

Abstract
DNA damage that leads to formation of DNA double-strand breaks (DSBs) induces phosphorylation of histone H2AX on Ser-139 at sites flanking the breakage. Immunocytochemical detection of phosphorylated H2AX (denoted as γH2AX) thus provides a marker of DSBs. The method presented in this chapter describes the detection of γH2AX for revealing the presence of DSBs, combined with differential staining of cellular DNA for revealing the cell cycle phase. The detection of γH2AX is based on indirect immunofluorescence using secondary antibody tagged with fluorescein isothiocyanate (FITC) while DNA is counterstained with propidium iodide (PI). Intensity of cellular green (FITC) and red (PI) fluorescence is measured by flow cytometry and bivariate analysis of the data is used to correlate the presence of DSBs with the cell cycle phase.
Keywords: Antitumor drugs, cell cycle phase, DNA damage, double-strand DNA breaks, flow cytometry, histone H2AX phosphorylation, immunofluorescence, ionizing radiation
Introduction
DNA damage that involves formation of DNA double-strand breaks (DSBs) triggers phosphorylation of histone H2AX (1) which is one of several variants of the nucleosome core histone H2A family (2). The phosphorylation, mediated either by ataxiatelangiectasia mutated (ATM), ataxia telangiectasia related (ATR) or DNA-dependent protein kinase (DNA-PK) (3–5), occurs on Ser-139 at the C-terminus of H2AX molecules flanking the DSBs in chromatin (2). The phosphorylated form of H2AX is defined as γH2AX (6). Antibodies that detect γH2AX have recently become commercially available. The appearance of γH2AX in chromatin in the form of discrete nuclear foci, each focus representing a single DSB, can be detected immunocytochemically shortly after induction of DSBs (7). Intensity of γH2AX immunofluorescence (IF) of the individual cell thus reports the extent of DNA damage (frequency of DSBs) in its nucleus. Flow or laser scanning cytometry offers the possibility to rapidly quantify γH2AX IF in large cell populations and multiparameter analysis of the cytometric data makes it possible to correlate DNA damage with other attributes of the cell, for example, cell cycle phase (8–13). This approach to measure DNA damage is rapid, more sensitive, and less cumbersome compared with the alternative, commonly used method, the comet assay (14).
The method presented in this chapter is designed to immunocytochemically detect γH2AX for revealing the presence of DSBs, combined with differential staining of DNA, to define the cell cycle phase of the cells in which DSBs are being detected (see Note 1). The detection of γH2AX is based on indirect immunofluorescence using the secondary antibody tagged with fluorescein isothiocyanate (FITC) while DNA is counterstained with propidium iodide (PI). The cells are briefly fixed in methanol-free formaldehyde and then transferred into 70% ethanol in which they can be stored at −20°C at least for 2 wk, perhaps longer. Ethanol treatment makes the plasma membrane permeable to the γH2AX antibody; further permeabilization is achieved by including the detergent Triton X-100 into a solution used to incubate cells with the antibody. After incubation with the primary γH2AX antibody, the cells are incubated with FITC-labeled secondary antibody and their DNA is then counterstained with PI in the presence of RNase A to remove RNA, which otherwise may also be stained with PI. Intensity of cellular green (FITC) and red (PI) fluorescence is measured by flow cytometry.
It should be noted that DSBs can also be intrinsic, occurring in healthy, nontreated cells, for example in the course of V(D)J and class-switch recombination during immune system development (15,16) or during DNA replication (8,9). Furthermore, DSBs are formed in the course of DNA fragmentation in apoptotic cells (12). Strategies are presented in this chapter to differentiate between DSBs induced by DNA damaging agents vs the intrinsically formed DSBs in untreated cells (see Note 2), or vs apoptosis-associated DSBs (see Note 3).
2. Materials
Cells to be analyzed: 106 – 5 × 106 cells, untreated (control) and treated with the DSB inducing agent(s), suspended in 1 mL of tissue culture medium.
70% Ethanol.
Phosphate-buffered saline (PBS).
Methanol-free formaldehyde fixative: Prepare 1% (v/v) solution of methanol-free formaldehyde (Polysciences, Warrington, PA) in PBS. This solution may be stored at 4°C for up to 2 wk.
BSA–T–PBS: Dissolve bovine serum albumin (BSA; Sigma) in PBS to obtain a 1% (w/v) BSA solution. Add Triton X-100 (Sigma) to obtain 0.2% (v/v) of its concentration. This solution may be stored at 4°C for up to 2 wk.
PI (Molecular Probes, Eugene, OR) stock solution: Dissolve PI in distilled water to obtain 1 mg/mL of solution. This solution can be stored at 4°C in the dark (e.g., in the tube wrapped in aluminum foil) for several months.
PI staining solution: Dissolve RNase A (DNase-free; Sigma) in PBS to obtain 0.1% (w/v; 100 mg/mL) solution. Add an appropriate aliquot of PI stock solution (e.g., 5 μL per 1 mL) to obtain its 5 μg/mL final concentration. Store the PI staining solution in the dark. This solution may be stored at 4°C for up to 2 wk.
Unconjugated primary antibody: Histone γH2AX antibody (murine monoclonal, available from Upstate Biotechnology, Lake Placid, NY; alternatively, rabbit polyclonal, available from Trevigen, Gaithersburg, MD).
FITC-conjugated secondary antibody, for example, either polyclonal goat anti-mouse, or antirabbit-F(ab′)2, depending on the source of the primary antibody, appropriately titered.
12 × 75 mm polypropylene tubes.
Centrifuge and rotor capable of 300g.
Flow cytometers of different types, offered by several manufacturers, can be used to measure cell fluorescence following staining according to the protocol given below. The most common flow cytometers are from Coulter Corporation (Miami, FL), Becton Dickinson Immunocytometry Systems (San Jose, CA), Cytomation/DAKO (Fort Collins, CO) and PARTEC (Zurich, Switzerland).
3. Methods
Centrifuge cells collected from tissue culture (suspended in culture medium) at 300g for 4 min at room temperature. Suspend the cell pellet (1–2 × 106 cells) in 0.5 mL of PBS.
With a Pasteur pipet transfer this cell suspension into a 6-mL polypropylene tube (see Note 4) containing 4.5 mL of ice-cold 1% methanol-free formaldehyde solution in PBS. Keep on ice for 15 min.
Centrifuge at 300g for 4 min at room temperature and suspend the cell pellet in 4.5 mL of PBS. Centrifuge again as in step 1 above and suspend the cell pellet in 0.5 mL of PBS. With a Pasteur pipet, transfer the suspension to a tube containing 4.5 mL of ice-cold 70% ethanol. The cells should be maintained in 70% ethanol at −20°C for at least 2 h, but may be stored under these conditions for up to 2 wk.
Centrifuge at 200g for 4 min at room temperature, remove the ethanol and suspend the cell pellet in 2 mL of BSA–T–PBS solution.
Centrifuge at 300g for 4 min at room temperature and suspend the cells again in 2 mL of BSA–T–PBS. Keep at room temperature for 5 min.
Centrifuge at 300g for 4 min at room temperature and suspend the cells in 100 μL of BSA–T–PBS containing 1 μg of the primary γH2AX antibody (see Note 5).
Cap the tubes to prevent drying and incubate them overnight at 4°C (see Note 6).
Add 2 mL of BSA–T–PBS and centrifuge at 300g for 4 min at room temperature.
Suspend the cells in 2 mL of BSA–T–PBS and centrifuge at 300g for 4 min at room temperature.
Suspend the cell pellet in 100 μL of BSA–T–PBS containing the appropriate (antimouse or antirabbit, depending on the source of the primary antibody) FITC-tagged secondary antibody (see Note 5).
Incubate for 1 h at room temperature, occasionally gently shaking. Add 5 mL of BSA–T–PBS and after 2 min centrifuge at 300g for 4 min at room temperature.
Suspend the cells in 1 mL of the PI staining solution. Incubate at room temperature for 30 min in the dark.
Set up and adjust the flow cytometer for excitation with light at blue wavelength (488-nm laser line or BG-12 excitation filter).
Measure the intensity of green (530 ± 20 nm) and red (>600 nm) fluorescence of the cells by flow cytometry. Record the data.
4. Notes
On the bivariate distributions (scatterplots), subpopulations of cells in G1 vs S vs G2/M are distinguished based on differences in their DNA content (intensity of PI fluorescence; see Fig. 1). To assess the mean extent of DNA damage (frequency of DSBs) for cells at a particular phase of the cycle, the mean values of γH2AX IF are calculated separately for G1, S, and G2/M cells by the computer-interactive “gating” analysis. It should be noted, however, that along with the doubling of DNA, histone content also doubles during the cell cycle. In fact the histone:DNA content ratio remains invariable throughout the cell cycle (17). Therefore, cells in S and G2/M with the same “degree” of H2AX phosphorylation (i.e., the “same percentage of phosphorylated H2AX molecules per total H2AX”) as cells in G1, will have (because of their higher histone content) 1.5 and 2.0 times higher γH2AX IF compared to cells in G1 phase. To assess the degree of H2AX phosphorylation, that is, to make γH2AX IF independent of histone doubling during the cycle, the data may be normalized by presenting per unit of DNA (histone) by dividing the mean γH2AX IF of S- and G2/M-phase cells by 1.5 and 2.0, respectively. After such normalization the intensity of γH2AX IF reflects the frequency of DSBs per unit of DNA rather than per nucleus, making this parameter independent of the cell cycle phase.
-
A low level of γH2AX IF seen in the cells that have not been treated with inducers of DSBs represents an intrinsic (“programmed”) H2AX phosphorylation, primarily associated with DNA replication (8). The level of intrinsic γH2AX IF varies between different cell lines. To quantify the γH2AX IF induced by external factors that damage DNA, this intrinsic component of γH2AX IF has to be subtracted. Towards this end the means of γH2AX IF of G1, S, and G2/M-phase untreated cells are subtracted from the respective means of the G1, S, and G2/M subpopulations of the radiation-, drug- or carcinogen-treated cells, respectively (ref. 11; and see Fig. 1). After the subtraction the extent of increase in intensity of γH2AX IF (Δ γH2AX IF) over the untreated sample represents the “treatment-induced phosphorylation” of this protein. There is no need, therefore, to use the isotype control to estimate the nonspecific antibody binding component, because it is expected that this component is similar for the untreated and treated cells, and thus is being subtracted while calculating Δ γH2AX IF.
DNA undergoes extensive fragmentation in apoptotic cells (18). It is often desirable, therefore, to distinguish between primary DSBs induced by DNA damaging agents vs DSBs generated during apoptosis. The following attributes of γH2AX IF allow one to distinguish cells with radiation-, drug-, or carcinogen-induced H2AX phosphorylation from cells that have phosphorylation of this histone triggered by apoptosis-associated (AA) DNA fragmentation:
The γH2AX IF induced by external DNA damaging agents is seen rather early during the treatment (10 min–2 h) whereas AA γH2AX IF is seen later (>3 h) (11,12).
The intensity AA γH2AX IF is generally much higher than that of DNA damage-induced γH2AX IF, unless the cells are at a very late stage of apoptosis (11).
The induction of AA γH2AX IF is prevented by cell treatment with the caspase inhibitor z-VAD-FMK, which precludes activation of endonuclease responsible for DNA fragmentation. In its presence the AA-γH2AX IF is suppressed (11).
-
AA H2AX phosphorylation occurs concurrently with activation of caspase-3 in the same cells. Multiparameter analysis (active caspase-3 vs γH2AX IF) thus provides the direct approach to distinguish cells in which DSBs were caused by inducers of DNA damage (active caspase-3 is undetectable) from the cells that have H2AX phosphorylation additionally triggered in response to apoptotic DNA fragmentation (active caspase-3 is present).
These strategies are discussed in more detail elsewhere (11).
If the sample initially contains a small number of cells, they may be lost during repeated centrifugations. To minimize cell loss, polypropylene or siliconized glass tubes are recommended. Since transferring cells from one tube to another causes electrostatic attachment of a large fraction of cells to the surface of each new tube, all steps of the procedure (including fixation) preferably should be done in the same tube. Addition of 1% BSA to rinsing solutions also decreases cell loss. When the sample contains very few cells, carrier cells (e.g., chick erythrocytes) may be included; they can be recognized during analysis based on differences in DNA content (intensity of PI fluorescence).
Quality of the primary and secondary antibodies is of particular importance. Their ability to detect γH2AX is often lost during improper transport or storage conditions. Also of importance is their use at optimal concentration. It is recommended that with the first use of every new batch of primary or secondary antibody they be tested at serial dilution (e.g., within the range between 0.2 and 2.0 μg/100 μL) to find their optimal titer for detection of γH2AX. The titer recommended by vendor is not always the optimal one.
Alternatively, incubate for 1 h at 22–24°C. The overnight incubation at 4°C, however, appears to yield somewhat higher intensity of γH2AX IF compared to a 1-h incubation.
Fig. 1.
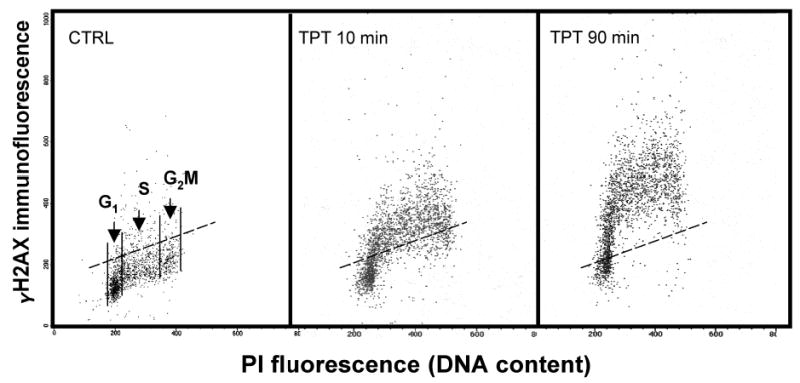
Detection of histone H2AX phosphorlation in relation to the cell position in the cell cycle. The bivariate (γH2AX IF vs DNA content) distribution (scatterplots) of the untreated cells (CTRL) and, to induce DSBs, of the cells treated with 0.15 μM of the DNA topisomerase I inhibitor topotecan (TPT) for 10 or 90 min (11). The cells were processed as described in Subheading 3., their fluorescence was measured by flow cytometry (FACScan; Becan Dickinson Immunochemistry, San Jose, CA). Notice increased γH2AX IF of TPT-treated cells. Based on difference in DNA content, subpopulations of cells in G1 vs S vs G2M phases of the cycle may be distinguished and gated as shown in the CTRL sample. The mean γH2AX IF may then be calculated for each subpopulation. To estimate the extent of treatment-induced H2AX phosphorylation the γH2AX IF means of the untreated cells have to be subtracted form the respective G1, S, G2M means of the treated samples. The dashed line(– – –) represents the γH2AX IF level which 95% of cells form the untreated culture (CTRL) express γH2AX.
Acknowledgments
This work was supported by NCI grant RO1 28704.
References
- 1.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 2.West MH, Bonner WM. Histone 2A, a heteromorphous family of eight protein species. Biochemistry. 1980;19:3238–3245. doi: 10.1021/bi00555a022. [DOI] [PubMed] [Google Scholar]
- 3.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42,462–42,467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 4.Furuta T, Takemura H, Liao ZY, et al. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian topoisomerase I cleavage complexes. J Biol Chem. 2003;278:20,303–20,312. doi: 10.1074/jbc.M300198200. [DOI] [PubMed] [Google Scholar]
- 5.Park EJ, Chan DW, Park JH, Oettinger MA, Kwon J. DNA-PK is activated by nucleosomes and phosphorylated H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res. 2003;31:6819–6827. doi: 10.1093/nar/gkg921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sedelnikova OA, Rogakou EP, Panuytin IG, Bonner W. Quantitive detection of 125IUdr-induced DNA double-strand breaks with γ-H2AX antibody. Radiation Res. 2002;158:486–492. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 8.MacPhail SH, Banath JP, Yu Y, Chu E, Olive PL. Cell cycle-dependent expression of phosphorylated histone H2AX: reduced expression in unirradiated but not X-irradiated G1-phase cells. Radiat Res. 2003;159:759–767. doi: 10.1667/rr3003. [DOI] [PubMed] [Google Scholar]
- 9.MacPhail SH, Banath JP, Yu TY, Chu EH, Lambur H, Olive PL. Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. Int J Radiat Biol. 2003;79:351–358. doi: 10.1080/0955300032000093128. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida K, Yoshida SH, Shimoda C, Morita T. Expression and radiation-induced phosphorylation of H2AX in mammalian cells. J Radiat Res (Tokyo) 2003;44:47–51. doi: 10.1269/jrr.44.47. [DOI] [PubMed] [Google Scholar]
- 11.Huang X, Okafuji M, Traganos F, Luther E, Holden E, Darzynkiewicz Z. Assessment of histone H2AX phosphorylation induced by DNA topoisomerase I and II inhibitors topotecan and mitoxantrone and by DNA cross-linking agent cisplatin. Cytometry. 2004;58A:99–110. doi: 10.1002/cyto.a.20018. [DOI] [PubMed] [Google Scholar]
- 12.Huang X, Traganos F, Darzynkiewicz Z. DNA damage induced by DNA topoisomerase I- or topoisomerase II- inhibitors detected by histone H2AX phosphorylation in relation to the cell cycle phase and apoptosis. Cell Cycle. 2003;2:614–619. [PubMed] [Google Scholar]
- 13.Albino AP, Huang X, Jorgensen E, et al. Induction of histone H2AX phosphorylation in A549 human pulmonary adenocarcinoma cells by tobacco smoke and in human bronchial epithelial cells by smoke condensate: a new assay to detect the presence of potential carcinogens in tobacco. Cell Cycle. 2004;3:1062–1068. [PubMed] [Google Scholar]
- 14.Speit G, Hartmann A. The comet assay (single-cell gel test): a sensitive genotoxicity test for detection of DNA damage and repair. Methods Mol Biol. 1999;113:203–212. doi: 10.1385/1-59259-675-4:203. [DOI] [PubMed] [Google Scholar]
- 15.Downs JA, Lowndes NF, Jackson SP. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature. 2000;408:1001–1004. doi: 10.1038/35050000. [DOI] [PubMed] [Google Scholar]
- 16.Jackson SP. DNA damage signaling and apoptosis. Biochem Soc Transactions. 2001;29:655–661. doi: 10.1042/0300-5127:0290655. [DOI] [PubMed] [Google Scholar]
- 17.Gorczyca W, Bruno S, Darzynkiewicz RJ, Gong J, Darzynkiewicz Z. DNA strand breaks occurring during apoptosis: their early in situ detection by the terminal-deoxynucleotidyl transferase and nick translation and prevention by serine protease inhibitors. Int J Oncol. 1992;1:639–648. doi: 10.3892/ijo.1.6.639. [DOI] [PubMed] [Google Scholar]
- 18.Marzluff WF, Duronio RJ. Histone mRNA expression: multiple levels of cell cycle regulation and important developmental consequences. Curr Opin Cell Biol. 2002;14:692–699. doi: 10.1016/s0955-0674(02)00387-3. [DOI] [PubMed] [Google Scholar]