

Abstract
We have constructed DNA microarrays containing 17,871 genes, representing about 94% of the 18,967 genes currently annotated in the Caenorhabditis elegans genome. These DNA microarrays can be used as a tool to define a nearly complete molecular profile of gene expression levels associated with different developmental stages, growth conditions, or worm strains. Here, we used these full-genome DNA microarrays to show the relative levels of gene expression for nearly every gene during development, from eggs through adulthood. These expression data can help reveal when a gene may act during development. We also compared gene expression in males to that of hermaphrodites and found a total of 2,171 sex-regulated genes (P < 0.05). The sex-regulated genes provide a global view of the differences between the sexes at a molecular level and identify many genes likely to be involved in sex-specific differentiation and behavior.
The completion of the Caenorhabditis elegans genome project has paved the way for the use of functional genomics approaches to begin to illuminate the function of every gene (1). Currently, only 8% of the predicted protein-coding genes in the genome (1,541/18,967 total) have been studied at the biochemical or genetic levels (2). Functional genomics approaches are beginning to generate data on a genome-wide level in C. elegans, greatly accelerating the rate of functional characterization of the remaining genes. Such approaches include generating deletion mutants for every gene or identifying binding interactions between all C. elegans proteins (R. Barstead, A. Coulson, and D. Moerman, personal communication; ref. (3).
Another genome-wide approach is to use DNA microarrays to characterize the expression profiles of large numbers of genes in parallel (4–6). DNA fragments for essentially all identified genes can be prepared and deposited onto DNA microarrays. Fluorescently labeled probes derived from RNAs from two different samples can be simultaneously hybridized to a DNA microarray, and the relative level of expression for every gene can be determined by comparing the signal intensities of each probe. C. elegans DNA microarrays containing 65% of the genome (11,917 genes) were recently produced and used to identify a total of 1,416 germ line enriched genes that could be further subdivided into those that were expressed predominantly in sperm (650 genes), in oocytes (258 genes), or in both sperm and oocytes (508 genes) (7).
In this paper, we report the construction and use of DNA microarrays containing nearly every gene in C. elegans. An important advantage of using DNA microarrays containing nearly the full genome is that they not only identify some, but can, in principle, identify all of the gene expression differences associated with different growth conditions or caused by genetic mutations. An important characteristic of every gene is its expression pattern throughout development. Housekeeping genes should be expressed at all times in development, genes involved in cell proliferation should be expressed in embryos and in the germ line, and genes with specific developmental functions should be expressed at specific developmental stages. Another important aspect of the expression profile of every gene is whether it is differentially expressed in males and hermaphrodites. We have used the full-genome C. elegans DNA microarrays to profile the expression of nearly all of the genes during development, from embryos through adulthood. We have also used the microarrays to characterize gene expression differences between males and hermaphrodites and have identified 2,171 sex-regulated genes.
Materials and Methods
Standard methods were used to culture C. elegans (variety Bristol, strain N2) at 20°C (8). Males were isolated from him-8(e1489) or him-5(e1490) strains as described (9). RNAs were isolated from staged animals as in Reinke et al. (7). The developmental stage of the animals was verified by observing a small sample of animals with Nomarski optics to score the size of the gonad and the development of the vulva.
The DNA microarrays have PCR fragments derived from genomic DNA corresponding to each protein-coding gene. The PCR fragments were 1–2 kb in length, containing at least 700 bp of predicted protein coding sequence, or they contained more than 90% of the predicted coding sequence of the gene. None of the PCR fragments have known C. elegans repetitive elements, although there could be crosshybridization between genes with highly similar sequences. Data were analyzed only from PCR fragments that gave one band of the predicted size and from DNA spots that were of high quality. The relative amount of DNA in each of the spots in the microarrays was not measured, and the length of hybridizable sequence present on each of the PCR fragments was not used in the data analysis. Therefore, differences in hybridization signals between two different DNA spots on the same microarray are not necessarily caused by gene expression levels; they could simply reflect spotting efficiency or length of hybridizable sequence.
RNA preparation, cDNA synthesis, microarray hybridization, and microarray scanning were performed as previously described (7). Cy3-dUTP was used to label cDNA from each of the developmental stages and from males. Cy5-dUTP was used to label cDNA from the mixed-stage reference RNA and from hermaphrodites.
Data Analysis.
It was not possible to directly determine the number of genes that show significant levels of expression in these experiments, because we did not have a set of negative control genes (that are known to have no expression in our experiments) to measure background hybridization levels. However, the microarrays and probes used in this paper are similar to those used by Reinke et al., in which it was estimated that 59% of genes were expressed at a detectable level by using probe derived from mixed-stage hermaphrodite poly(A)+ RNA (7). To obtain an average expression ratio from repeats of the same experiment, the average of the log2 of the expression ratio was used.
The global standard deviation was determined by using the data from 43 separate experiments that were each repeated between two and five times. These experiments include 7 experiments reported here, 5 experiments reported by Reinke et al. (7), and 31 experiments that are currently stored in the Stanford Microarray Database. The measurement of the global standard deviation is derived from a large number of repeated hybridizations and so should not have aberrantly large or small values that might occur with small sample size. To calculate the SD from all of the experiments, the following formula was used:
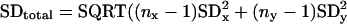 |
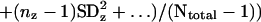 |
where x, y, z represent individual experiments, n is the number of repeats in each experiment, and N is the total number of repeats. A one-way ANOVA was used to compare data over the developmental time course, and a Student's t test was used to compare male and hermaphrodite expression.
Each statistical method was done twice, first using the standard deviation resulting from the specific experiments presented in this paper and then using the global standard deviation derived from a large set of combined experiments. Genes were selected only if they were significant using both measurements of standard deviation.
Genes were sorted into groups on the basis of their expression patterns by using a self-organizing map (10), or they were clustered by using a hierarchical clustering algorithm (4). Of the 17,871 genes on the microarray, 5,385 do not show at least a 2-fold expression ratio in at least 1 experiment. These genes form one expression group with relatively even expression levels throughout development and in the two sexes. They were not included in the self-organizing map, as they would form one large node. The hybridization signals of these 5,385 genes (reflecting both the quality of the DNA spots and the expression levels) from two randomly chosen hybridizations do not differ significantly from those of the remaining 12,486 genes that show more than a 2-fold change in expression. The overall distribution of hybridization signals for the two sets of genes using probe derived from mixed-stage poly(A)+ RNA was similar, and their median levels of expression were within 10% of each other in two experiments examined (data not shown). These data indicate that neither DNA spot quality nor levels of expression were strong factors for exclusion of the 5,385 genes from the self-organizing map. The self-organizing map was constructed using the 12,486 genes that show more than a 2-fold change in at least 1 experiment. As described above, there are no negative controls for the seven experiments reported in this paper to measure background levels, and so we cannot determine which genes are significantly expressed in the experiments presented here.
The self-organizing map includes all 12,486 genes regardless of their expression level or whether the observed level of regulation is statistically significant. Each of the 12,486 genes was placed into the most appropriate expression group (node on the self-organizing map) for that gene, regardless of the statistical significance of its regulation. Every gene is present in only one expression group, although some genes may overlap substantially with genes of other expression groups.
Title lines for the predicted proteins were provided by Proteome (version as of January 2000; ref. 2). In figures 2–6 reported here, the expression ratios during development were normalized, such that the expression level of each gene at every developmental stage was compared with the stage showing the lowest level of expression. The data for this article can be found as supplemental material on the PNAS web site, www.pnas.org.
Results
Whole-Genome Analysis of C. elegans Gene Expression.
We constructed DNA microarrays containing nearly all of the 18,967 genes in the C. elegans genome. We previously designed primer sets for each gene and used them to construct DNA microarrays containing 11,917 genes (7). Here, we amplified the rest of the genes from genomic DNA by PCR. We used only data from PCR reactions that yielded a single band of the predicted size for each gene. We used the new DNA fragments along with the previous PCR fragments to construct DNA microarrays containing 17,871 genes, corresponding to 94% of the genome.
We used these C. elegans DNA microarrays to determine changes in expression throughout development and between the two sexes. To profile gene expression patterns during development, poly(A)+ was isolated from populations of synchronized hermaphrodites at each of six developmental stages: eggs, L1, L2, L3, L4, and young adults (with no eggs). We isolated four RNA samples from independent cultures of eggs or L2-staged worms and three samples from independent cultures of L1-, L3-, L4-stage, or young adult worms. In these experiments, we compared expression at each of the developmental stages to a single preparation of reference RNA [mixed-stage hermaphrodite poly(A)+ RNA].
To determine which genes are developmentally regulated, we first determined an average expression ratio from the three to four repeats for each of the developmental stages (staged RNAs vs. reference RNA; see Materials and Methods). Because each staged RNA was compared with the same reference RNA, we could compare the expression levels of one stage to those of any other (see Materials and Methods). We plotted a developmental profile for each gene: egg/reference, L1/reference, L2/reference, L3/reference, L4/reference, and adult/reference. The complete developmental profile for each of these genes can be viewed at http://cmgm.stanford.edu/∼kimlab/dev.
We used two separate approaches to determine which genes show significant levels of regulation and kept only those genes that were significant according to both. First, we used the standard deviation observed in the developmental time course experiments (local SD) to calculate which genes are developmentally regulated using one-way ANOVA. This approach identified genes that vary over the time course more than they vary within repeats of time points. However, because the time points were repeated only three or four times, there were a large number of genes in which all of the repeats for a given time point were similar, fortuitously resulting in a very low variance. Because of the low observed variance, low levels of developmental regulation for these genes appeared to be significant, and they would have been falsely included as being developmentally regulated.
We screened out these false positives by using a second approach in which we measured the standard deviation for each gene using the pooled data from independent repeats of 43 different experiments involving 100–200 DNA microarray hybridizations (global SD; see Materials and Methods and http://cmgm.stanford.edu/∼kimlab/dev). This approach reduces variability in the measured standard deviations by using a large number of experiments. It also determines an independent error measurement for each gene, partially reflecting variable quality of DNA spots on the microarrays as well as variation in different RNA preparations. The median standard deviation of the log2 (ratio) for all of the genes was 0.57 (average, 0.61), and the range was 0.21 to 1.56. Thus, for genes with the median standard deviation, gene expression ratios of 2.2-fold or more are significant at 2 standard deviations. Genes that show statistically significant levels of developmental regulation in these DNA microarray experiments were identified by using ANOVA and the global standard deviation calculated above. At the 95% confidence level, there are 1,815 genes whose expression changes during development over a range of 1.6- to 300-fold.
To identify sex-regulated genes, we directly compared labeled cDNAs from either adult him-8 or him-5 males to labeled cDNA from adult N2 hermaphrodite RNAs. DNA microarray experiments were repeated four times, using three independent preparations of RNA from him-8 males, one preparation of him-5 male RNA, and four preparations of adult hermaphrodite RNAs. We determined the average male/hermaphrodite expression ratio for every gene and calculated the average expression ratio in a manner similar to before. As before, we first used a Student's t test to select only those genes that were significantly regulated using both the observed standard deviation (local SD) and the global standard deviation compiled from a large number of different DNA microarray experiments (global SD). We selected genes that are sex regulated above the 95% confidence level and found 1,651 male-enriched genes (with expression ratios ranging from 1.5- to 110-fold). Nine genes were previously known to be enriched in the male soma relative to the hermaphrodite soma. Six of the male positive controls were enriched at least 2-fold and as much as 6-fold in our microarray experiments, and the remaining 3 did not show significant expression above background (Table 1). Five hundred and twenty genes are expressed more abundantly in hermaphrodites than in males (with expression ratios ranging from 1.5- to 12-fold; see data at http://cmgm.stanford.edu/∼kimlab/dev).
Table 1.
Positive control male genes
| Gene name | Ratio* | N† | Reference |
|---|---|---|---|
| mab-3 | 6.3 | 3 | 25 |
| mab-9 | 5.7 | 1 | 26 |
| her-1 | 2.8 | 2 | 27 |
| vab-3 | 2.6 | 4 | 28 |
| sra-6 | 2.4 | 1 | 29 |
| srd-1 | 2.0 | 3 | 29 |
| pkd-2 | ND‡ | 2 | 30 |
| sra-1 | ND | 4 | 29 |
| lov-1 | ND | 2 | 30 |
Average male/hermaphrodite expression ratio.
Number of experiments.
Not detected. The expression signals were below background and did not give reliable expression ratios.
A Survey of Gene Expression Patterns.
The data presented above serve as a resource of gene expression patterns for nearly all of the genes throughout development. It is useful to divide these genes into smaller groups according to their expression patterns; for example, genes could be grouped according to whether they are male or hermaphrodite enriched, or according to when they are most abundantly expressed during development. To do this, we used a self-organizing map to group the genes according to how they are expressed (see Materials and Methods; ref. 10). Of the 17,871 genes on the microarray, there are 5,385 genes that do not show at least a 2-fold expression ratio at some time in development (compared with reference RNA) or between males and hermaphrodites. The remaining 12,486 genes were used to form a self-organizing map with 25 nodes (Fig. 1). Each gene was placed into the expression node that has the most similar developmental profile. Fig. 1 shows the number of genes in each expression node, and the entire list of genes can be found at http://cmgm.stanford.edu/∼kimlab/dev. By trial and error, we found that 25 expression groups (i.e., nodes in the self-organizing map) were sufficient to divide the set of developmentally regulated genes such that each expression group appeared significantly different; maps with less nodes (16 nodes) were not sufficient to adequately capture the entire diversity of the observed gene expression patterns and maps with more nodes (36 nodes) contained instances in which similar expression patterns were present in different nodes. The expression map in Fig. 1 shows the best placement for each gene into one of the expression groups. As statistical significance was not evaluated in generating Fig. 1, the map does not imply that all of the genes in an expression group are significantly related nor does it imply that a gene is significantly similar to genes in only one expression group.
Figure 1.

A global profile of gene expression during development. Of the entire set of 17,871 genes, 12,486 showed at least a 2-fold difference in expression during development or in the two sexes. The noncentered Pearson correlation was calculated between each pair-wise combination of these genes and used to place them into a self-organizing map with 25 nodes (a 5 × 5 grid) using 105 reiterations. Details about the self-organizing map and its source code can be found at http://genome-www.stanford.edu/∼sherlock/cluster.html. The total number of genes, the number of sperm-enriched genes, and the number of oocyte-enriched genes in each group are shown. A detailed list of the genes in each expression group can be found at http://cmgm.stanford.edu/∼kimlab/dev. For the developmental time course, samples from developmental stages are compared with reference RNA, and for sex regulation, samples from males are compared with those from hermaphrodites. Red indicates more abundant than the reference RNA or enriched in males. Green indicates less abundant than the reference RNA or hermaphrodite enriched. Scale shows level of expression (fold expression) as varying shades of color.
The 25 expression groups shown in Fig. 1 represent the most common patterns of expression in development. Genes that function in males can be found in expression groups 10–11, 13–20, and 24–25, whereas hermaphrodite-enriched genes are found in groups 2, 4–7, and 9. In some cases, the specific gene expression profile is highly indicative of the function for the genes in that group. For example, the expression profile represented by expression groups 10 and 11 is often associated with sperm-enriched genes; 91 of the 368 genes in group 10 (25%) and 261 of the 576 genes in group 11 (45%) were previously identified as being sperm enriched (7). The sperm-enriched genes in groups 10, 11, and 16 are expressed with different developmental kinetics and may have different functions in sperm; group 10 is expressed primarily in the adult stage, group 11 is expressed in the L4 and adult, and group 16 is expressed primarily in the L4 stage.
Developmentally Regulated Genes.
Gene expression patterns often correlate with the sites and times in which they function, and a clear example of this is seen in the expression of the cyclin genes (Fig. 2). Almost all cell divisions occur either in the embryo when the single cell divides to generate about 700 out of a total of 959 somatic cells or in the L4 stage and adult when there is extensive cell proliferation of the germ line cells (11, 12). There are seven cyclin genes in C. elegans currently identified in the C. elegans genome sequence (2), and these genes show strikingly similar patterns of expression during development. They show peak expression levels in embryos, the L4 stage, and adults, as would be expected for genes involved in cell proliferation. Four of the cyclin genes (T06E6.2, F48E8.7, F43D2.1, C37A2.4) are hermaphrodite enriched, indicating that these may be involved in germ line proliferation in hermaphrodites. One cyclin gene (ZC168.4) is enriched in adult males, suggesting that it is involved in male germ line proliferation.
Figure 2.

Developmental expression of cyclin genes. blast searches of the current sequence in the C. elegans genome identified seven cyclin genes. Developmental expression levels are normalized such that each stage is compared with the stage with the lowest level. Scale shows level of expression.
Another use of the gene expression data is to study coordinate expression of genes that function together in a protein complex or genetic pathway. Proteins that work together in a group (for example, as subunits in a protein complex or as successive steps in a signaling pathway) function only when every protein in the complex or pathway is expressed, and so these proteins may show coordinate patterns of expression. An example is the retinoblastoma Rb tumor suppressor complex, which controls progression through the cell cycle by regulating gene expression (13). The mammalian Rb tumor suppressor protein complex consists of three core proteins (Rb, RbAP48, and histone deacetylase) and a peripheral protein (the E2F transcription factor) that associates with the Rb complex under certain conditions and that can function independently of the Rb complex. Fig. 3 shows the expression data from the microarray experiments of the worm Rb complex genes: lin-35 (Rb), hda-1 (histone deacetylase), rba-1 and rba-2 (both RbAP48). The genes are listed according to their similarity in expression, as determined by using a hierarchical clustering algorithm (4). Three genes encoding proteins in the core complex (rba-2, hda-1, lin-35) are clustered tightly, indicating similar patterns of expression during development. A fourth gene (rba-1) has a similar pattern of expression but is expressed at a lower level than the other Rb complex genes. The two E2F genes (F49E12.6 and Y102A5C.18) show different expression profiles than the core Rb complex genes, as would be expected for peripheral components of the Rb complex.
Figure 3.

Developmental expression of Rb complex genes. Genes that encode proteins similar to mammalian proteins in the core Rb complex are shown in blue. The hierarchical tree shows the degree of similarity of the expression patterns. Developmental expression levels are normalized, such that each stage is compared with the stage with the lowest level. Scale shows level of expression.
Genes that are C. elegans homologs of Rb tumor suppressor complex genes were genetically identified in screens for excessive growth of vulval cells. In addition to homologs of known mammalian Rb complex genes, these screens identified several genes (lin-9, lin-15, and lin-36) that were not similar to previously identified genes (14). These new genes encode novel proteins, and how they function with the Rb complex is currently unknown. The expression profile of lin-9 is highly similar to the Rb complex genes rba-2, hda-1, and lin-35, suggesting that lin-9 might interact closely with the Rb complex genes to repress cell growth (Fig. 3). In contrast, the expression profiles of lin-15 and lin-36 are less similar to the core Rb complex genes, suggesting that these genes may not interact tightly with the Rb complex genes.
Because lin-35 is the only Rb gene in C. elegans, it should be expressed whenever the Rb complex functions. In contrast, there are many other cases of protein complexes or genetic pathways in which more than one gene is capable of encoding a particular part or step. For example, there are five genes that encode Wnt ligands and two genes that encode Fz receptors in C. elegans. Expression of any of the Wnt or Fz homologs could allow the Wnt signaling pathway to function, and experimental data are required to determine which ligands and which receptors act together in a specific cellular context. Because genes that function together should be expressed at the same developmental time, developmental expression data could be used to group members of gene families according to their temporal pattern of expression during development.
We used the Wnt signaling pathway to demonstrate this point, because there is independent confirmation from genetic studies showing which genes interact with each other. In the embryo, Wnt signaling is responsible for polarizing the EMS blastomere, and this pathway includes the maternal effect genes mom-2 (Wnt), mom-5 (Frizzled), wrm-1 (Armadillo), apr-1 (APC), and pop-1 (Tcf/Lef) (15). In larvae, a Wnt signaling pathway is responsible for cell polarity of certain hypodermal blast cells, for vulval fate specification, and for regulating cell migration. The larval Wnt signaling genes include egl-20 (Wnt), lin-44 (Wnt), bar-1 (Arm), apr-1, and pop-1 (15–19).
Fig. 4 shows the expression patterns of those Wnt signaling genes that are represented on the DNA microarray. The Wnt signaling genes are listed on the basis of similarity of their expression patterns, and the hierarchical clustering tree shows that the Wnt genes can be placed into three expression groups. One group is expressed at highest levels in the embryo and in adult hermaphrodites. This group includes genes known to function to polarize the EMS cell in the embryo [mom-2 (Wnt), mom-5 (Fz), wrm-1 (Arm) and apr-1 (APC)] (15). As previously noted in ref. 7, two disheveled genes (mig-5 and C27A2.6) are included in the embryonic expression cluster, suggesting that these two disheveled genes might also function to transduce the Wnt signal during EMS polarization. These genes were not previously identified in genetic screens for EMS polarity defects, most likely because they may act redundantly in EMS polarity and may not show a strong EMS polarity phenotype in single mutants.
Figure 4.

Developmental expression of Wnt signaling genes. The hierarchical tree shows the degree of similarity of the expression patterns. The embryonic expression group is shown in blue, and the larval expression group is shown in green. Developmental expression levels are normalized, such that each stage is compared with the stage with the lowest level. Scale shows level of expression.
A second group is expressed more abundantly in larvae than in embryos or adults. This expression group includes three genes known to function during larval development [egl-20 (Wnt), lin-44 (Wnt) and bar-1 (Arm)] (16, 17, 19). The final group is separate from the embryonic and larval groups and contains the single gene pop-1 (Tcf/Lef), which is known to act in both the embryonic and larval Wnt signaling pathways (16). Thus, the genes in the Wnt signaling pathway can be placed into three groups according to their expression patterns (embryonic, larval, or both), and these expression groups correlate closely with their function in embryogenesis or larval development. In contrast to the Wnt signaling pathway, there are little or no data about the function of many other signaling pathways and protein complexes in C. elegans. Expression data from these microarray experiments could be used to group genes in these poorly understood pathways on the basis of developmental kinetics by showing which genes may act together in embryonic, larval, or sex-regulated pathways.
Sex-Regulated Genes.
Previous microarray experiments identified 1,416 germ line-enriched genes among the set of 11,917 genes on the microarrays (7). We used these data to divide the sex-regulated genes into those expressed in the germ line and those expressed in the soma. Of the 1,651 male-enriched genes identified above, 619 were not analyzed in the previous experiments, and so there are no data regarding whether these genes are germ line enriched. The remaining 1,032 male-enriched genes include 554 genes (54%) that were previously found to be sperm enriched at the 95% confidence level. The remaining 478 male-enriched genes (46%) are not significantly sperm enriched and hence are presumably expressed in the male soma. The list of 478 somatic male genes contains 379 that encode proteins that show significant similarity to proteins in other organisms, including: 7 transcription factors, 21 protein kinases and phosphatases, 20 neuronal proteins, and 13 other types of cell-signaling components (see http://cmgm.stanford.edu/∼kimlab/dev).
Three hundred and seventy-six hermaphrodite-enriched genes were analyzed in the previous sperm/oocyte experiment, and 259 of these were found to be germ line enriched (69%). The remaining 117 hermaphrodite-enriched genes (31%) are presumably expressed in the soma, and 89 of these are similar to other known genes (see http://cmgm.stanford.edu/∼kimlab/dev). Genes that were not examined in the sperm/oocyte experiment cannot yet be assigned to either the germ line-enriched or somatically enriched classes.
We wished to identify the sex-regulated transcription factors that define the regulatory network controlling sexually dimorphic gene expression. C. elegans sexual fate is regulated by the TRA-1 transcription factor; in XX animals, TRA-1 is active and animals develop as hermaphrodites, whereas in XO animals, TRA-1 is inactive and animals differentiate as males (20). TRA-1 is the master regulator that initiates the entire sex differentiation cascade, ultimately affecting the expression of all of the sex-regulated genes. Some of the sex-enriched genes are directly regulated by TRA-1, whereas others are regulated by transcription factors that lie downstream of TRA-1. Analysis of the DNA sequence located upstream of every gene has identified five genes that have three or more TRA-1-binding sites, predicting that these genes may be directly regulated by TRA-1 (21). Data from the microarray experiments showed that three of the five genes are indeed enriched in males vs. hermaphrodites by at least 4.3-fold or more (C03C11.2, Y95B8A 79.A and mab-3I). The combination of the sequence analysis and microarray expression data suggests that these three genes may be directly regulated by TRA-1.
The sex-regulated genes include 37 that encode putative transcription factors, and these are likely to constitute the regulatory network that controls sexually dimorphic gene expression patterns. Twenty-three of the sex-regulated transcription factors are enriched in hermaphrodites, and fourteen are enriched in males; the expression data for these genes are available at http://cmgm.stanford.edu/∼kimlab/dev. The hermaphrodite-regulated transcription factors include sex-1 (nuclear hormone receptor), which was previously known to act as an X chromosome signal that determines sex (22). Other than sex-1, none of the other 36 genes were previously known to be involved in regulating sex differentiation. Interestingly, one of the male-enriched genes (ham-2) acts in hermaphrodites to specify the fate of the hermaphrodite specific neurons (23). The male-enriched expression pattern of ham-2 suggests that it may also function in the male.
Males have an elaborate mating behavior that allows them to sense hermaphrodites, to find the vulva with their tails, and then ejaculate sperm into the hermaphrodite. Males have 79 neurons and 36 neuronal support cells that are not present in hermaphrodites (11, 12). In contrast, hermaphrodites do not overtly respond to males or mating and have only eight sex-specific neurons. Forty-two of the sex-regulated genes encode proteins with known neuronal functions in other organisms and are thus likely to have neuronal functions in C. elegans; the expression data for these genes are available at http://cmgm.stanford.edu/∼kimlab/dev. Forty of the genes are enriched in males, and two are enriched in hermaphrodites. Five genes encode neuropeptides, twenty-five encode neuronal receptors, seven encode proteins associated with synaptic vesicles, and one encodes a protein similar to the human survival motor neuron protein (CeSMN). These sex-regulated neuronal genes may be involved in male- or hermaphrodite-specific neuroanatomy, physiology, or behavior.
Discussion
This paper presents the construction of DNA microarrays containing nearly every gene in C. elegans. The full genome DNA microarrays are produced by the C. elegans microarray resource at Stanford University and are available to be used by the entire C. elegans academic community.
We used the DNA microarrays to profile gene expression during development and in the two sexes, as these are fundamental properties of all genes in metazoans. The experiments in this paper show the developmental expression profiles of nearly all of the genes from the embryonic stage through adulthood, and these data are an important resource to help define the function of specific genes currently being studied. These experiments also identified 1,651 male-enriched genes and 520 hermaphrodite-enriched genes with expression ratios that are significant at least at the 95% confidence level.
The expression data from the DNA microarray experiments were verified in several ways. First, genes were identified only if they showed consistent changes in their expression levels from three or four independent microarray hybridizations. Second, six of nine genes that were previously known to exhibit higher expression in the male soma than in the hermaphrodite soma were found to be male enriched at least 2-fold (Table 3). Third, there was a high degree of overlap between the sex-regulated genes identified in this paper and the sperm- or oocyte-enriched genes identified previously. Fourth, the expression patterns are internally consistent. Male-enriched genes include known sperm-enriched but not oocyte-enriched genes. Wnt signaling genes, Rb complex genes, and cyclin genes show expression patterns that are consistent with times that they are known to function.
Nevertheless, there are likely to be some false positives and false negatives. One would expect false positives at the 95% confidence level using 17,871 genes. Some genes may be falsely included because they crosshybridize with other genes. The DNA microarray lacks 6% of the genes, and genes that are expressed at a low level could be missed. These experiments do not measure developmental changes during embryogenesis and would miss genes that only show developmental regulation in the embryo. Finally, these experiments measure expression in the entire animal and could miss genes with tissue-specific expression patterns.
Gene Expression Profiles During Development.
One example of using the expression data is to analyze expression of proteins that function together in a complex, such as the Rb tumor suppressor complex (13). Genes that encode the core proteins in the Rb tumor suppressor protein complex (lin-35, rba-2, hda-1) were highly coregulated. The developmental profile of lin-9 expression is highly similar to the Rb complex genes, suggesting that lin-9 might interact with the Rb complex genes.
In other cases, multiple genes encode similar proteins, and expression data can be used to determine which members of a gene family function together at different times of development. For example, we used expression data to separate the Wnt signaling genes into an embryonic group, a larval group, and a group expressed throughout development. These expression groups correlated closely with the known functions of these genes in embryonic and larval signaling pathways. There are a large number of genes in other genetic pathways that have not yet been functionally characterized, but that might interact together because they encode proteins that are known to interact in other animals. Expression of these genes during development and in the two sexes could be used to show which are expressed in the same time or sex and thus indicate which might interact together.
The transcription factor TRA-1 acts as a master regulator of sexual identity in C. elegans (20). The sex-regulated genes identified in this paper define the genes acting downstream of TRA-1, comprising a molecular definition of the differences between the two sexes. This study identified 37 sex-regulated transcription factors that likely act downstream of TRA-1 and upstream of the terminal sex-specific execution genes, thereby forming the regulatory hierarchy controlling sexually dimorphic gene expression. The network of transcription factors controls the expression of the terminal execution genes, such as the 42 sex-regulated neuronal genes identified in these microarray experiments.
C. elegans Functional Genomics.
The gene expression data derived from DNA microarray experiments can be combined with data from other functional genomics projects such as the global knockout project to determine mutant phenotypes or the global yeast two-hybrid project to determine protein-binding interactions (R. Barstead, A. Coulson, and D. Moerman, personal communication; ref. 3). The combination of protein sequences, gene expression profiles, mutant phenotypes, and protein-binding interactions will be a powerful resource to characterize genetic functions on a global scale.
Supplementary Material
Acknowledgments
The authors thank Kathy Mach and Peter Roy for critical reading of the manuscript, D. Zarkower for insightful advice, and Proteome for annotation of the C. elegans sequence. We are especially grateful to Mike Cherry, Gavin Sherlock, and the programmers and curators at the Stanford Microarray Database. This work was supported by grants from the National Center for Research Resources, Merck Genome Research Institute, and Rhone–Poulenc Rorer/Gencell.
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.011520898.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.011520898
References
- 1.C. elegans Sequencing Consortium. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- 2.Costanzo M C, Hogan J D, Cusick M E, Davis B P, Fancher A M, Hodges P E, Kondu P, Lengieza C, Lew-Smith J E, Lingner C, et al. Nucleic Acids Res. 2000;28:73–76. doi: 10.1093/nar/28.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walhout A J, Sordella R, Lu X, Hartley J L, Temple G F, Brasch M A, Thierry M N, Vidal M. Science. 2000;287:116–122. doi: 10.1126/science.287.5450.116. [DOI] [PubMed] [Google Scholar]
- 4.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schena M, Shalon D, Davis R W, Brown P O. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 6.Shalon D, Smith S J, Brown P O. Genome Res. 1996;6:639–645. doi: 10.1101/gr.6.7.639. [DOI] [PubMed] [Google Scholar]
- 7.Reinke V, Smith H E, Nance J, Wang J, Van Doren C, Begley R, Jones S J M, Davis E B, Scherer S, Ward S, Kim S K. Mol Cell. 2000;6:605–616. doi: 10.1016/s1097-2765(00)00059-9. [DOI] [PubMed] [Google Scholar]
- 8.Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klass M, Hirsh D. Dev Biol. 1981;84:299–312. doi: 10.1016/0012-1606(81)90398-5. [DOI] [PubMed] [Google Scholar]
- 10.Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander E S, Golub T R. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sulston J, Horvitz H R. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 12.Sulston J E, Schierenberg E, White J G, Thomson J N. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 13.Dyson N. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 14.Ferguson E L, Horvitz H R. Genetics. 1989;123:109–121. doi: 10.1093/genetics/123.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thorpe C J, Schlesinger A, Bowerman B. Trends Cell Biol. 2000;10:10–17. doi: 10.1016/s0962-8924(99)01672-4. [DOI] [PubMed] [Google Scholar]
- 16.Eisenmann D M, Maloof J N, Simske J S, Kenyon C, Kim S K. Development (Cambridge, UK) 1998;125:3667–3680. doi: 10.1242/dev.125.18.3667. [DOI] [PubMed] [Google Scholar]
- 17.Herman M A, Vassilieva L L, Horvitz H R, Shaw J E, Herman R K. Cell. 1995;83:101–110. doi: 10.1016/0092-8674(95)90238-4. [DOI] [PubMed] [Google Scholar]
- 18.Hoier E F, Mohler W A, Kim S K, Hajnal A. Genes Dev. 2000;14:874–886. [PMC free article] [PubMed] [Google Scholar]
- 19.Whangbo J, Kenyon C. Mol Cell. 1999;4:851–858. doi: 10.1016/s1097-2765(00)80394-9. [DOI] [PubMed] [Google Scholar]
- 20.Cline T W, Meyer B J. Annu Rev Genet. 1996;30:637–702. doi: 10.1146/annurev.genet.30.1.637. [DOI] [PubMed] [Google Scholar]
- 21.Clarke N D, Berg J M. Science. 1998;282:2018–2022. doi: 10.1126/science.282.5396.2018. [DOI] [PubMed] [Google Scholar]
- 22.Carmi I, Kopczynski J B, Meyer B J. Nature (London) 1998;396:168–173. doi: 10.1038/24164. [DOI] [PubMed] [Google Scholar]
- 23.Baum P D, Guenther C, Frank C A, Pham B V, Garriga G. Genes Dev. 1999;13:472–483. doi: 10.1101/gad.13.4.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raymond C S, Shamu C E, Shen M M, Seifert K J, Hirsch B, Hodgkin J, Zarkower D. Nature (London) 1998;391:691–695. doi: 10.1038/35618. [DOI] [PubMed] [Google Scholar]
- 25.Woollard A, Hodgkin J. Genes Dev. 2000;14:596–603. [PMC free article] [PubMed] [Google Scholar]
- 26.Trent C, Purnell B, Gavinski S, Hageman J, Chamblin C, Wood W B. Mech Dev. 1991;34:43–55. doi: 10.1016/0925-4773(91)90090-s. [DOI] [PubMed] [Google Scholar]
- 27.Chisholm A D, Horvitz H R. Nature (London) 1995;377:52–55. doi: 10.1038/377052a0. [DOI] [PubMed] [Google Scholar]
- 28.Troemel E R, Chou J H, Dwyer N D, Colbert H A, Bargmann C I. Cell. 1995;83:207–218. doi: 10.1016/0092-8674(95)90162-0. [DOI] [PubMed] [Google Scholar]
- 29.Barr M M, Sternberg P W. Nature (London) 1999;401:386–389. doi: 10.1038/43913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.