

Abstract
Drugs of abuse are known to cause persistent modification of neural circuits, leading to addictive behaviours1-5. Changes in synaptic plasticity in dopamine neurons of the ventral tegmental area (VTA) may contribute to circuit modification induced by many drugs of abuse, including cocaine6-13. Here we report that, following repeated cocaine exposure in vivo, excitatory synapses to VTA dopamine neurons become highly susceptible to the induction of long-term potentiation (LTP) by correlated pre- and postsynaptic activity, and this facilitated LTP induction is caused by cocaine-induced reduction of GABAA receptor-mediated inhibition of these dopamine neurons. In midbrain slices from saline- or single cocaine-treated rats LTP could not be induced in VTA dopamine neurons unless GABAergic inhibition was reduced by bicuculline or picrotoxin. In slices from repeated cocaine-treated rats, however, LTP became readily inducible, but was prevented by enhancing GABAergic inhibition with diazepam. Furthermore, repeated cocaine exposure reduced the amplitude of GABAergic synaptic currents and increased the probability of spike initiation in these dopamine neurons. This cocaine-induced enhancement of synaptic plasticity in VTA may be important for the formation of drug-associated memory.
To determine the impact of in vivo cocaine exposure on synaptic plasticity in VTA dopamine neurons, we examined LTP induction in these neurons in midbrain slices prepared from rats that were given single (1 d) or repeated (5-7 d) daily intraperitoneal injections of saline or cocaine. The effectiveness of the cocaine treatment was shown by the sensitization of locomotor activity (Supplementary Fig. 1). Dopamine neurons were identified by the presence of large Ih currents and distinct firing characterics14,15 (Supplementary Fig. 2). Extracellular stimulation was applied to the rostral region of VTA and evoked excitatory postsynaptic potentials (EPSPs) were monitored by whole-cell recordings from these dopamine neurons at −70 mV, near the reversal potential (−69.7 ± 1.5 mV, n = 5) of inhibitory postsynaptic currents (IPSCs). These EPSPs were mediated by the activation of glutamate receptors, since they were completely abolished by CNQX (6-cyaon-7-nitroquinoxaline-2,3-dione, 20 μM) and AP5 (D-2-amino-5-phosphonopentanoic acid, 50 μM), the antagonist of α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptors (AMPARs) and N-methyl-d-aspartate receptors (NMDARs), respectively. To induce LTP, we used a spike-timing protocol consisting of bursts of EPSP-spike pairs, with the onset of EPSPs preceding the peak of the postsynaptic spike by ∼5 ms (Fig. 1a, see Methods). This pattern of stimulation was used to mimic bursts of spikes observed in VTA dopamine neurons of behaving rats or monkeys in response to reward-related stimuli16,17. We found that repetitive EPSP-spike pairing induced a long-lasting increase in the amplitude of EPSPs in VTA dopamine neurons in slices obtained from rats treated with cocaine for 5-7 d (Fig. 1c), but not from rats treated with a single injection (1 d) of cocaine (Fig. 1e). No LTP was induced in slices from rats that were given time-matched saline injections (Fig. 1b,d). Thus repeated but not single cocaine exposure facilitated LTP induction in VTA dopamine neurons.
Figure 1.

Repeated cocaine exposure facilitates LTP induction in VTA dopamine neurons. a, Left, the protocol for LTP induction consists of 20 bursts of EPSP-spike pairs. Right, a typical postsynaptic response during one burst of paired stimuli. Scales: 30 mV, 100 ms. b,c, examples (top) and summary (below) of normalized EPSP amplitude before and after the pairing stimulation for 5-7 d saline- and cocaine-treated rats. Arrow, induction. Scales: 5 mV, 50 ms. d,e, The effect of the pairing stimulation on the EPSP amplitude for 1 d saline- or cocaine-treated rats. Error bars indicate s.e.m.
It is known that GABAergic inhibition suppresses LTP induction at many excitatory synapses18-21. To determine whether the presence of GABAergic inhibition is responsible for the absence of LTP induction in VTA dopamine neurons, we examined LTP induction in the presence of GABAA receptor blockers bicuculline or picrotoxin. We found that, in the presence of bicuculline methiodide (“BMI”, 20 μM, Fig. 2a,e) or picrotoxin (“PTX”, 100 μM, Fig. 2e), the EPSP-spike pairing induced robust LTP in these dopamine neurons in slices from 5-7 d saline-treated rats, to a similar extent as that observed for 5-7 d cocaine-treated rats (Fig. 2b,e). This LTP induction depends on the activation of NMDARs, because LTP was absent in the presence of AP5 (50 μM, Fig. 2e). Interestingly, the presence of bicuculline did not further increase the extent of LTP in 5-7 d cocaine-treated rats (Fig. 2e). Similarly, we found that in the presence of picrotoxin the pairing stimulation induced LTP in dopamine neurons in slices from naïve rats, and from 1 d saline- or cocaine-injected rats (Fig. 2c-e). However, application of bicuculline without or immediately after the pairing stimulation at −70 mV did not increase the amplitude of EPSPs in 1 d saline-treated rats (Supplementary Fig. 3), suggesting that the increase in EPSP amplitude following the pairing stimulation was not caused by changes in shunting inhibition but due to LTP induction. Blocking GABAergic inhibition is known to enhance temporal summation of EPSPs and NMDAR activation during repetitive afferent stimulation22,23 and to enhance action potential back-propagation into dendrites24. These mechanisms are likely to be responsible for the facilitatory effect of bicuculline (or picrotoxin) on LTP induction.
Figure 2.
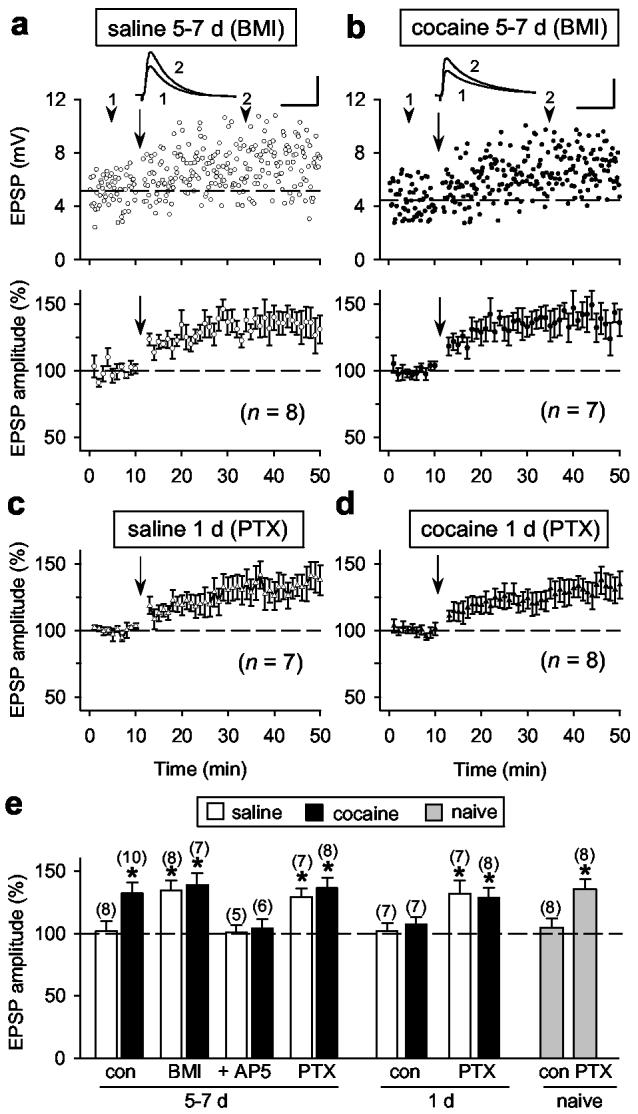
The effect of blocking GABAergic inhibition on LTP induction in dopamine neurons. a-d, In the presence of bicuculline (BMI, 20 μM) or picrotoxin (PTX, 100 μM), the pairing stimulation induced LTP for all saline- or cocaine-treated rats. Data are presented in the same manner as those in Fig. 1. Scales: 5 mV, 50 ms. e, Summary of LTP induction under various conditions. “Con”, without any blockers. “+AP5”, BMI and AP5. The total number of neurons examined is shown in the parenthesis. Data significantly different from the baseline value of the same experiment are marked by the asterisk (p < 0.01; paired Student's t-test). Error bars, s.e.m.
To examine whether the increased susceptibility to LTP induction following repeated cocaine exposure is caused by reduced GABAergic inhibition, we recorded IPSCs in VTA dopamine neurons in saline- and cocaine-injected rats. Monosynaptic IPSCs were evoked by extracellular stimulation and recorded at Vc = −20 mV, in the presence of CNQX (20 μM) and AP5 (50 μM). The stimulation intensity was gradually increased until the maximal IPSC amplitude was attained19 (Fig. 3a). The mean amplitude of maximal IPSCs was significantly reduced in slices from cocaine-injected rats than those from saline-injected rats after 5 d of treatment and this reduction persisted for at least 12 d after withdrawal from the 7 d cocaine injection (Fig. 3a). The change in IPSCs was not due to differences in the stimulation intensity (data not shown). The passive membrane properties of these dopamine neurons also remained unchanged following 5-7 d of cocaine treatment (Supplementary Table 1). Furthermore, the cocaine-induced reduction in the IPSC amplitude was cell-specific, since the average maximal IPSC amplitude in non-dopamine neurons was not changed (Fig. 3b). The cocaine-induced reduction in the IPSC amplitude is of postsynaptic origin, because local puffing of GABAA receptor agonist muscimol (20 μM) to the soma of dopamine neurons produced smaller currents (Fig. 3c) and the amplitude of miniature IPSCs (mIPSCs) recorded was smaller for 5-7 d cocaine-treated rats than those from saline-treated rats, while the mean frequency of mIPSCs was not significantly different (Fig. 3d). Together, these findings indicate that repeated cocaine exposure selectively reduces postsynaptic GABA responsiveness in dopamine neurons.
Figure 3.

Effects of cocaine exposure on GABAergic inhibition of VTA neurons. a, Repeated cocaine treatment reduced the amplitude of maximal IPSCs in dopamine neurons. Top, Sample IPSCs in response to stimuli of incremental intensities (20 to 140 μA). Scales: 100 pA, 15 ms. Bottom, the mean amplitude of maximal IPSCs after 1-7 d saline or cocaine treatment, and 12 d withdrawal from 7 d cocaine treatment (19 d, n = 17-25 for each bar). b, Mean maximal IPSC amplitudes in non-dopamine neurons (n = 15-19). Scales: 150 pA, 15 ms. c, Samples (top) and average amplitudes (below) of currents induced by puffing muscimol (20 μM, arrow) at the soma of dopamine neurons. (* p < 0.01, t-test). Scales: 500 pA, 3s. d, Repeated cocaine treatment (5-7 d) had no effect on the mean mIPSC frequency but decreased the mean mIPSC amplitude (*p < 0.01, t-test) and significantly shifted the mIPSC amplitude distribution (bin size 2 pA) towards smaller values (p < 0.001, Kolmogorov-Smirnov test). Sample traces of mIPSCs are shown at the top. Scales: 80 pA, 250 ms. e, Effect of repeated cocaine exposure on the probability for spike initiation in dopamine neurons. Top, Examples of APs induced by 80 μA stimulation for 5 d saline-treated rat before (left) and after (right) BMI (20 μM). Scales: 30 pA, 10 ms. Bottom, Input-output curves (fit with the Boltzmann function) of probability of APs induced by graded stimuli for 5-7 d saline- and cocaine-treated rats. Error bars, s.e.m (n = 8-10).
Consistent with a reduced inhibition of VTA dopamine neurons, we found that the probability of action potential (AP) initiation in response to presynaptic stimuli was increased following repeated cocaine exposure. We constructed the input-output curves for APs by varying the stimulation intensity and measuring the probability for AP firing using cell-attached patch recording and found that the slope of the input-output curve for 5-7 d cocaine-treated rats was significantly higher than that for 5-7 d saline-treated rats. Furthermore, the slopes for both groups were increased and converged to the same level after bath addition of bicuculline (20 μM, Fig. 3e and Supplementary Table 2). Similarly, the input-output relationship for evoked excitatory postsynaptic currents (EPSCs) was also examined by voltage-clamping the dopamine neuron at the mean resting membrane potential (Vc = −54 mV). We found that slope of the input-output curve for EPSCs was significant higher in cocaine-treated rats than that in 5 d saline-treated rats and bicuculline increased the slope for both groups (Supplementary Fig. 4). Thus repeated cocaine exposure increases spike output primarily through the reduction of GABAergic inhibition, although potential changes in the intrinsic excitability may also contribute. Such hyper-excitability might endow VTA dopamine neurons with an additional capacity to promote reward-dependent learning, besides the enhanced LTP induction described above.
To further test the idea that reduced GABAergic inhibition is responsible for the facilitation of LTP induction in dopamine neurons, we examined LTP induction in slices from 5-7 d saline-treated rats in the presence of different concentrations of bicuculline. Consistent with findings that LTP induction is highly sensitive to the level of GABAergic inhibition19-21,25, we found that LTP was induced in the presence of 1 μM but not 0.3 μM bicuculline (Fig. 4a,c). Interestingly, more complete blockade of GABAergic inhibition with 20 μM bicuculline did not further increase the extent of potentiation (Fig. 4c, Supplementary Fig. 5a-b). Furthermore, application of diazepam, a benzodiazepine agonist known to enhance the activation of GABAA receptors26, prevented LTP induction in 5-7 d cocaine-treated rats at a concentration of 1 μM, but not at 0.1 μM, and the effect of diazepam (1 μM) on LTP induction was abolished by bicuculline (Fig. 4 b,c). The effectiveness of diazepam in enhancing the activation of GABAA receptors was shown by the dose-dependent increase in the amplitude and decay time constant of IPSCs (Supplementary Fig. 5c-f). Taken together, these results indicate that GABAergic inhibition normally suppresses LTP induction in VTA dopamine neurons, and repeated cocaine exposure facilitates LTP induction by reducing GABAergic inhibition below a critical level.
Figure 4.
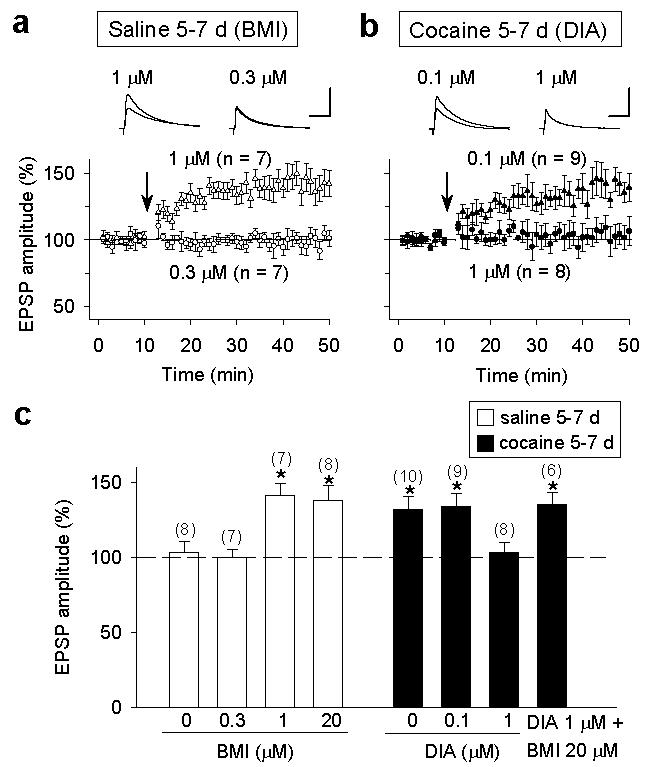
A critical level of GABAergic inhibition regulates LTP induction in dopamine neurons. a, The effects of partial blocking of GABAA receptor with bicuculline (BMI, 0.3 and 1 μM) on LTP induction in 5-7 d saline-treated rats. Scales: 5 mV, 50 ms. b, The effects of enhancing GABAA receptor activation with diazepam (DIA, 0.1 and 1 μM) on LTP induction in 5-7 d cocaine-treated rats. Scales: 5 mV, 50 ms. c, Summary graph showing concentration-dependent effects of bicuculline and diazepam on LTP induction in saline- and cocaine-treated rats. Error bars, s.e.m.
Previous studies have shown that single cocaine exposure in vivo resulted in an increase in the ratio of AMPAR- to NMDAR-mediated EPSCs in VTA dopamine neurons in midbrain slices8,9 and repeated cocaine exposure produced no further increase in this ratio10. Furthermore, single cocaine exposure occluded LTP induction in these neurons in vitro in the presence of picrotoxin, using an induction protocol of pairing presynaptic stimulation with steady postsynaptic depolarization8. These results suggest that single cocaine exposure has already produced saturated potentiation of excitatory inputs onto dopamine neurons. We note, however, that the latter induction protocol was not efficient for LTP induction even in the saline-treated group8 (Fig. 5a). In the present study, we confirmed that the AMPAR/NMDAR ratio was increased following single cocaine exposure (Fig.5b,c). However, LTP could readily be induced in the presence of picrotoxin (or bicuculline) in slices from rats exposed to single or repeated cocaine treatment using the spike timing induction protocol. Thus the discrepancy of LTP induction may be attributed in part to the induction protocol used. The finding that cocaine-induced increase in the AMPAR/NMDAR ratio did not preclude LTP induction indicates that either only a subset of synapses are potentiated in vivo by the cocaine exposure, or alternatively, LTP at these synapses is expressed by mechanisms independent of the increased AMPAR/NMDAR ratio, e.g., an increase in presynaptic transmitter release27,28. Regardless of the mechanism, our finding suggests that the strength of excitatory synaptic inputs to dopamine neurons still remains within a modifiable range, and drug-associated stimuli can exert persistent imprint on VTA circuits following repeated cocaine exposure.
Figure 5.
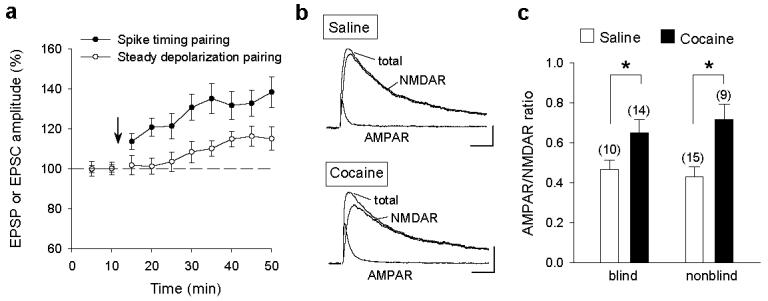
Comparison of LTP induced by two different protocols and cocaine-induced changes in the AMPAR/NMDAR ratio. a, Effects of spike-timing and steady-depolarization pairing protocols on the amplitude of EPSPs (spike-timing, same data of Fig.2c but averaged at 5 min interval) and EPSCs (steady depolarization) for 1 d saline-treated rats (n = 7). c, Examples of total, AMPAR and NMDAR-mediated EPSCs in slices from 1 d saline- and cocaine-injected rats. Scales: 50 pA, 15 ms. c, Summary of mean AMPAR/NMDAR ratios obtained by the experimenters who were either “blind” or “nonblind” to the treatment history of the rats. * p < 0.05, t-test. Error bars, s.e.m.
By inducing LTP in VTA dopamine neurons in the absence of GABAA receptor blockers, we were able to uncover the cocaine-induced reduction in GABAergic inhibition and its facilitatory effect on LTP induction. The reduced inhibition may help to transform drug-associated stimuli into incentive stimuli by allowing such stimuli to induce persistent synaptic modification in VTA dopamine neurons. Such incentive sensitization may underlie drug craving and compulsive drug seeking behaviour3. On the other hand, we note that the reduced inhibition and in vivo synaptic potentiation are unlikely to be responsible for locomotor sensitization, because the locomotor sensitization precedes the reduction in inhibition (Fig 3a) and there is no correlation between the increase in AMPAR/NMDAR ratio and locomotor sensitization10,11 (Supplementary Fig.1). It has been reported that γ-vinyl-GABA, which enhances GABAergic inhibition, reduces cocaine-seeking behaviour in rats29 and cocaine craving/taking in cocaine addicts in clinical trials30, but the underlying cellular mechanism is largely unknown. Our findings suggest that preventing drug-induced synaptic plasticity through enhancement of GABAergic inhibition might in part account for the effectiveness of γ-vinyl-GABA in diminishing cocaine addiction.
Methods
Cocaine treatment and slice preparation
Male Sprague Dawley rats (21-40 days old) were given intraperitoneal injections daily with either saline (0.9% NaCl, 1 ml kg−1) or cocaine (15 mg kg−1) dissolved in saline for 1-7 d. Rats were anaesthetized by halothane and killed by decapitation 20-24 h or 12 d (for Fig. 3a only) after the last injection, in accordance with institutional guidelines. Horizontal midbrain slices (250 μm thick) were cut using a vibratome (Vibratome Co.). Slices were prepared at 4-6°C in a solution containing (in mM): 110 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 7 MgSO4, 26 NaHCO3, 25 glucose, 11.6 sodium ascorbate, and 3.1 sodium pyruvate. The slices were incubated in artificial cerebrospinal fluid (ACSF) containing (in mM): 125 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose. The solutions were saturated with 95% O2 and 5% CO2.
Electrophysiology
Whole-cell or cell-attached recordings were made using a patch clamp amplifier (Multiclamp 700B, Axon Ins.) under infrared-DIC microscopy. Data acquisition and analysis were performed using a digitizer (DigiData 1322A, Axon Ins.) and analysis softwares pClamp 9 (Axon Ins.) and Mini Analysis 6 (Synaptosoft). Signals were filtered at 2 Hz and sampled at 10 Hz. For presynaptic stimulation, a bipolar tungsten stimulation electrode (WPI) placed ∼150 μm rostral to the recording electrode was used to stimulate synaptic inputs, using stimulation pulse of duration 40 μs and frequency 0.1 Hz. For recording of EPSPs, neurons were current-clamped at −70 mV. For recording IPSCs, neurons were voltage-clamped at −20 mV, in the presence of CNQX (20 μM) and AP5 (50 μM). For recording of EPSC-IPSC sequences, neurons were clamped near the resting membrane potential (−54 mV). For recording muscimol-induced currents, muscimol (20 μM) was ejected through a glass micropipette (tip resistance 8-10 MΩ) with a picospritzer (100 ms, 30 psi) to the soma of the recorded dopamine neuron (Vc = −20 mV), with the pipette tip locating at ∼20 μm from the soma surface. The whole-cell recording pipette (tip resistance 3-5 MΩ) was filled with a solution containing (in mM): potassium gluconate 140, KCl 5, HEPES 10, EGTA 0.2, MgCl2 2, MgATP 4, Na2GTP 0.3, and Na2-phosphocreatine 10 at pH 7.2 (with KOH). For recording evoked IPSCs and muscimol-induced currents, the experimenter was blind to the treatment history of the rat. For recording of mIPSCs, potassium gluconate was replaced by KCl (140 mM) in the internal solution, and TTX (0.5 μM), CNQX (10 μM), and AP5 (25 μM) were added in the bath. Series resistance (15-30 MΩ) or input resistance (300-500 MΩ) was monitored throughout the whole-cell recording and data were discarded if the resistance changed by more than 20%. All recordings were performed at 33 ± 1°C by using an automatic temperature controller (Warner Ins.).
The spike timing protocol for LTP induction consists of 20 bursts of EPSP-spike pairs, with each burst consisting of 5 paired stimuli delivered at 100-ms intervals (10 Hz) and an inter-burst interval of about 5 s. The postsynaptic spikes were evoked by injection of depolarizing current pulses (1-2 nA, 2-3 ms), with the onset of EPSPs preceding the peak of postsynaptic spikes by ∼5 ms. Evoked EPSPs were sampled at 0.1 Hz before and after LTP induction. For quantifying the magnitude of LTP, 60 consecutive EPSPs were averaged 10 min before and 20 min after the end of the induction protocol. Data are given as mean ± s.e.m. For data comparison, paired or unpaired Student's t-test and oneway ANOVA were used. The steady-depolarization pairing protocol for LTP induction consists of 200 stimuli of synaptic inputs (1 Hz) paired with a steady postsynaptic depolarization (+10 mV), as described previously8.
For measurements of the AMPAR/NMDAR ratio, the dopamine neuron was voltage-clamped at +40 mV. First, a stable baseline recording of total EPSCs was obtained, NMDA receptor antagonist AP5 (50 μM) was then applied in the bath for 5-10 min to isolate fast AMPAR-mediated EPSCs. Digital subtraction of AMPAR-EPSCs from the total EPSCs from the same neuron yielded NMDAR-EPSCs. An average of 12-20 EPSCs were collected for the each type of EPSCs. The bath solution contained picrotoxin (100 μM). The intracellular solution contained (in mM): 120 CsCH3SO3, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 TEA-Cl, MgCl2, 2.5 MgATP, 0.3 GTP (pH 7.2 with CsOH).
Acknowledgement
This work was supported by grants from USNIH.
References
- 1.Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–28. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- 2.Hyman SE, Malenka RC. Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci. 2001;2:695–703. doi: 10.1038/35094560. [DOI] [PubMed] [Google Scholar]
- 3.Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–91. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- 4.White FJ, Kalivas PW. Neuroadaptations involved in amphetamine and cocaine addiction. Drug Alcohol Depend. 1998;51:141–53. doi: 10.1016/s0376-8716(98)00072-6. [DOI] [PubMed] [Google Scholar]
- 5.Robinson TE, Kolb B. Structural plasticity associated with exposure to drugs of abuse. Neuropharmacology. 2004;47(Suppl 1):33–46. doi: 10.1016/j.neuropharm.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 6.Kauer JA. Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol. 2004;66:447–75. doi: 10.1146/annurev.physiol.66.032102.112534. [DOI] [PubMed] [Google Scholar]
- 7.Faleiro LJ, Jones S, Kauer JA. Rapid synaptic plasticity of glutamatergic synapses on dopamine neurons in the ventral tegmental area in response to acute amphetamine injection. Neuropsychopharmacology. 2004;29:2115–25. doi: 10.1038/sj.npp.1300495. [DOI] [PubMed] [Google Scholar]
- 8.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–7. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 9.Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–82. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 10.Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–90. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong Y, et al. Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(−/−) mice. Proc Natl Acad Sci U S A. 2004;101:14282–7. doi: 10.1073/pnas.0401553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolf ME, Sun X, Mangiavacchi S, Chao SZ. Psychomotor stimulants and neuronal plasticity. Neuropharmacology. 2004;47(Suppl 1):61–79. doi: 10.1016/j.neuropharm.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–79. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 14.Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol. 1992;450:455–68. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones S, Kauer JA. Amphetamine depresses excitatory synaptic transmission via serotonin receptors in the ventral tegmental area. J Neurosci. 1999;19:9780–7. doi: 10.1523/JNEUROSCI.19-22-09780.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyland BI, Reynolds JN, Hay J, Perk CG, Miller R. Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience. 2002;114:475–92. doi: 10.1016/s0306-4522(02)00267-1. [DOI] [PubMed] [Google Scholar]
- 17.Schultz W, Apicella P, Ljungberg T. Responses of monkey dopamine neurons to reward and conditioned stimuli during successive steps of learning a delayed response task. J Neurosci. 1993;13:900–13. doi: 10.1523/JNEUROSCI.13-03-00900.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wigstrom H, Gustafsson B. Facilitated induction of hippocampal long-lasting potentiation during blockade of inhibition. Nature. 1983;301:603–4. doi: 10.1038/301603a0. [DOI] [PubMed] [Google Scholar]
- 19.Huang ZJ, et al. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–55. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- 20.Bissiere S, Humeau Y, Luthi A. Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat Neurosci. 2003;6:587–92. doi: 10.1038/nn1058. [DOI] [PubMed] [Google Scholar]
- 21.Meredith RM, Floyer-Lea AM, Paulsen O. Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. J Neurosci. 2003;23:11142–6. doi: 10.1523/JNEUROSCI.23-35-11142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–63. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- 23.Herron CE, Williamson R, Collingridge GL. A selective N-methyl-Daspartate antagonist depresses epileptiform activity in rat hippocampal slices. Neurosci Lett. 1985;61:255–60. doi: 10.1016/0304-3940(85)90473-2. [DOI] [PubMed] [Google Scholar]
- 24.Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999;398:338–41. doi: 10.1038/18686. [DOI] [PubMed] [Google Scholar]
- 25.Artola A, Brocher S, Singer W. Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature. 1990;347:69–72. doi: 10.1038/347069a0. [DOI] [PubMed] [Google Scholar]
- 26.Eghbali M, Curmi JP, Birnir B, Gage PW. Hippocampal GABA(A) channel conductance increased by diazepam. Nature. 1997;388:71–5. doi: 10.1038/40404. [DOI] [PubMed] [Google Scholar]
- 27.Stevens CF, Wang Y. Changes in reliability of synaptic function as a mechanism for plasticity. Nature. 1994;371:704–7. doi: 10.1038/371704a0. [DOI] [PubMed] [Google Scholar]
- 28.Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation. Science. 1995;269:1730–4. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- 29.Gardner EL, et al. Gamma-vinyl GABA, an irreversible inhibitor of GABA transaminase, alters the acquisition and expression of cocaine-induced sensitization in male rats. Synapse. 2002;46:240–50. doi: 10.1002/syn.10138. [DOI] [PubMed] [Google Scholar]
- 30.Brodie JD, Figueroa E, Dewey SL. Treating cocaine addiction: from preclinical to clinical trial experience with gamma-vinyl GABA. Synapse. 2003;50:261–5. doi: 10.1002/syn.10278. [DOI] [PubMed] [Google Scholar]