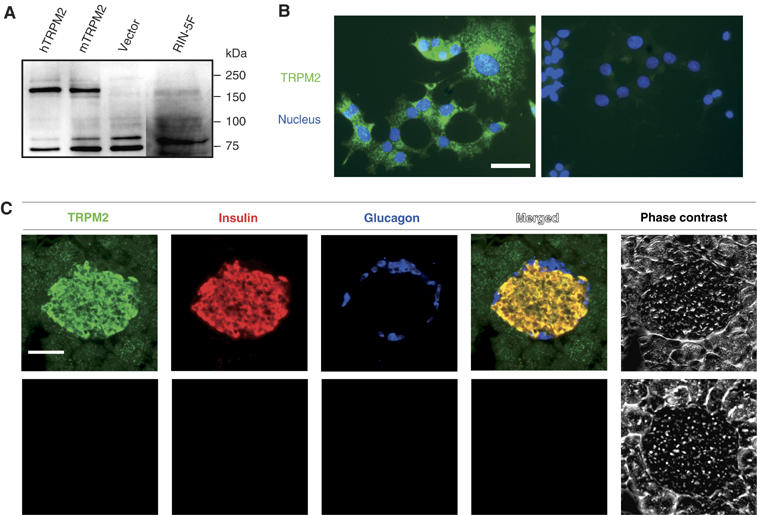
TRPM2 expression in insulin-secreting cells. (A) Immunoblot analysis reveals a specific band (around 171 kDa) in lysates from hTRPM2-HEK, mTRPM2-HEK and RIN-5F cells. (B) TRPM2-like immunoreactivity in RIN-5F cells but not in control cells (right, without anti-TRPM2 antibody). Scale bar, 50 μm. (C) Triple immunofluorescent analysis reveals coexpression of TRPM2 with insulin but not with glucagon in mouse pancreas. Lower panels indicate negative controls without primary antibodies. Scale bar, 50 μm.
Heat-evoked responses in RIN-5F cells
We next investigated heat-evoked responses in RIN-5F cells, using both Ca2+-imaging and patch-clamp methods. In these cells, as in hTRPM2-HEK, [Ca2+]i was increased upon heat stimulation with a temperature threshold of about 40°C in the presence of extracellular Ca2+ (Supplementary Figures 4A and B). Significant and desensitizing inward currents were observed in the presence or absence of cADPR when temperature ramps were applied at −60 mV (19.3±1.9 pA/pF for heat alone, n=4; 8.1±1.9 and 144.2±26.7 pA/pF before and after heat in cADPR-exposed cells, P<0.05, n=6) (Figure 5A, and data hot shown). The temperature threshold for heat-evoked responses in RIN-5F cells with cADPR was 34.0±1.0°C (n=5) (Figure 5B) and the whole-cell currents showed a linear I–V relationship with a reversal potential near 0 mV (+3.4 mV±1.2, n=3) (Figure 5C). It should be noted that the threshold temperature for heat-evoked activation was not changed, regardless of the heat-stimulus sequence, a quite different phenomenon from those reported for TRPV1 and TRPV2, whose temperature thresholds for activation decrease upon repetitive heat stimulation (Caterina et al, 1999). Heat-evoked currents in the RIN-5F cells decreased upon further temperature increases over 40–42°C, suggesting the existence of an optimal temperature for activation. These electrophysiological properties are almost identical to those obtained from hTRPM2-HEK, suggesting that endogenous TRPM2 functions as a thermosensor and a target of cADPR. To further prove the involvement of endogenous TRPM2 in the heat-evoked responses, we treated RIN-5F cells with a TRPM2-specific siRNA (siTRPM2). This intervention reduced expression of TRPM2 both at protein and mRNA levels, whereas treatment with control siRNA did not (Figure 5D). Heat failed to increase [Ca2+]i upon treatment with siTRPM2, but evoked normal responses with control siRNA (Figures 5E and F), indicating that endogenous TRPM2 is responsible for heat-evoked [Ca2+]i increase in RIN-5F cells.
Figure 5.
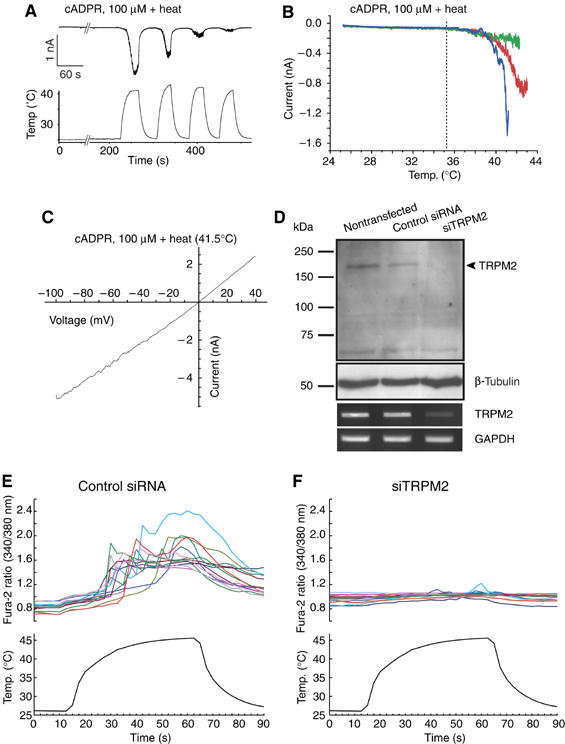
Heat-evoked responses in RIN-5F cells through TRPM2 activation. (A) A representative heat-evoked whole-cell current trace in RIN-5F cells in the presence of cADPR (100 μM). A lower panel shows bath temperature. Vh: −60 mV. (B) Temperature–response profiles of heat-evoked currents in the presence of cADPR (100 μM) shown in (A). Blue, red and green lines indicate the profiles obtained in the first, the second and the third heat stimuli, respectively. Vh: −60 mV. (C) Current–voltage relationship for heat-evoked currents in the presence of cADPR (100 μM). (D) Reduction of TRPM2 protein (upper) and mRNA (lower) expression by treatment with TRPM2-specific siRNA (siTRPM2) but not with control siRNA. (E, F) Increase of [Ca2+]i by heat in RIN-5F cells treated with control siRNA (E) but not with siTRPM2 (F). Lower panels show bath temperature.
Heat-evoked responses in pancreatic β-cells
To assay for heat-evoked responses in the rat primary pancreatic β-cells, we utilized Ca2+ imaging in dissociated rat pancreatic cells. Isolated cells (49.6%) were insulin positive and 24.5% were glucagon positive. TRPM2 was highly colocalized with insulin but not with glucagon (Figure 6A), a phenomenon similar to that observed in mouse pancreas sections. Almost all TRPM2-positive cells costained with anti-insulin antibody (91.7%). Upon exposure to heat, 55.3% of the isolated pancreatic cells showed a [Ca2+]i increase in the presence of extracellular Ca2+ (Figures 6B–D) like those seen in hTRPM2-HEK and RIN-5F cells. The percentage of heat-sensitive cells in the isolated pancreatic cells (55.3%) was almost equal to that of TRPM2-positive cells (53.1%), suggesting that TRPM2 is involved in the heat-evoked responses.
Figure 6.
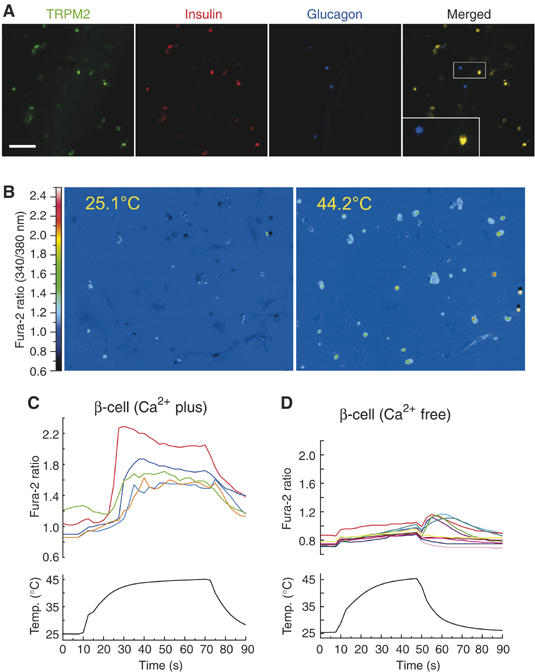
Expression of TRPM2 and heat-evoked responses in isolated pancreatic cells. (A) Expression of TRPM2 (green), insulin (red) and glucagon (blue) in the isolated rat pancreatic cells. An inset indicates the high magnification image of the square box area. (B) Change of cytosolic Ca2+ concentration indicated by the fura-2 ratio with pseudo-color expression in response to heat stimulus. (C, D) Representative traces of [Ca2+]i change by heat in the presence (C) or absence (D) of extracellular Ca2+.
Insulin release involving endogenous TRPM2 in pancreatic islets
Finally, we examined the temperature effects on insulin release from pancreatic islets. We examined the effects of heat stimulation (40°C, 5 min) on insulin release from islets. In this experiment, pancreatic islets were cultured overnight at 33°C and incubated for 60 min at 29°C to minimize TRPM2 activation at 37°C before measurement. Even such short heat stimulation caused a significant increase in insulin release from islets incubated with either 3.3 mM glucose (G3.3) or 16.7 mM glucose (G16.7) (Figure 7A). Furthermore, the amount of insulin released from the islets heated for 5 min (40°C) after 29°C incubation was comparable to that released from islets incubated for 60 min at 37°C (Figure 7A), suggesting that exposure to body temperature causes insulin release, probably through TRPM2 activation. To prove the involvement of endogenous TRPM2 in the heat-evoked insulin release, we first examined the effects of known TRPM2 inhibitors on insulin release. Both econazole (Eco, 10 μM) and flufenamic acid (FFA, 200 μM) significantly inhibited glucose-induced insulin release from pancreatic islets maintained at 37°C (49.7 and 20.1% of the control, respectively, n=4 each) (Figure 7B). Next, we treated the pancreatic islets with siTRPM2. siTRPM2 but not control siRNA caused a significant reduction of TRPM2 mRNA (Figure 7C, inset). Control siRNA-treated islets exhibited an insulin release pattern similar to that observed in naïve islets (Figure 7C, left). On the other hand, heat-induced insulin release was significantly reduced in siTRPM2-treated pancreatic islets (Figure 7C, right). These results clearly indicate the involvement of endogenous TRPM2 activation in insulin release from pancreatic islets.
Figure 7.
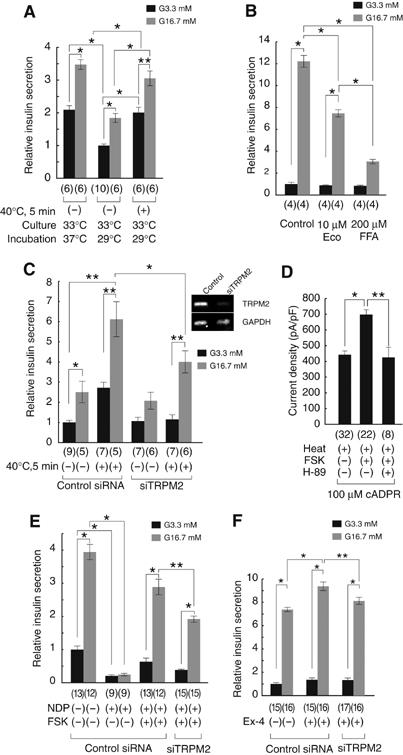
Body temperature-evoked insulin release from pancreatic islets. (A) Heat stimulus (40°C for 5 min) causes insulin release from the islets. Data from the islets incubated at 37 or 29°C (1 h) with (+) or without (−) heat stimulation (±s.e.m.) in the presence of 3.3 mM (G3.3) or 16.7 mM (G16.7) glucose after culture at 33°C (16 h). *P<0.01; **P<0.05. Numbers in parenthesis indicate samples tested. (B) Reduction of insulin release by Eco (10 μM) or FFA (200 μM) at 37°C. *P<0.01. (C) Insulin release by heat stimulus (+) was inhibited by treatment with siTRPM2 but not with control siRNA (±s.e.m.). *P<0.01; **P<0.05. An inset indicates amounts of RT–PCR products from the pancreatic islets treated with control siRNA or siTRPM2. (D) cADPR-induced TRPM2-mediated whole-cell currents were potentiated by FSK (2 μM), and the potentiation was inhibited by H-89 (1 μM). *P<0.01. (E) Increase in insulin release by FSK (2 μM) in the presence of nimodipine (NDP, 2 μM, +) was significantly reduced in the islets treated with siTRPM2 but not with control siRNA. *P<0.01; **P<0.05. Numbers in parenthesis indicate samples tested. (F) Increase in insulin release by exendin-4 (10 nM) was significantly reduced in the islets treated with siTRPM2 but not with control siRNA. *P<0.01; **P<0.05.
It is well documented that both KATP channel-dependent and -independent mechanisms are involved in insulin secretion in pancreatic β-cells and that the former mechanism involves L-type voltage-gated Ca2+ channels, whereas the latter involves several events, including cAMP production, which causes insulin release through protein kinase A (PKA)-dependent and -independent mechanisms (Henquin, 2004; Seino and Shibasaki, 2005). Therefore, it was examined which mechanism is more related to the TRPM2-mediated response. TRPM2 currents in HEK293 cells were significantly potentiated by forskolin (FSK, 2 μM) treatment (Figure 7D). The FSK-induced potentiation of TRPM2 currents was almost completely inhibited by H-89 (1 μM), a PKA inhibitor, suggesting that TRPM2 activity is enhanced by phosphorylation of TRPM2 or its closely related protein by PKA. Then, we examined the involvement of cAMP-dependent mechanism in insulin release at more physiological condition at 37°C for both culture and incubation steps. In the presence of nimodipine (NDP, 2 μM), an inhibitor of L-type voltage-gated Ca2+ channel, the FSK-induced increase in insulin release was significantly reduced in siTRPM2-treated islets in G16.7 conditions (Figure 7E), suggesting that endogenous TRPM2 is involved in the glucose-dependent and KATP channel-independent insulin release mediated by cAMP to some extent. One of the likely candidate stimuli causing insulin release through a cAMP-dependent mechanism is glucagon-like peptide 1 (GLP-1) (Lund, 2005; Meier and Nauck, 2005). Therefore, we examined GLP-1 receptor-mediated insulin release in our system. Exendin-4, a GLP-1 receptor agonist, significantly increased insulin release at 37°C, which was inhibited by both H-89 (Supplementary Figure 5) and siTRPM2 treatment (Figure 7F). These results suggest that insulin release from pancreatic β-cells involves PKA-dependent phosphorylation of TRPM2 or its closely related protein in the downstream of GLP-1 receptor activation.
Discussion
A common feature among the known thermo-TRPs is that they are expressed in sensory neurons and therefore are believed to be involved in temperature sensation by neurons, although TRPV3 and TRPV4 have been reported to be involved in thermosensation in skin epithelial cells, as well. On the other hand, TRPM2 was found to be expressed in many tissues including pancreas but not in DRG neurons, making this new thermo-TRP unique. The fact that the threshold and optimal temperatures for TRPM2 activation are near normal mammalian body temperature is in good agreement with this broad expression pattern. Our study suggests that body temperature functions as ‘an endogenous coactivator' of TRPM2, allowing this channel to act as a molecular integrator of chemical and physical signals.
We demonstrated that cADPR could activate TRPM2 in a warm environment, despite a negligible ability to activate TRPM2 at 25°C. Ca2+ release in response to cADPR was initially reported for sea urchin eggs (Clapper et al, 1987). Since then, cADPR has been increasingly recognized as a pivotal signaling molecule in such diverse functions as cell cycle regulation, egg fertilization, insulin secretion and cell proliferation in mammalian and nonmammalian cells (Guse, 2000; Lee, 2002; Berridge et al, 2003). Many of these responses have been proposed to result from cADPR action on ryanodine receptors expressed on the ER membrane. To date, however, it is not completely clear if this is the only mechanism by which cADPR exerts its Ca2+-mobilizing effect. For example, a direct effect of cADPR on Ca2+ entry was reported in human T-lymphocytes (Guse et al, 1997), and cADPR was found to regulate Ca2+ influx through plasma membrane Ca2+ channel in neutrophils (Partida-Sanchez et al, 2001). It has been reported that a temperature signaling cascade in marine sponges involves cADPR and a heat-gated cation channel, although the molecular identity of this channel is not known (Zocchi et al, 2001). This result suggests an ancient role for cADPR as an intracellular signal relating cell function to temperature, one that appears to be evolutionally conserved in more complex organisms. Together with the recent work showing cADPR and hydrogen peroxide synergize in the activation of TRPM2 (Kolisek et al, 2005), our study reveals TRPM2 as the ion channel activated by cADPR, and indicates that TRPM2 may play important functions in [Ca2+]i homeostasis in the warm environment of our body. Thirty to 100 μM of cADPR seems to be necessary to activate TRPM2 with heat, although it is not known how much amount of cADPR is produced in the native cells and concentrated beneath the plasma membrane. However, the facts that β-NAD+, ADPR and cADPR, all of which have ability to activate TRPM2, are in the same complex metabolic network and that cADPR synergizes with ADPR in the activation of TRPM2 (Kolisek et al, 2005) suggest that much less amount of each ligand is needed to activate TRPM2 in the physiological condition.
The findings reported in this study also provide evidence that the interaction of body temperature and cADPR at TRPM2 may have physiological significance in insulin-secreting pancreatic β-cells. In these cells, TRP channels have previously been proposed as candidate targets of cADPR action (Qian et al, 2002). The results in this study clearly indicate that endogenous TRPM2 is involved in insulin release from the pancreatic islets in a KATP channel-independent cAMP-mediated (probably through PKA-dependent phosphorylation) manner at body temperature, and are consistent with a previous report that cooling dissociates glucose-induced insulin release in rodent pancreatic islets although glucose metabolism is also thought to be temperature dependent (Atwater et al, 1984). What might be the relevance of TRPM2 activation to glucose-stimulated insulin release at 37°C? Body temperatures around 37°C could render TRPM2 in pancreatic islets maximally susceptible to activation by the cADPR that is produced following glucose uptake depending on metabolism (Takasawa et al, 1998). The in vivo relevance of TRPM2-mediated insulin release remains unclear. Mice lacking KATP channel (Kir 6.2) exhibit mild impairment in glucose tolerance (Miki et al, 1998), suggesting the existence of other regulators of insulin secretion such as TRPM2, although increased insulin sensitivity in the mice also causes the phenotype as well. Interestingly, it has been reported that serum from patients with diabetes that contains autoantibodies against CD38 (an ADP-ribosyl cyclase/cADPR hydrolase that catalyzes β-NAD+ to cADPR) inhibits insulin secretion from pancreatic islets by glucose (Ikehata et al, 1998). In light of this observation and the findings of our study, it is therefore tempting to speculate that a TRPM2-dependent mechanism for insulin secretion might be related to the pathophysiology of diabetes.
Materials and methods
Cell culture
HEK293 cells or RIN-5F cells were maintained in Dulbecco's modified Eagle's medium or RPMI-1640 medium, respectively. HEK293 cells were transfected with 1.0 μg of hTRPM2 or mTRPM2 cDNA using Lipofectamine and Plus Reagent (Invitrogen), and kept at 33°C. RIN-5F cells were also kept at 33°C for 24–48 h before experiments. Pancreatic islets were isolated from male Wistar rats by the method modified from one described before (Sutton et al, 1986; Ban et al, 2000). Islets were dispersed in RPMI-1640 medium, and then cultured over night at 33 or 37°C. Dissociated pancreatic islet cells were reseeded on coverslips coated with poly-D-lysine (Sigma).
Fluorescent measurements and electrophysiology
The fura-2 fluorescence was measured in standard bath solution containing (in mM) 140 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES and 10 glucose, pH 7.4. The 340:380 nm ratio was shown. Standard bath solution for the patch-clamp experiments is same as that used in fluorescent measurements. For divalent cation permeability experiments, the bath solution was changed to 110 MgCl2 (or 110 CaCl2 or 140 NaCl), 10 glucose, 10 HEPES or Tris, pH 7.4, adjusted with Mg(OH)2 (or Ca(OH)2 or NaOH), and the reversal potential was measured using voltage ramps (−100 to +40 mV in 100 ms). Bath solution for inside-out patch recordings (Figure 3C) contained 140 KCl, 10 HEPES and 5 EGTA, pH 7.4. Pipette solution for whole-cell recordings contained 140 CsCl, 5 EGTA and 10 HEPES, pH 7.4. Pipette solution and bath solution for other inside-out patch recordings (Figure 3E) contained 140 NaCl, 1 CaCl2 and 10 HEPES, pH 7.4. Whole-cell or single-channel recording data were sampled at 10 or 2 kHz and filtered at 5 or 1 kHz for analysis, respectively. Whole-cell or single-channel patch-clamp recordings and Ca2+ imagings were performed 1 day after transfection to HEK293 cells, 3–4 days after replating of RIN-5F cells or 1 day after isolation of rat pancreatic cells. Liquid junction potentials were measured directly in separate experiments, and significant changes (>5 mV) were observed and corrections made for membrane potentials and reversal potentials. All patch-clamp experiments were performed at room temperature (25°C), except for heat stimulus. In an effort to observe a maximal TRPM2 response to heat, we decided to apply heat ramps at about 2–3 min (180, 96±8, 567±79 or 180 s for β-NAD+-, ADPR- or cADPR-exposed cells, respectively) after establishing the whole-cell configuration with a pipette solution containing β-NAD+, ADPR or cADPR. As ADPR-evoked currents developed at various time points (about 3 min) after getting exposed, heat stimuli were applied just after showing significant inward currents. Thermal stimulation was applied by increasing the bath temperature at a rate of 1.0°C/s with a preheated solution. When the heat-activated currents started to inactivate, the preheated solution was changed to a 25°C one. FSK with or without H-89 was extracellularly applied for 2 min before heat stimulation (Figure 7D). Chamber temperature was monitored with a thermocouple placed within 100 μm of the patch-clamped cell. Permeability ratio for cations was calculated as described previously (Adams et al, 1980; Caterina et al, 1997). In brief, permeability ratio for Na to Cs (PNa/PCs) was calculated as follows: Px/PCs=exp(ΔVrev F/RT), where Vrev is the reversal potential, F is Faraday's constant, R is the universal gas constant and T is absolute temperature. For measurement of divalent cation permeability, PY/PCs was calculated as follows:
![]()
where the bracketed terms are activities. NP0 values were obtained using Fetchan software (Axon). Linear regression analyses were conducted using Origin (Microcal).
The temperature coefficient Q10 was used to characterize the temperature dependence of the membrane current. The absolute current values were plotted on a log scale against the reciprocal of the absolute temperature (T) (Arrhenius plot). Q10 values were calculated from QΔT=(Q10)ΔT/10 for an arbitrary temperature ΔT.
Immunofluorescence staining
Anti-mouse TRPM2 rabbit antiserum (anti-TRPM2-C1) was directed against the C-terminus 1488–1506 (YANHKTILQKVASLFGAHF) (Hara et al, 2002). Cells were fixed with 4% paraformaldehyde, and then blocked and incubated with the anti-TRPM2-C1. After washing, cells were incubated with Alexa 488-conjugated anti-rabbit IgG (Molecular probes), DAPI (Amersham Pharmacia) and Texas Red–phalloidin (Molecular probes). Adult mouse (C57BL/6) was perfused transcardially with 2% paraformaldehyde in 0.1 M sodium phosphate (pH 7.3). Then, organs were removed and frozen, and the frozen tissue was cut on a cryostat. The sections were incubated with the anti-TRPM2-C1 or the anti-rat TRPV1 antibody (Oncogene). In some experiments, the sections were incubated with mixture of the anti-TRPM2-C1, guinea-pig anti-porcine insulin antibody (DAKO) and anti-porcine glucagon mouse monoclonal antibody (Sigma). Slides were washed with PBS, followed by incubation with Alexa 488-conjugated anti-rabbit IgG, Alexa 350-conjugated anti-mouse IgG (Molecular Probes) and Cy3-conjugated anti-guinea-pig IgG antibody (Jackson ImmnunoResearch).
Immunoblot analysis
Immunoblotting was performed by using whole-cell lysates from the cells. The protein-transferred PVDF membranes were blotted with the anti-TRPM2-C1 or monoclonal anti-β-tubulin (Sigma) antibody, followed by blotting with HRP-conjugated anti-rabbit or anti-mouse IgG (Cell Signaling technology) antibody, respectively. In some experiments, the anti-TRPM2-C1 was preincubated with immunogenic peptide described above.
β-NAD+ binding and cADPR competition assay
The template DNA was pCI-neo-TRPM2 or ΔNudix (Hara et al, 2002). The fusion proteins immobilized with His-Bind (Novagen) resins were used for β-NAD+-binding assay. The resins were incubated with [32P]-NAD+ (Amersham) and were washed with binding buffer. In cADPR competition assay, 10 μM cADPR (Sigma) was added in reaction mixture. The primers used for the TRPM2 C-terminus were 5′-AAAGAATTCGCGGAGGAGCCGGATGCTG-3′ (forward) and T3 primer (reverse). The resins were incubated with [32P]-NAD+ (1000 Ci/mmol; Amersham) in 0.3 ml of binding buffer for 30 min and were washed three times with binding buffer at room temperature.
Measurement of insulin release from islets
Insulin release from islets was measured after 16 h culture at 33°C (Figures 7A and C) or 37°C (Figures 7B, E and F) after isolation as described (Fujimoto et al, 1998). The amount of immunoreactive insulin was determined by ELISA (Morinaga). Incubation temperature was 37°C, except for the experiments in Figures 7A or C (29°C). Amount of insulin was normalized to the values released from the islets incubated at 37°C (G3.3) (Figure 7A), the islets cultured and incubated at 37°C (G3.3) (Figure 7B and Supplementary Figure 5), the islets treated with control siRNA without heat stimulation (G3.3) (Figures 7C, E and F), respectively. Eco (10 μM), FFA (200 μM), FSK (2 μM), NDP (2 μM), H-89 (10 μM) or exendin-4 (10 nM) was applied in the step of incubation (60 min). Groups of islets were incubated for 60 min with Krebs–Ringer bicarbonate buffer with additives. At the end of the incubation period, islets were pelletted by centrifugation and aliquots of the buffer were sampled.
siRNA treatment
siRNA designed to against rat TRPM2 was synthesized using Silencer siRNA Construction Kit (Ambion) and control siRNA (Silencer Negative Control #1 siRNA) was obtained from Ambion. siRNA was transfected using Lipofectamine 2000 (Invitrogen). Cells and pancreatic islets were treated the siRNAs for 46 h. The primers to construct for siTRPM2 were 5′-AACCAGAUUGUGGAAUGGAtt-3′ and 5′-UCCAUUCCACAAUCUGGUUtt-3′. To estimate the transfection efficiency, Cy-3-labeled siRNA was used for fluorescent measurements by using Label IT siRNA Tracker Intracellular Localization Kit (Mirus). Primer sequences for RT–PCR detecting TRPM2 message were 5′-AAGTATGTCCGGGTCTCCC-3′ and 5′-TAACGGCCCAAAT GAGAAGGTCACG-3′.
Chemicals
All chemicals were from Sigma, except for H-89 (from Upstate).
Statistical analysis
Data are analyzed using an unpaired (* or **) or a paired (#) t-test. Values are shown as mean±s.e.m. P-values <0.05 were considered significant.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Acknowledgments
We thank Drs MJ Caterina (Johns Hopkins University), N Inagaki (Akita University), T Miki (Kobe University) and Y Minokoshi (National Institute for Physiological Sciences) for their critical reading of the manuscript, Drs A Mizoguchi and T Kayahara (Mie University) for their help in immunofluorescence study, Ms E Shimanuki for her help in immunoblotting study, Drs Y Ando-Akatsuka, T Iida and M Numazaki (Mie University) for their encouragement, Dr S Hayashi (Nippon Shinyaku Co., Ltd) for his advice for getting pancreatic β-cells, and Dr N Ban (Akita University) for his advice on experiments of insulin secretion and pancreatic β-cell staining. We also thank NIBB Center for Analytical Instruments for their help for the measurement of insulin release. This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology in Japan, Japan Brain Foundation, AstraZeneca Foundation, Naito Foundation, Uehara Memorial Foundation and Japan Diabetes Foundation to MT.
References
- Adams DJ, Dwyer TM, Hille B (1980) The permeability of endplate channels to monovalent and divalent metal cations. J Gen Physiol 75: 493–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwater I, Goncalves A, Herchuelz A, Lebrun P, Malaisse WJ, Rojas E, Scott A (1984) Cooling dissociates glucose-induced insulin release from electrical activity and cation fluxes in rodent pancreatic islets. J Physiol 348: 615–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N, Yamada Y, Someya Y, Ihara Y, Adachi T, Kubota A, Watanabe R, Kuroe A, Inada A, Miyawaki K, Sunaga Y, Shen ZP, Iwakura T, Tsukiyama K, Toyokuni S, Tsuda K, Seino Y (2000) Activating transcription factor-2 is a positive regulator in CaM kinase IV-induced human insulin gene expression. Diabetes 49: 1142–1148 [DOI] [PubMed] [Google Scholar]
- Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, Patapoutian A (2004) Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41: 849–857 [DOI] [PubMed] [Google Scholar]
- Benham CD, Gunthorpe MJ, Davis JB (2003) TRPV channels as temperature sensors. Cell Calcium 33: 479–487 [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4: 517–529 [DOI] [PubMed] [Google Scholar]
- Brauchi S, Orio P, Latorre R (2004) Clues to understanding cold sensation: thermodynamics and electrophysiological analysis of the cold receptor TRPM8. Proc Natl Acad Sci USA 101: 15494–15499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Julius D (2001) The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci 24: 487–517 [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D (1999) A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398: 436–441 [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389: 816–824 [DOI] [PubMed] [Google Scholar]
- Clapham DE (2003) TRP channels as cellular sensors. Nature 426: 517–524 [DOI] [PubMed] [Google Scholar]
- Clapper DL, Walseth TF, Dargie PJ, Lee HC (1987) Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J Biol Chem 262: 9561–9568 [PubMed] [Google Scholar]
- Fujimoto S, Ishida H, Kato S, Okamoto Y, Tsuji K, Mizuno N, Ueda S, Mukai E, Seino Y (1998) The novel insulinotropic mechanism of pimobendan: direct enhancement of the exocytotic process of insulin secretory granules by increased Ca2+ sensitivity in beta-cells. Endocrinology 139: 1133–1140 [DOI] [PubMed] [Google Scholar]
- Gasser A, Glassmeier G, Fliegert R, Langhorst MF, Meinke S, Hein D, Kruger S, Weber K, Heiner I, Oppenheimer N, Schwarz JR, Guse AH (2006) Activation of T cell calcium influx by the second messenger ADP-ribose. J Biol Chem 281: 2489–2496 [DOI] [PubMed] [Google Scholar]
- Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, Caterina M (2002) Heat-evoked activation of the ion channel, TRPV4. J Neurosci 22: 6408–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse AH (2000) Cyclic ADP-ribose. J Mol Med 78: 26–35 [DOI] [PubMed] [Google Scholar]
- Guse AH, Berg I, da Silva CP, Potter BV, Mayr GW (1997) Ca2+ entry induced by cyclic ADP-ribose in intact T-lymphocytes. J Biol Chem 272: 8546–8550 [DOI] [PubMed] [Google Scholar]
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y (2002) LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 9: 163–173 [DOI] [PubMed] [Google Scholar]
- Heiner I, Eisfeld J, Halaszovich CR, Wehage E, Jungling E, Zitt C, Luckhoff A (2003) Expression profile of the transient receptor potential (TRP) family in neutrophil granulocytes: evidence for currents through long TRP channel 2 induced by ADP-ribose and NAD. Biochem J 371: 1045–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC (2004) Pathways in beta-cell stimulus-secretion coupling as targets for therapeutic insulin secretagogues. Diabetes 53 (Suppl 3): S48–S58 [DOI] [PubMed] [Google Scholar]
- Ikehata F, Satoh J, Nata K, Tohgo A, Nakazawa T, Kato I, Kobayashi S, Akiyama T, Takasawa S, Toyota T, Okamoto H (1998) Autoantibodies against CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) that impair glucose-induced insulin secretion in noninsulin-dependent diabetes patients. J Clin Invest 102: 395–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamura K, Sano Y, Mochizuki S, Yokoi H, Miyake A, Nozawa K, Kitada C, Matsushime H, Furuichi K (2003) Response to ADP-ribose by activation of TRPM2 in the CRI-G1 insulinoma cell line. J Membr Biol 191: 201–207 [DOI] [PubMed] [Google Scholar]
- Jordt SE, Bautista DM, Chuang HH, McKemy DD, Zygmunt PM, Hogestatt ED, Meng ID, Julius D (2004) Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427: 260–265 [DOI] [PubMed] [Google Scholar]
- Jordt SE, McKemy DD, Julius D (2003) Lessons from peppers and peppermint: the molecular logic of thermosensation. Curr Opin Neurobiol 13: 487–492 [DOI] [PubMed] [Google Scholar]
- Kolisek M, Beck A, Fleig A, Penner R (2005) Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell 18: 61–69 [DOI] [PubMed] [Google Scholar]
- Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G, Harteneck C (2004) Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol 286: C129–C137 [DOI] [PubMed] [Google Scholar]
- Lee HC (2002) Cyclic ADP-Ribose and NAADP: A Story of Two Calcium Messengers. Norwell: Kluwer Academic Publishers [Google Scholar]
- Lund PK (2005) The discovery of glucagon-like peptide 1. Regul Pept 128: 93–96 [DOI] [PubMed] [Google Scholar]
- McKemy DD, Neuhausser WM, Julius D (2002) Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 416: 52–58 [DOI] [PubMed] [Google Scholar]
- Meier JJ, Nauck MA (2005) Glucagon-like peptide 1(GLP-1) in biology and pathology. Diabetes Metab Res Rev 21: 91–117 [DOI] [PubMed] [Google Scholar]
- Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S (1998) Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci USA 95: 10402–10406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minke B, Cook B (2002) TRP channel proteins and signal transduction. Physiol Rev 82: 429–472 [DOI] [PubMed] [Google Scholar]
- Montell C (2005) The TRP superfamily of cation channels. Sci STKE 2005: re3. [DOI] [PubMed] [Google Scholar]
- Montell C, Rubin GM (1989) Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron 2: 1313–1323 [DOI] [PubMed] [Google Scholar]
- Moqrich A, Hwang SW, Earley TJ, Petrus MJ, Murray AN, Spencer KS, Andahazy M, Story GM, Patapoutian A (2005) Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science 307: 1468–1472 [DOI] [PubMed] [Google Scholar]
- Nilius B, Talavera K, Owsianik G, Prenen J, Droogmans G, Voets T (2005) Gating of TRP channels: a voltage connection? J Physiol 567: 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partida-Sanchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, Kusser K, Goodrich S, Howard M, Harmsen A, Randall TD, Lund FE (2001) Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med 7: 1209–1216 [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Peier AM, Story GM, Viswanath V (2003) ThermoTRP channels and beyond: mechanisms of temperature sensation. Nat Rev Neurosci 4: 529–539 [DOI] [PubMed] [Google Scholar]
- Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, Patapoutian A (2002a) A TRP channel that senses cold stimuli and menthol. Cell 108: 705–715 [DOI] [PubMed] [Google Scholar]
- Peier AM, Reeve AJ, Andersson DA, Moqrich A, Earley TJ, Hergarden AC, Story GM, Colley S, Hogenesch JB, McIntyre P, Bevan S, Patapoutian A (2002b) A heat-sensitive TRP channel expressed in keratinocytes. Science 296: 2046–2049 [DOI] [PubMed] [Google Scholar]
- Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM (2001) ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411: 595–599 [DOI] [PubMed] [Google Scholar]
- Qian F, Huang P, Ma L, Kuznetsov A, Tamarina N, Philipson LH (2002) TRP genes: candidates for nonselective cation channels and store-operated channels in insulin-secreting cells. Diabetes 51 (Suppl 1): S183–S189 [DOI] [PubMed] [Google Scholar]
- Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, Furuichi K (2001) Immunocyte Ca2+ influx system mediated by LTRPC2. Science 293: 1327–1330 [DOI] [PubMed] [Google Scholar]
- Seino S, Shibasaki T (2005) PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev 85: 1303–1342 [DOI] [PubMed] [Google Scholar]
- Smith GD, Gunthorpe MJ, Kelsell RE, Hayes PD, Reilly P, Facer P, Wright JE, Jerman JC, Walhin JP, Ooi L, Egerton J, Charles KJ, Smart D, Randall AD, Anand P, Davis JB (2002) TRPV3 is a temperature-sensitive vanilloid receptor-like protein. Nature 418: 186–190 [DOI] [PubMed] [Google Scholar]
- Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S, Patapoutian A (2003) ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112: 819–829 [DOI] [PubMed] [Google Scholar]
- Sutton R, Peters M, McShane P, Gray DW, Morris PJ (1986) Isolation of rat pancreatic islets by ductal injection of collagenase. Transplantation 42: 689–691 [DOI] [PubMed] [Google Scholar]
- Takasawa S, Akiyama T, Nata K, Kuroki M, Tohgo A, Noguchi N, Kobayashi S, Kato I, Katada T, Okamoto H (1998) Cyclic ADP-ribose and inositol 1,4,5-trisphosphate as alternate second messengers for intracellular Ca2+ mobilization in normal and diabetic beta-cells. J Biol Chem 273: 2497–2500 [DOI] [PubMed] [Google Scholar]
- Takasawa S, Nata K, Yonekura H, Okamoto H (1993) Cyclic ADP-ribose in insulin secretion from pancreatic beta cells. Science 259: 370–373 [DOI] [PubMed] [Google Scholar]
- Takasawa S, Okamoto H (2002) The CD38–Cyclic ADP-Ribose Signal System in Pancreatic Beta-Cells. Norwell: Kluwer Academic Publishers [Google Scholar]
- Talavera K, Yasumatsu K, Voets T, Droogmans G, Shigemura N, Ninomiya Y, Margolskee RF, Nilius B (2005) Heat activation of TRPM5 underlies thermal sensitivity of sweet taste. Nature 438: 1022–1025 [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ (2004) Thermosensation and pain. J Neurobiol 61: 3–12 [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D (1998) The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21: 531–543 [DOI] [PubMed] [Google Scholar]
- Voets T, Droogmans G, Wissenbach U, Janssens A, Flockerzi V, Nilius B (2004) The principle of temperature-dependent gating in cold- and heat-sensitive TRP channels. Nature 430: 748–754 [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Suh SH, Benham CD, Droogmans G, Nilius B (2002) Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J Biol Chem 277: 47044–47051 [DOI] [PubMed] [Google Scholar]
- Xu H, Ramsey IS, Kotecha SA, Moran MM, Chong JA, Lawson D, Ge P, Lilly J, Silos-Santiago I, Xie Y, DiStefano PS, Curtis R, Clapham DE (2002) TRPV3 is a calcium-permeable temperature-sensitive cation channel. Nature 418: 181–186 [DOI] [PubMed] [Google Scholar]
- Zocchi E, Carpaneto A, Cerrano C, Bavestrello G, Giovine M, Bruzzone S, Guida L, Franco L, Usai C (2001) The temperature-signaling cascade in sponges involves a heat-gated cation channel, abscisic acid, and cyclic ADP-ribose. Proc Natl Acad Sci USA 98: 14859–14864 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5