

Abstract
Current estimates indicate that nearly a third of the world's population is latently infected with Mycobacterium tuberculosis. Reduced oxygen tension and nitric oxide exposure are two conditions encountered by bacilli in vivo that may promote latency. In vitro exposure to hypoxia or nitric oxide results in bacterial stasis with concomitant induction of a 47-gene regulon controlled by the transcription factor DosR. In this report we demonstrate that both the dosS gene adjacent to dosR and another gene, dosT (Rv2027c), encode sensor kinases, each of which can autophosphorylate at a conserved histidine and then transfer phosphate to an aspartate residue of DosR. Mutant bacteria lacking both sensors are unable to activate expression of DosR-regulated genes. These data indicate that DosR/DosS/DosT comprise a two-component signaling system that is required for the M. tuberculosis genetic response to hypoxia and nitric oxide, two conditions that produce reversible growth arrest in vitro and may contribute to latency in vivo.
Tuberculosis (TB)1 has placed a heavy burden on the global community for centuries, earning such morbid nicknames as The White Plague and The Captain of the Men of Death (1). The causative agent, Mycobacterium tuberculosis (MTB), kills about 2 million people annually making it a leading cause of infectious death worldwide (2, 3). The success of MTB as a pathogen is closely linked with its capacity to persist for years or decades in humans in the absence of any clinical disease symptoms. Current estimates place the number of people latently infected with MTB at nearly 2 billion, or one-third of the Earth's population (3, 4). Eradicating this enormous reservoir of latently infected carriers is complicated by several factors, including the availability, cost, and length of drug therapy required for successful treatment of latent TB.
Although TB has been studied for centuries, the triggers that promote and maintain latent infections are still obscure. Two conditions frequently associated with latent TB in vivo are reduced oxygen tension and nitric oxide (NO) exposure (5, 6). Both of these stimuli can induce reversible bacterial stasis in vitro (7, 8), and both are encountered by bacilli in vivo (5, 9, 10). Further, although MTB requires oxygen for growth, it can survive without oxygen for surprisingly long periods of time (11, 12). Still the evidence linking hypoxia and NO to latent TB in vivo remains circumstantial. Analysis of the MTB response to these factors is needed to define the role they may play in promoting and maintaining TB latency in humans.
Previous reports identified a set of 47 MTB genes that are rapidly up-regulated in response to reduced oxygen tension or NO (8, 13). Among the MTB genes induced by hypoxia or exposure to NO is the putative two-component regulatory system dosR-dosS (also called devR-devS, Rv3133c/Rv3132c2) (8, 13). In bacteria, two-component response regulator systems are an important means by which a variety of environmental signals are transduced into a phenotypic response. These systems typically consist of a membrane-bound sensor kinase and soluble response regulator that is activated by a histidine-aspartate phosphorelay to bind upstream of specific genes and alter their expression (14). We hypothesized that dosR and dosS may form a signaling system involved in the initial adaptation of bacilli to conditions within the host (13). We showed previously that DosR binds upstream of hypoxic response genes (15). Further, nearly all MTB genes rapidly up-regulated in response to low doses of NO (8) or by hypoxia require DosR for their induction (15).
In this report, we demonstrate that DosS (Rv3132c) is a functional kinase of the two-component class and that it can transfer phosphate to DosR in vitro. We show that a second putative kinase, DosT (Rv2027c), also phosphorylates DosR. We demonstrate that mutants lacking both DosS and DosT can no longer activate DosR-dependent MTB gene expression in response to reduced oxygen tension.
EXPERIMENTAL PROCEDURES
Targeted Gene Inactivation
Inactivation of the dosS gene was performed by gene replacement as described previously (13). Inactivation of the dosT gene also was by gene replacement. We amplified a portion of the wild type gene by PCR using mutagenic primers that introduced three in-frame stop codons and a PacI site for screening and eliminated the putative histidine phosphorylation site (primers RPL8 to RPL11; see Table I). The 952-bp PCR product then was cloned into the vector pKO2 containing a hygromycin resistance marker and a sacB counterselection marker. The resulting plasmid then was electroporated into MTB isolates H37Rv and H37Rv:ΔdosS. Homologous recombination and gene replacement was detected stepwise using hygromycin resistance followed by sacB counterselection and loss of hygromycin resistance as described previously (13). Gene replacement was confirmed by PCR using mutagenic and wild type dosT-specific primers along with primers specific for sequences flanking the site of integration (RPL12 and RPL13; see Table I) (data not shown). Digestion of the resulting amplicons with PacI also was performed to confirm the PCR screen results (data not shown).
Table I.
Primer sequences Primers used in the experiments are listed with sequences and a brief description (see text for details). Underlined sequences represent non-native nucleotide additions including ligase independent cloning (LIC) sequences, introduced restriction sites, and artificial ribosome binding sites. Nucleotides in bold represent base substitutions in mutagenic primers.
| Name | Sequence | Description |
|---|---|---|
| RPL8 | 5′-gaggagaagcccggtttatgcgacaacggtgctgaccatc-3′ | 5′ dosT |
| RPL9 | 5′-ttaattaactacctacctatgcgatccggtcgcgatc-3′ | 5′ mutagenic dosT |
| RPL10 | 5′-taggtaggtagttaattaaatccagcggctcttcg-3′ | 3′ mutagenic dosT |
| RPL11 | 5′-taataagcttcaggccgctttcggtgatgtc-3′ | 3′ dosT |
| RPL12 | 5′-gcggccgggattgccgttga-3′ | 5′ dosT flank |
| RPL13 | 5′-ctccggtcggcatgttctcga-3′ | 3′ dosT flank |
| DMR28 | 5′-gttgacaagcttaggaggagctatgtgacacaccc-3′ | 5′ dosT with RBS |
| DMR29 | 5′-cgtaacggtacccatcaatccatcagcgcagcg-3′ | 3′ dosT |
| DMR005 | 5′-gacgacgacaagatgacacaccctgacagg-3′ | 5′ LIC-dosT |
| DMR006 | 5′-gaggagaagcccggtcatcaatccatcagcgcagcgg-3′ | 3′ LIC-dosT |
| DMR007 | 5′-gacgacgacaagatgacaacagggggcctcgtc-3′ | 5′ LIC-dosS |
| DMR008 | 5′-gaggagaagcccggtttatgcgacaacggtgctgaccatc-3′ | 3′ LIC-dosS |
| DMR024 | 5′-cgtgatctgaaagacaaagtcatccag-3′ | 5′ mutagenic dosT |
| DMR025 | 5′-ctggatgactttgtctttcagatcacg-3′ | 3′ mutagenic dosT |
| DMR026 | 5′-cgtgacctcaaagacaaagtcatccag-3′ | 5′ mutagenic dosS |
| DMR027 | 5′-ctggatgactttgtctttgaggtcacg-3′ | 3′ mutagenic dosS |
| GW1 | 5′-ccatggtaaaggtcttcttggtcgatgacc-3′ | 5′ dosR |
| GW2 | 5′-ctcgagtggtccatcaccgggtgg-3′ | 3′ dosR |
Luciferase Reporter Assay
H37Rv, H37Rv:ΔdosS, H37Rv:ΔdosT, and H37Rv:ΔdosS:ΔdosT isolates were transformed with the plasmid pMH108 (16, 17) containing the firefly luciferase reporter gene (luc) under the control of the DosR-activated acr (hspX, Rv2031c) promoter. This plasmid integrates into the L5 phage attachment site within the Gly tRNA locus of the MTB chromosome (18). In three separate experiments transformants were grown in 7H9 medium in triplicate to an A600 of 0.3–0.4. 0.5 ml of each culture was removed to 13-ml tubes, which were sealed tightly with rubber septa. 0.2% O2 was infused through an 18-gauge needle for 30 s, and tubes then were incubated on a rotator for 2 h at 37 °C. Promoter activity was measured by combining a 100-μl aliquot of MTB isolate culture with 100 μl of luciferase assay reagent (Promega) in the wells of a 96-well plate, and incubation was continued for 5 min at room temperature. Luciferase activity was measured using a TD2020 luminometer.
dosT Complementation
For complementation studies, a pMH108-based construct was developed in which dosT is driven by mycobacterial optimal promoter (19). The dosT upstream region was amplified using primers DMR28 and DMR29 (see Table I). This construct was electroporated into H37Rv, H37Rv:ΔdosS, H37Rv:ΔdosT, and H37Rv:ΔdosS:ΔdosT with the DNA integrating into the MTB chromosome at the L5 phage attachment site. Luciferase reporter activity was measured as indicated above.
Cloning and Expression of dosS and dosT Genes
Full-length dosS and dosT were PCR-amplified from H37Rv genomic DNA using the primers DMR005 to DMR008 (see Table I) and were cloned into the pET32 Ek/LIC vector (Novagen). In addition, dosS and dosT mutants encoding amino acid substitutions DosS (H395K/H397K) and DosT (H392K/H394K) were generated by mutagenic fusion PCR with primers DMR024 to DMR027 (see Table I) and were cloned into pET32 Ek/LIC. Recombinant proteins were overexpressed as S-Tag fusion proteins in Escherichia coli BL21(DE3) and localized within inclusion bodies. The proteins were extracted from the inclusion bodies using the protein refolding kit from Novagen. Briefly, E. coli cells were disrupted using the BugBuster protein extraction reagent (Novagen), and inclusion bodies were recovered by centrifugation. After several washes in dilute BugBuster reagent, the inclusion bodies were solubilized in 500 mm CAPS, pH 11.0, supplemented with 0.3% N-lauroylsarcosine. After centrifugation, the supernatant containing the proteins was dialyzed at 4 °C against 20 mm Tris-HCl, pH 8.5, and 0.1 mm dithiothreitol. After several dialysis buffer changes, dithiothreitol was omitted, and final overnight dialysis was performed in the presence of oxidized and reduced glutathione (1 and 0.2 mm, respectively) to promote proper protein folding. Samples were concentrated using Centricon Plus-20 spin filters (Amicon).
Cloning and Expression of DosR and DosR (D54E) Mutant
The plasmids pDosR and pDosR (D54E) were obtained by insertion of the wild type DosR and the DosR (D54E) mutant coding sequences (15) into pET-21d(+) vector (Novagen) via NcoI and XhoI sites. DNA was amplified with primers GW1 and GW2 (see Table I). The Rosetta (DE3) (Novagen) and the BL21-Gold(DE3) (Stratagene) E. coli strains were used as hosts for plasmids pDosR and pDosR (D54E), respectively. Cells were grown in LB media with 100 mg/liter ampicillin (with additional 34 mg/liter chloramphenicol for cells containing plasmid pDosR (D54E)) at 37 °C to A600 = 0.6 and then were induced with 1 mm isopropyl-1-thio-•-d-galactopyranoside. Growth was continued for 3 h at 30 °C after induction. The cells were harvested by centrifugation at 5,000 rpm for 20 min at 4 °C and stored at −80 °C before use.
DosR and DosR (D54E) Protein Purification
Cell pellets containing wild type DosR and DosR (D54E) mutant were resuspended in 300 mm NaCl, 10% glycerol, 1 mm 2-mercaptoethanol, 1 mm phenylmethylsulfonyl fluoride, 20 mm Tris-HCl buffer, pH 8.0, and were lysed by French press. Protamine sulfate was added, and the lysate was incubated on ice for 30 min. The lysate was centrifuged at 20,000 × g for 40 min, and the supernatant was clarified by filtration and then was applied to a nickel-nitrilotriacetic acid affinity column. The nonspecifically bound proteins were eluted with 30 mm imidazole in 300 mm NaCl, 10% glycerol, 20 mm Tris-HCl buffer, pH 8.0. DosR and DosR (D54E) mutant were eluted from the column with 200 mm imidazole in the same buffer. Protein fractions were pooled and dialyzed against a buffer containing 20 mm MES buffer, pH 6.0, 50 mm NaCl, 10% glycerol, 1 mm EDTA, and 1 mm tri[2-carboxyethyl]phosphine hydrochloride. The proteins then were purified by cation exchange chromatography using a 20HS column (Per-Septive Biosystems) and were eluted with 50 mm to 1 m NaCl gradient. Peak fractions were combined and concentrated using Amicon Centriplus spin concentrators.
In Vitro Phosphorylation Assay
4 μg of the recombinant S-Tag fusion protein DosT or DosT (H392K/H394K) was assayed for the ability to autophosphorylate in a reaction mixture containing 50 μCi [γ-32P]ATP (6000 Ci/mmol, PerkinElmer Life Sciences), 100 mm Tris-HCl, pH 8.0, 5 mm MgCl2, 50 mm KCl2 in a final volume of 20 γl. The reaction was incubated at room temperature for 60 min. 4-γl aliquots were removed from the autophosphorylation reaction at 0 and 60 min and were stopped with the addition of an equal volume of SDS-PAGE (Laemmli) buffer and immediate immersion in a dry ice/EtOH bath. An additional 4-γl aliquot (800 ng) was removed and added to reactions containing 4 μg of either recombinant DosR or DosR (D54E) in the same reaction buffer as above (excluding isotope) for a final volume of 20 μl. 4-μl aliquots were removed for analysis at 0, 30 s, 1 min, 5 min, and 30 min. All aliquots were analyzed by SDS-PAGE and blotted to polyvinylidene difluoride membrane. The membranes were exposed to film overnight at −80 °C. Studies with DosS and DosS (H395K/H397K) were performed in the same manner except that a 10-μl aliquot (2 μg) was carried over from the autophosphorylation reaction to the reaction containing either wild type or mutant DosR, and membranes were exposed to film for 72 h at −80 °C.
Western Analysis
All blots were screened first with HRP-conjugated S-protein (Novagen), which binds the S-Tag with high affinity, at a 1:5000 dilution. ECL analysis was performed with exposure times varying from 1 to 10 s. Blots were then stripped in 200 mm glycine, pH 2.5, 0.2% Tween 20 followed by incubation in HRP color substrate to confirm loss of signal. The blots were rescreened with rabbit anti-rDosR polyclonal IgG antibody at a 1:5000 dilution followed by incubation with goat anti-rabbit IgG-HRP-conjugated secondary antibody (Pierce), and ECL analysis was performed again.
Electrophoretic Mobility Shift Assay
A double-stranded oligonucleotide containing the palindromic consensus promoter sequence of hypoxic response genes (15) was used as a DNA probe. The binding of DosR and DosR (D54E) was carried out by incubation at room temperature for 30 min in 10 μl of reaction mixtures composed of 12.5 μm DNA, 25 μm protein, 24 mm Tris-HCl buffer, pH 7.5, 20 mm MgCl2 in the presence or absence of the phosphate donor lithium potassium acetyl phosphate (20 mm). Following incubation the entire reaction volume was electrophoresed in a 15% non-denaturing Tris borate EDTA polyacrylamide gel (Bio-Rad). The gel was stained with 1 μg/ml ethidium bromide and visualized with a 312 nm transilluminator (Fisher Scientific).
RESULTS
Sensor Kinase Mutagenesis
Two-component response regulators activate gene transcription following phosphorylation by a sensor kinase (14). When the putative phosphorylation site of the regulator DosR is mutated, it no longer activates transcription of the MTB genes normally induced under hypoxic conditions (15). DosS is an obvious candidate to carry out the phosphorylation and activation of DosR. The dosS gene is adjacent to dosR in the genome, is co-transcribed, and is predicted to encode a sensor kinase (13, 20). However, the role of the putative kinase and its interaction with DosR was unclear as targeted deletion of dosS had little effect on the ability of DosR to activate gene transcription (13), and disruption of dosS had relatively little effect on survival under hypoxic conditions (21). These results led to speculation that a second putative sensor kinase gene in the MTB genome with homology to DosS, DosT (Rv2027c), may complement the function of DosS (13, 20). To assess the role of the putative histidine sensor kinases DosS and DosT in the phosphorylation of DosR, we targeted these genes for deletion. The dosS gene was inactivated in isolate H37Rv by gene replacement as reported previously (13). We then inactivated the dosT gene in both H37Rv and H37Rv:ΔdosS. A mutated copy of dosT was inserted that contained premature stop codons, introduced a PacI restriction site, and lacked the sequence encoding the histidine-containing putative signaling region. Confirmation of the dosT gene replacement was determined by performing PCR on transformants and amplicon digestion with PacI (data not shown).
Effects of Sensor Kinase Gene Mutagenesis
One of the MTB genes powerfully induced under hypoxic conditions is acr (also called Rv2031c or hspX) encoding an α-crystallin-like heat shock protein (13, 22). The acr promoter contains two high affinity DosR binding sequences (15, 23). Mutation of these sequences abolished DosR binding and resulted in dramatically reduced levels of expression (15). To determine the roles of DosS and DosT, both wild type and mutant bacteria were transformed with plasmid pMH108 in which the acr promoter is fused upstream of the firefly luciferase (luc) reporter gene. Isolates were subjected to hypoxic conditions, and levels of luciferase activity were measured (Fig. 1). In the wild type strain H37Rv, luc gene expression was induced about 115-fold (Fig. 1). In both the H37Rv:ΔdosS and H37Rv:ΔdosT mutants luc induction was reduced to 40–45% of wild type values. When both dosS and dosT genes were inactivated, reporter gene expression levels dropped to base-line values, indicating that DosR no longer is able to activate transcription (Fig. 1). These results demonstrate that both DosS and DosT are required for full activation of DosR under conditions of reduced oxygen tension.
Fig. 1.
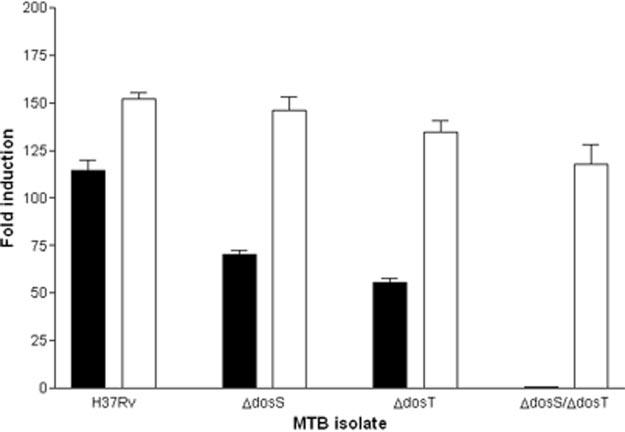
Effects of sensor kinase mutagenesis and complementation of dosT on reporter gene expression under hypoxic conditions. Wild type H37Rv, H37Rv:ΔdosS, H37Rv:ΔdosT, and H37Rv:ΔdosS:ΔdosT were transformed with the acr-luc reporter constructs pMH108 (black bars) or pMH108 with dosT driven by the constitutive mycobacterial optimal promoter (white bars). The isolates were subjected to hypoxic conditions, and luciferase activity was measured. Data are shown from one experiment (representative of three), reported as -fold induction of the acr promoter relative to expression in an aerated, log-phase culture. The differences observed between H37Rv, H37Rv:ΔdosS, H37Rv:ΔdosT, and H37Rv:ΔdosS:ΔdosT are statistically significant (paired Student's t test, p ≤ 0.0012).
dosT Complementation Analyses
To determine whether the interruption of reporter gene expression was caused by the targeted disruption of dosS and dosT, we restored a functional dosT gene to the mutants H37Rv:ΔdosS, H37Rv:ΔdosT, and H37Rv:ΔdosS:ΔdosT. The wild type dosT gene was PCR-amplified and cloned into the reporter construct pMH108 under the control of the constitutive mycobacterial optimal promoter. Each MTB isolate was transformed with the pMH108:dosT complementation constructs, incubated under hypoxic conditions, and tested for luciferase activity (Fig. 1). When dosT was reintroduced into each isolate, reporter gene expression was restored to levels comparable with or greater than those observed with wild type H37Rv, indicating that loss of hypoxic responsiveness in the mutants was caused by the lack of an appropriate sensor kinase.
In Vitro Phosphorylation Assay
The first steps in two-component signal transduction are generally signal recognition by the sensor kinase followed by dimerization and autophosphorylation at a specific His residue (14). Once autophosphorylated, the kinase transfers the phosphate to a second protein, the response regulator. We utilized recombinant DosS, DosT, and DosR in an in vitro phosphorylation assay to determine whether they functioned as a two-component system. In addition to testing wild type proteins, mutant DosS and DosT were generated via mutagenic fusion PCR. The mutations encoded amino acid substitutions intended to disrupt the histidine kinase phosphorylation motifs, giving rise to DosS (H395K/H397K) and DosT (H392K/H394K). Recombinant proteins were overexpressed in E. coli as S-Tag fusion proteins. The production of the recombinant S-Tag proteins and their purification from inclusion bodies was verified via SDS-PAGE Coomassie Blue staining and Western analysis with S-protein HRP conjugate (data not shown). In both instances a protein slightly larger than the 75-kDa marker was detected, corresponding to the molecular mass of both DosS and DosT (∼62 kDa) fused to the 16-kDa S-Tag.
First we analyzed whether DosT had the ability to autophosphorylate and then transfer the phosphate to DosR. DosT was incubated at room temperature with 50 μCi [γ-32P]ATP, and aliquots of the reaction were removed for analysis at 0 and 60 min. An additional aliquot from the autophosphorylation reaction was removed at 60 min and put into a relay reaction containing purified DosR. Aliquots of the relay reaction were removed at 0, 0.5, 1, 5, and 30 min. The aliquots from both reactions were fractionated by SDS-PAGE, blotted, and visualized by autoradiography (Fig. 2). The DosT (H392K/H394K) mutant also was analyzed for its ability to autophosphorylate in the same manner.
Fig. 2.

In vitro phosphorelay reaction between DosT and DosR or DosR (D54E). Purified DosT was incubated with [γ-32P]ATP for 60 min. An aliquot was transferred to a reaction containing either DosR (A–C) or DosR (D54E) (D–F) and incubated for 30 min. Aliquots were removed and analyzed at the indicated time points. A and D, autoradiographs of reactions; B and E, S-Tag antibody immunoblot to detect DosT; C and F, DosR antibody immunoblot. Gray and black arrows indicate predicted size of DosT-S-Tag and DosR, respectively.
The autoradiograph of the blot corresponding to the DosT autophosphorylation/phosphorelay reaction is shown in Fig. 2A. By 60 min a radiolabeled protein was detected consistent with the predicted ∼78-kDa DosT fusion protein size. Upon transfer to the relay reaction, this same protein was observed at time 0, but the radiolabel in this band waned with time until it was barely detectable at 30 min. In contrast, a radiolabeled protein consistent with the 26-kDa size of DosR was barely detectable at time 0 of the phosphorelay reaction yet became more intense as the 30-min mark approached, indicating that phosphotransfer is occurring between the two proteins. To confirm the identity of the differentially labeled proteins, immunoblot analyses were performed with an S-protein HRP conjugate to detect DosT (Fig. 2B) and after stripping with an anti-DosR antibody (Fig. 2C). Reactivity with the anti-DosR antibody was observed only in the phosphorelay lanes with the radiolabeled 26-kDa protein confirming its identity as DosR, whereas reactivity with the S-protein HRP conjugate was observed with the radiolabeled protein at ∼78 kDa confirming it as DosT. The additional bands in Fig. 2 also reacted with the S-protein HRP conjugate indicating that they are truncated forms of DosT that no longer have the ability to transfer phosphate (data not shown). When the DosT (H392K/H394K) mutant replaced the wild type DosT in the assay, there was no detectable autophosphorylation after 60 min and thus no subsequent phosphate transfer to DosR (data not shown). In addition, no labeling was observed in control phosphotransfer reactions from which DosT was omitted (data not shown), demonstrating that labeling of DosR was caused by phosphotransfer from DosT as opposed to DosR autophosphorylation.
To determine whether the phosphorelay proceeded in a manner consistent with two-component response regulators, we performed the same reaction with a DosR mutant harboring an amino acid substitution at position 54, which replaces the putative phosphorylation site aspartate residue with a glutamate incapable of receiving the donor phosphate. The autoradiograph of the reaction again showed that DosT undergoes autophosphorylation, but when DosT was removed to the relay reaction containing DosR (D54E) there was no observable decrease in its label throughout the 30-min time course (Fig. 2D). Similarly, there was no protein consistent with DosR showing an increase in radiolabel, unlike when wild type DosR is the substrate. Immunoblot analyses confirm the presence and identities of both DosT (Fig. 2E) and DosR (D54E) (Fig. 2F). These results demonstrate that the phosphotransfer observed between DosT and DosR occurs at a specific site consistent with their function as a two-component signal transduction system.
DosS was shown previously to undergo autophosphorylation (24), but its ability to transfer phosphate to DosR was not established. To address this issue, a second set of in vitro phosphorylation experiments was conducted using S-Tagged DosS and DosS (H395K/H397K). As shown in Fig. 3, DosS underwent autophosphorylation and was able to transfer the phosphate to DosR (Fig. 3A) but not to DosR (D54E) (Fig. 3B). The mutant DosS (H395K/H397K) was unable to autophosphorylate (data not shown). The identities of DosS and DosR were confirmed by immunoblotting as above (data not shown). Results with DosS were strikingly similar to those with DosT (Fig. 2). However, under identical experimental conditions DosT was more efficient at autophosphorylation and phosphotransfer than DosS. To visualize DosS phosphoactivity comparable with that observed with DosT, we needed to transfer more of the autophosphorylation reaction to the relay reaction and expose the blots for longer time (see “Experimental Procedures”).
Fig. 3.

In vitro phosphorelay reaction between DosS and DosR or DosR (D54E). Purified DosS was incubated with [γ-32P]ATP for 60 min. An aliquot was transferred to a reaction containing either DosR (A) or DosR (D54E) (B) and incubated for 30 min. Aliquots were removed and analyzed at the indicated time points. Gray and black arrows indicate the predicted size of DosS-S-Tag and DosR, respectively. The 1-min time point in A contains an artifact at ∼30 kDa that did not appear in replicate blots and should be disregarded.
Electrophoretic Mobility Shift Assay
Collectively, the results of these analyses demonstrate that both DosS and DosT can autophosphorylate and then transfer phosphate to DosR in the fashion of a two-component signal transduction system, resulting in regulation of MTB genes involved in the response to hypoxia or NO. To probe the effects of DosR phosphorylation more closely, we examined the DNA binding efficiency of the phosphorylated DosR versus the inactive, unphosphorylated form using an electrophoretic mobility shift assay. We and others (15, 23) determined previously that DosR recognizes variations of a 20-bp palindromic sequence (5′-TTGGGGACTAAAGTCCCCAA-3′) that precedes nearly all of the MTB genes initially up-regulated by hypoxia. We exploited the DosR consensus binding sequence as the DNA probe in the electrophoretic mobility shift assay (Fig. 4). For each reaction, equal amounts of DNA and protein were incubated and electrophoresed. DosR bound to the DNA even in the absence of phosphorylation. However, when DosR was incubated with the consensus sequence in the presence of the phosphate donor acetyl phosphate, enhanced DosR binding was observed. Densitometry of the gel image indicated that phosphorylation increased affinity of DosR for its binding site by about 3-fold (data not shown). Even though it cannot phosphorylate or activate gene transcription (15), the DosR (D54E) mutant bound to the consensus sequence nearly as well as phosphorylated wild type protein, and co-incubation with the phosphate donor did not affect this binding. In at least one other response regulator (the NtrC protein of enteric bacteria), a D54E mutant bound to its cognate DNA nearly as well as fully activated wild type protein (25). Glutamate at position 54 may partially resemble aspartyl phosphorylation and modestly activate the protein (25).
Fig. 4.

DosR binding to DNA is enhanced by phosphorylation. An electrophoretic mobility shift assay was performed using purified DosR or DosR (D54E) and a DNA probe containing a DosR binding motif in the presence or absence of the phosphate donor acetyl phosphate (Acetyl-P).
DISCUSSION
Given the postulated role of hypoxia and NO in promoting and maintaining TB latency (5, 6), the effects of these stimuli on MTB physiology need to be better understood. We have shown in this study that the MTB dosS and dosT genes encode functional histidine sensor kinases that upon exposure to hypoxia activate the response regulator DosR through phosphorylation. Phosphorylated DosR binds to specific DNA sequences and activates transcription of the great majority of genes rapidly induced by reduced oxygen tension. When the kinases are inactivated through mutagenesis, DosR no longer activates gene expression. These data demonstrate a role for the DosR/DosS/DosT signaling system in the adaptation of MTB to conditions that trigger reversible bacterial stasis in vitro and may contribute to latency in vivo. Additionally, it was interesting to note that along with dosS, which lies adjacent to and is co-transcribed with dosR, dosT also plays a significant role in the MTB hypoxic response. DosT is constitutively expressed in response to hypoxia (13), so this kinase may respond initially to the hypoxic signal, leading to increased levels of DosS and amplification of the hypoxic response.
That hypoxia and NO activate the same set of MTB genes suggests the DosS and DosT kinases may be able to recognize multiple inputs. Alternatively these stimuli may each alter a common factor that can then interact with the kinases. Consistent with this possibility, hypoxia and NO are additive in their effects on MTB growth and gene expression (8). Analogous sensor systems have been described in other microbes. For example, the E. coli sensor kinase ArcB detects changes in oxygen levels through interaction with the pool of cytosolic quinones (26). Oxidized quinones generated during oxygen availability inhibit ArcB autophosphorylation and subsequent ArcA regulator activation. In contrast, reduced quinones generated under oxygen limitation do not inhibit ArcB autophosphorylation, leading to phosphorylated ArcA and differential gene expression. Similarly, the Rex repressor of Streptomyces coelicolor alters gene expression in response to changes in cellular oxygen levels through competitive binding of NAD+ and NADH (27). Finally, cytochrome oxidase, a hemeprotein that can bind both oxygen and NO, has already been proposed as a mediator of DosR signal transduction (8).
The DosR/DosS/DosT two-component system has been implicated in the adaptation of mycobacteria other than MTB to hypoxic conditions. Mutant Mycobacterium smegmatis lacking dosR no longer could activate a universal stress response gene required for survival under hypoxia (28). Similarly, when Mycobacterium bovis BCG was tested in a hypoxic model of nonreplicating persistence, survival of a mutant lacking dosR was reduced 1500-fold by day 40 (21). Our experiments to define phenotypes for the ΔdosS, ΔdosT, and ΔdosS/dosT mutants of MTB under hypoxic conditions in culture have been highly variable thus far, but none of our experiments (n = 4) have yielded a phenotype nearly as dramatic as that reported in M. bovis BCG by Boon and Dick (21). We currently are investigating whether the difference in these results stems from genetic differences in MTB and M. bovis BCG.
In addition to the data from culture models, the DosR/DosS/DosT two-component system also has been studied in animals. In a recent study, a ΔdosR mutant of MTB was reported to be hypervirulent in both severe combined immunodeficient and DBA mice (29). In contrast, another recent report indicated that a ΔdosR mutant of MTB was attenuated for virulence in guinea pigs (30). Thus, although both studies implicate the DosR response in MTB virulence, the precise role of this regulon in vivo still must be determined.
Detailed analysis of the DosR/DosS/DosT two-component signaling system is an important step in characterizing the MTB response to hypoxia and NO, two stimuli that promote non-replicating persistence in culture. By determining the molecular processes involved in the adaptation of MTB to non-replicating persistence, we hope to provide insight into the etiology of TB latency. Data are accumulating that DosR response genes are induced during infections (8, 29, 31-33). Careful dissection of the DosR response should help to establish whether it plays a role in TB latency in vivo and may facilitate a more focused approach to the discovery of new MTB targets for drug and vaccine development.
Acknowledgments
We thank Marcel Behr and members of the Sherman laboratory for helpful discussions and for critical review of the data. We also thank Konstantin V. Korotkov for providing the expression systems for DosR and DosR (D54E).
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: TB, tuberculosis; MTB, Mycobacterium tuberculosis; CAPS, 3-(cyclohexylamino)propanesulfonic acid; MES, 4-morpholineethanesulfonic acid; HRP, horseradish peroxidase.
Genes are denoted both by name and the designation assigned by the TubercuList Web site: http://genolist.pasteur.fr/TubercuList/.
REFERENCES
- 1.Dock W. Am. Rev. Tuberc. 1946;53:297–305. doi: 10.1164/art.1946.53.4.297. [DOI] [PubMed] [Google Scholar]
- 2.Bloom BR, Small PM. N. Engl. J. Med. 1998;338:677–678. doi: 10.1056/NEJM199803053381008. [DOI] [PubMed] [Google Scholar]
- 3.Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC. J. Am. Med. Assoc. 1999;282:677–686. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 4.Enarson DA, Murray JF. In: Tuberculosis. Rom WN, Garay S, editors. Little, Brown and Co.; Boston: 1996. pp. 55–75. [Google Scholar]
- 5.Wayne LG, Sohaskey CD. Annu. Rev. Microbiol. 2001;55:139–163. doi: 10.1146/annurev.micro.55.1.139. [DOI] [PubMed] [Google Scholar]
- 6.Nathan C, Shiloh MU. Proc. Natl. Acad. Sci. U. S. A. 2000;97:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wayne LG. Infect. Immun. 1977;17:528–530. doi: 10.1128/iai.17.3.528-530.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voskuil MI, Schnappinger D, Harrell MI, Visconti KC, Dolganov G, Sherman DR, Schoolnik GK. J. Exp. Med. 2003;198:705–713. doi: 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi HS, Rai PR, Chu HW, Cool C, Chan ED. Am. J. Respir. Crit. Care Med. 2002;166:178–186. doi: 10.1164/rccm.2201023. [DOI] [PubMed] [Google Scholar]
- 10.Canetti G. The Tubercle Bacillus in the Pulmonary Lesion of Man. Springer Publishing Co.; New York: 1955. pp. 111–126. [Google Scholar]
- 11.Corper HJ, Cohn ML. Am. Rev. Tuberc. 1933;28:856–874. [Google Scholar]
- 12.Wayne LG, Lin KY. Infect. Immun. 1982;37:1042–1049. doi: 10.1128/iai.37.3.1042-1049.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, Schoolnik GK. Proc. Natl. Acad. Sci. U. S. A. 2001;98:7534–7539. doi: 10.1073/pnas.121172498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoch JA, Silhavy TJ. Two-component Signal Transduction. ASM Press; Washington, D. C: 1995. [Google Scholar]
- 15.Park HD, Guinn KM, Harrell MI, Liao R, Voskuil MI, Tompa M, Schoolnik GK, Sherman DR. Mol. Microbiol. 2003;48:833–843. doi: 10.1046/j.1365-2958.2003.03474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mdluli K, Sherman DR, Hickey MJ, Kreiswirth BN, Morris S, Stover CK, Barry CE., III J. Infect. Dis. 1996;174:1085–1090. doi: 10.1093/infdis/174.5.1085. [DOI] [PubMed] [Google Scholar]
- 17.Yuan Y, Crane DD, Simpson RM, Zhu Y, Hickey MJ, Sherman DR, Barry CE., III Proc. Natl. Acad. Sci. U. S. A. 1998;95:9578–9583. doi: 10.1073/pnas.95.16.9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee MH, Pascopella L, Jacobs WRJ, Hatfull GF. Proc. Natl. Acad. Sci. U. S. A. 1991;88:3111–3115. doi: 10.1073/pnas.88.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.George KM, Yuan Y, Sherman DR, Barry CE., III J. Biol. Chem. 1995;270:27292–27298. doi: 10.1074/jbc.270.45.27292. [DOI] [PubMed] [Google Scholar]
- 20.Dasgupta N, Kapur V, Singh KK, Das TK, Sachdeva S, Jyothisri K, Tyagi JS. Tuber. Lung Dis. 2000;80:141–159. doi: 10.1054/tuld.2000.0240. [DOI] [PubMed] [Google Scholar]
- 21.Boon C, Dick T. J. Bacteriol. 2002;184:6760–6767. doi: 10.1128/JB.184.24.6760-6767.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan Y, Crane DD, Barry CE., III J. Bacteriol. 1996;178:4484–4492. doi: 10.1128/jb.178.15.4484-4492.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Florczyk MA, McCue LA, Purkayastha A, Currenti E, Wolin MJ, McDonough KA. Infect. Immun. 2003;71:5332–5343. doi: 10.1128/IAI.71.9.5332-5343.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saini DK, Pant N, Das TK, Tyagi JS. Protein Expression Purif. 2002;25:203–208. doi: 10.1006/prep.2002.1628. [DOI] [PubMed] [Google Scholar]
- 25.Klose KE, Weiss DS, Kustu S. J. Mol. Biol. 1993;232:67–78. doi: 10.1006/jmbi.1993.1370. [DOI] [PubMed] [Google Scholar]
- 26.Georgellis D, Kwon O, Lin EC. Science. 2001;292:2314–2316. doi: 10.1126/science.1059361. [DOI] [PubMed] [Google Scholar]
- 27.Brekasis D, Paget MS. EMBO J. 2003;22:4856–4865. doi: 10.1093/emboj/cdg453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Toole R, Williams HD. Res. Microbiol. 2003;154:387–392. doi: 10.1016/S0923-2508(03)00081-0. [DOI] [PubMed] [Google Scholar]
- 29.Parish T, Smith DA, Kendall S, Casali N, Bancroft GJ, Stoker NG. Infect. Immun. 2003;71:1134–1140. doi: 10.1128/IAI.71.3.1134-1140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malhotra V, Sharma D, Ramanathan VD, Shakila H, Saini DK, Chakravorty S, Das TK, Li Q, Silver RF, Narayanan PR, Tyagi JS. FEMS Microbiol. Lett. 2004;231:237–245. doi: 10.1016/S0378-1097(04)00002-3. [DOI] [PubMed] [Google Scholar]
- 31.Fenhalls G, Stevens L, Moses L, Bezuidenhout J, Betts JC, van Helden P, Lukey PT, Duncan K. Infect. Immun. 2002;70:6330–6338. doi: 10.1128/IAI.70.11.6330-6338.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohno H, Zhu G, Mohan VP, Chu D, Kohno S, Jacobs WR, Jr., Chan J. Cell. Microbiol. 2003;5:637–648. doi: 10.1046/j.1462-5822.2003.00307.x. [DOI] [PubMed] [Google Scholar]
- 33.Shi L, Jung YJ, Tyagi S, Gennaro ML, North RJ. Proc. Natl. Acad. Sci. U. S. A. 2003;100:241–246. doi: 10.1073/pnas.0136863100. [DOI] [PMC free article] [PubMed] [Google Scholar]