


Abstract
Telomeres are composed of repetitive G-rich sequence and an abundance of associated proteins that together form a dynamic cap that protects chromosome ends and allows them to be distinguished from deleterious DSBs. Telomere-associated proteins also function to regulate telomerase, the ribonucleoprtotein responsible for addition of the species-specific terminal repeat sequence. Loss of telomere function is an important mechanism for the chromosome instability commonly found in cancer. Dysfunctional telomeres can result either from alterations in the telomere-associated proteins required for end-capping function, or from alterations that promote the gradual or sudden loss of sufficient repeat sequence necessary to maintain proper telomere structure. Regardless of the mechanism, loss of telomere function can result in sister chromatid fusion and prolonged breakage/fusion/bridge (B/F/B) cycles, leading to extensive DNA amplification and large terminal deletions. B/F/B cycles terminate primarily when the unstable chromosome acquires a new telomere, most often by translocation of the ends of other chromosomes, thereby providing a mechanism for transfer of instability from one chromosome to another. Thus, the loss of a single telomere can result in on-going instability, affect multiple chromosomes, and generate many of the types of rearrangements commonly associated with human cancer.
TELOMERES AND THEIR FUNCTIONS
Telomeres, unique structures at the physical ends of linear eukaryotic chromosomes, were first described almost 70 yearsago by Hermann Muller in his classic studies of the fruit fly Drosophilia melanogaster (1). He noted that chromosomal inversions resulting from ionizing radiation (IR)-induced double-strand breaks (DSBs) never involved the very end of a chromosome rejoining with some other part of a chromosome. Muller coined the term ‘telomere’, which comes from Greek—telos meaning end and meros meaning part—based on this chromosome end protection phenomenon. Shortly thereafter, Barbara McClintock observed that while broken ends of maize chromosome fused, forming dicentric chromosomes, unbroken chromosomes rarely fused; i.e. natural chromosomal termini are not ‘sticky’ (2). The absence of interstitial telomere sequence within IR-induced dicentrics was later verified in human cells (3). These studies demonstrate that normally cells accurately distinguish telomeric ends from random DSB ends and protect the former from illegitimate end-joining reactions. How cells make this critical distinction continues to be an active area of research today, especially as the dividing lines between the two types of ends have become less, rather than more clear. Recent discoveries that certain DSB repair proteins act to preserve—rather than to join—the natural ends of mammalian chromosomes (4–7), have provided impetus for the union of two seemingly disparate scientific fields, DNA repair and telomere biology. Here, we focus on the creation of dysfunctional mammalian telomeres in various repair deficient backgrounds that result from either the loss of end-capping structure or the loss of terminal sequence (shortening), and the consequences of this loss of function.
Telomeres serve multiple functions in preserving chromosome stability, including protecting the ends of chromosomes from degradation and preventing chromosomal end fusion. The DNA component of telomeres consists of tandem arrays of short, repetitive G-rich sequence [TTAGGG in vertebrates, (8,9)], oriented 5′-to-3′ towards the end of the chromosome (10), ending in an essential 3′ single-stranded overhang that ranges in length from ∼50 to 400 nt (11–13). Electron microscopy studies suggest this overhang can loop back and integrate into the duplex repeat tract, forming a ‘t-loop’ (14), an attractive, although not necessarily exclusive, architectural solution to the end-capping dilemma.
Telomeres gradually shorten in replicating cells due to end-processing and the ‘end replication problem’ (15–18), so in order for continuous cell division to occur, they must be replenished. The addition of telomeric repeats de novo is accomplished by the reverse transcriptase telomerase (19,20). Thus, telomere length maintenance is a state of equilibrium between telomere loss and re-addition. Precisely how telomere length regulation occurs is unknown, but it does appear that the shortest telomeres are preferentially targeted for elongation by telomerase (21). Telomere length is maintained in germ line cells, however, most human somatic cells do not express sufficient telomerase activity to prevent telomere loss as they divide. As a result, telomeres eventually shorten to the point where they initiate a cell cycle arrest in G1, a state termed replicative senescence (22). Telomere shortening can, therefore, limit the number of times somatic cells divide, contributing not only to aging phenotypes, but also providing an effective tumor suppressor mechanism. Cells that lose the ability to senesce because of mutations in p53 protein continue to divide, eventually entering ‘crisis’ where extensive telomere shortening results in chromosomal fusion and cell death. In contrast, cells that constitutively express telomerase can continue to divide almost indefinitely (23,24). Consistent with the requirement for telomere maintenance as a step in carcinogenesis, most tumor cells express telomerase (25). However, an alternative mechanism for telomere maintenance has also been described (26,27), which is observed in some tumors (28) and involves recombination (26,29).
Although telomerase is responsible for addition of telomeric sequence with every cell cycle, a plethora of other proteins also play important roles in the regulation of telomere length maintenance and in the formation of a protective end-cap that prevents chromosome fusion (30,31). Proteins that directly bind the double-stranded telomeric repeats include the TTAGGG Repeat Factors TRF1 (32,33) and TRF2 (34,35). POT1 (Protection Of Telomeres 1) specifically recognizes telomeric single-stranded DNA, belongs to a family of oligosaccharide/oligonucleotide-binding (OB)-fold-containing proteins and is highly conserved among eukaryotes (36,37). Telomere-associated proteins that do not bind DNA directly include TIN2 (TRF1-Interacting Nuclear Protein 2), which associates with TRF1 (38) and TRF2 (39,40). The TRF1 complex contains both TIN2 and POT1 and acts to regulate telomere-length homeostasis. TPP1, the recently proposed name (31) for a POT1-interacting protein identified independently in three laboratories [TINT1 (41), PTOP (42) and PIP1 (43)], recruits POT1 to telomeres and so is also involved in telomere length regulation. Rap1 (human repressor activator protein) (44) is recruited to telomeres by TRF2 and has been shown to negatively regulate telomere length in vivo (45). Six proteins have been proposed to form an essential, dynamic complex at human telomeres: TRF1, TRF2, POT1, TIN2, TPP1 and Rap1. This mammalian telomeric core complex serves to form and protect the telomere, and has been termed both the telosome (42) and alternately, Shelterin (31).
Other proteins, many of which are more commonly associated with DNA repair, are also found at telomeric ends [reviewed in (46)]. Examples include DNA-PK (Ku70/Ku86/DNA-PKcs), the MRN complex (MRE11/RAD50/NBS1), PARP1/2, Tankyrase 1/2, ATM, ERCC1/XPF, RAD51D, WRN and BLM. TRF2 has been shown to bind to ATM, blocking a damage response at telomeres (47). Interestingly, it has recently been proposed that normally, functional human telomeres must be recognized as DNA damage in the G2 phase of the cell cycle, in order to recruit the processing machinery necessary for formation of a functional telomere (48). The interplay between telomeres and DSBs may be better understood not by viewing the striking differences between them, but instead by viewing the obvious similarity—both are DNA ends, the very substrate telomere and damage response/repair proteins specifically recognize and bind.
FAILURE OF TELOMERIC FUNCTION DUE TO LOSS OF END-CAPPING STRUCTURE
Functional telomeres are essential for continuous cellular proliferation, and therefore loss of chromosomal end-capping has consequences in both aging and carcinogenesis (49,50). We have shown that effective end-capping of mammalian telomeres requires the non-homologous end-joining (NHEJ) protein DNA-dependent protein kinase (DNA-PK) (4). Mutation of any of the genes comprising DNA-PK, i.e. Ku70, Ku86, or the catalytic subunit, DNA-PKcs (51), leads to spontaneous chromosomal end-to-end fusions that maintain large blocks of telomeric sequence at the points of fusion (Figure 1). DNA-PK has since been shown to associate with human telomeric DNA in vivo (52,53). Events at the extreme terminus of the chromosome that normally serve to create a functionally protected telomere fail in the absence of DNA-PK, resulting in inappropriate end-to-end fusion events of uncapped telomeres fusing not only to each other, but also to IR-induced DSBs (54). We have also shown that the kinase activity of DNA-PKcs is required for effective telomere protection, just as it is for NHEJ (55,56). However, as with NHEJ, the critical in vivo substrates are not known. The in vivo consequences of inappropriate interstitial blocks of telomere sequence are also unknown, but they most likely affect chromatin stability, and in vitro studies have demonstrated they increase chromosomal instability (57).
Figure 1.

Strand-specific CO-FISH detection of leading- (red) and lagging- (green) strand telomeres demonstrating chromosomal telomere-telomere fusion in a DNA-PKcs deficient background, indicative of end-capping failure owing to loss of structure, not loss of sequence.
It is of interest to note that a ‘free end’ remains following a telomere–DSB fusion, thus providing a means of generating on-going instability. We find that telomere fusions contribute significantly to the background level of chromosomal aberrations, and that they occur despite the presence of ample telomere sequence. Thus, they are obviously not a consequence of telomere shortening, nor are they telomere associations [defined as distinct telomere signals separated by less than approximately one-third the width of a chromatid, (58)]. Banding studies demonstrated that any chromosome could be involved in the telomere fusions (end-capping failure is not chromosome specific), and further, that some of the telomere fusions were clonal, suggestive of covalent linkages (S. Bouffler, unpublished data). Utilizing the strand-specific CO-FISH technique (59,60), we found that the telomere fusions in DNA-PKcs-deficient cells exclusively involved telomeres synthesized via leading-strand DNA synthesis, suggesting a crucial difference in the post-replicative protection of telomeres that is linked to their mode of replication (61). Additional support for a model of end-specific differences in telomeric end-protection was provided by the demonstration of preferential loss of lagging-strand telomeres with WRN deficiency in human cell lines (62).
TRF2 directly binds the duplex telomere repeat tract as a homodimer (63,64). It protects the 3′ single-stranded G-rich overhang and is involved in t-loop formation, perhaps facilitating invasion of the 3′ single-stranded overhang (14). Inhibition of TRF2 induces a dramatic telomere fusion phenotype (65) resulting from failure of end-protection preferentially at leading-strand telomeres (61). End-capping failure occurs after replication, as evidenced by the presence of numerous chromatid-type telomere fusions (61) (Figure 2). It has also been shown that DNA damage foci form at telomeres uncapped by TRF2 inhibition (telomere dysfunction-induced foci; TIFs), and consistent with the cytogenetic results, uncapping of telomeres occurs in late S/G2, i.e. after replication (66).
Figure 2.
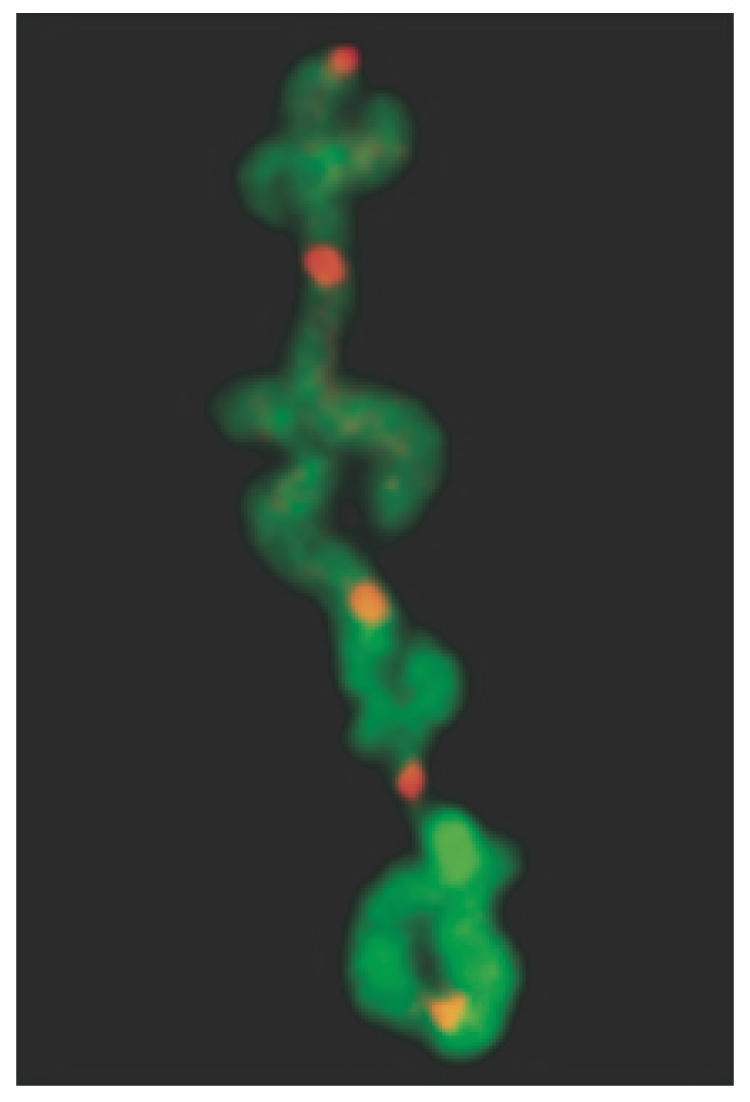
CO-FISH detection of chromatid-type telomere fusions in TRF2 deficient cells demonstrating preferential failure of end-capping at leading-strand (red) telomeres.
FAILURE OF TELOMERE FUNCTION DUE TO LOSS OF TELOMERIC REPEAT SEQUENCES
In addition to loss of end-capping function, chromosomal fusion can also result from the absence of sufficient telomeric repeat sequences to form a functional telomere. The most obvious mechanism for the loss of telomeric repeat sequences is attrition due to the failure to compensate for the gradual loss of telomeric repeat sequences during cell division. This failure to maintain telomere homeostasis can result from either insufficient telomerase activity or alterations in other telomere-associated proteins required for the recruitment of telomerase to the telomere. The gradual loss of telomeric repeat sequence due to insufficient telomerase activity is exemplified by the telomere shortening that occurs in telomerase-deficient somatic cells with each cell division, although due to cell senescence or apoptosis, this does not normally result in chromosome instability. In addition to gradual loss, telomeric repeat sequences can also be lost through stochastic processes, in which large blocks of telomeric repeat sequences are lost in single events. Stochastic events leading to telomere loss can occur through a variety of different mechanisms, the most obvious being large deletions involving recombination, problems encountered during DNA synthesis or inefficient DNA repair. Replication forks stall near telomeres in yeast, and require the Rrm3 helicase, which promotes replication through regions of non-histone chromatin (67,68). Mammalian telomeres may also pose problems for DNA replication, since the mammalian telomere-binding proteins TRF1 and TRF2 can inhibit replication fork movement (69). Studies in yeast have also demonstrated that telomeric regions are deficient in repair of DSBs. The introduction of DSBs at different sites along a chromosome with the I-SceI endonuclease demonstrated that DSBs near telomeres are not repaired efficiently by NHEJ, but instead result in complex chromosome rearrangements (70). Mammalian telomeres have been shown to be deficient in repair of single-strand breaks (71) and get damaged from ultraviolet light (72). Studies with mouse ES cells (73,74) and human tumor cell lines (J. P. Murnane, unpublished data) also show that a single DSB generated by I-SceI often results in complex chromosome rearrangements not commonly observed at I-SceI-induced DSBs at other locations within chromosomes (75–77).
Cells containing mutations in various proteins known to be involved in telomere maintenance have provided valuable insights into the mechanisms of telomere loss. One such protein is WRN, which is responsible for the human genetic disease Werner Syndrome (WS), an autosomal recessive genetic disease presenting a wide range of phenotypic abnormalities, including characteristics of premature aging (78). WRN is a member of the RecQ DNA helicase family and has both a 3′ to 5′ helicase activity and a 3′ to 5′ exonuclease activity (79–81). Cells isolated from individuals with WS exhibit shortened life span in culture (82), as well as an increased rate of DNA rearrangements, including translocations, deletions and dicentrics (83–88). WRN binds to a number of proteins involved in recombination, including replication protein A (RPA) (89,90), PCNA and topoisomerase I (91), DNA polymerase delta (92), and co-localizes with RAD51 (93) and the Mre11/Rad50/Nbs1 (MRN) complex in response to DSBs (94). Consistent with these protein interactions, WS cells have been shown to have a defect in homologous recombination (HR) (95,96). In addition, the observations that WS cells have a prolonged S-phase (97) and WRN co-localizes with RPA in cells arrested in S-phase with hydroxyurea (98), suggest that WS cells have a defect in resolving stalled replication forks (80,98,99). WRN has also been demonstrated to bind the DNA-PK complex (94,100–103) and FEN-1 (104), proteins involved in NHEJ. Moreover, the activity of WRN is influenced by its binding to the DNA-PK complex (101,102). Thus, in addition to a role in HR, WRN also appears to have a role in the NHEJ pathway for DSB repair.
WRN is also important in telomere maintenance. The role of telomeres in cellular senescence initially led to the proposal that the shortened life-span of WS cells in culture might be due to accelerated telomere shortening (105). WS cells were found to have accelerated telomere shortening, although the premature senescence in WS cells was found to occur when telomeres were longer than in senescent normal cells (106). However, a subsequent study found that WS cells at senescence have telomeres that are similar in length to normal senescent cells (107). Combined with the fact that WS cells can be immortalized by expression of telomerase (107–109), these results suggest that WS cells have a defect in telomere maintenance that leads to premature senescence. This conclusion was confirmed by studies in mice deficient in both WRN and telomerase. Although due to their long telomeres, mice deficient in WRN alone do not demonstrate the premature aging phenotype observed in humans with WS, later generations of telomerase-deficient mice with shortened telomeres demonstrate classic WS-like premature aging, accelerated replicative senescence, and genomic instability (110).
The mechanism responsible for telomere shortening in WRN-deficient cells has yet to be determined. One study found that cells expressing dominant-negative WRN showed an increase in chromosome ends without detectable telomeres even though no difference in average telomere length was observed (111), indicating that telomere loss in WS is due to a stochastic process. The association of WRN with the DNA-PK complex involved in NHEJ would suggest that telomere loss in WS cells is due to a deficiency in repair of DSBs near telomeres. However, the DNA-PK complex also has a role in telomere capping (4–6,53,112,113). In addition, WRN has been found to bind to TRF2 (114), which is essential for maintaining the cap on the end of the chromosome (65), and WRN is required for D-loop resolution regulated by TRF1 and TRF2 (114). Based on these observations, another possible mechanism for telomere loss in WRN-deficient cells would involve t-loop deletions similar to those observed in cells deficient in TRF2 (115). However, loss of end-capping structure would not lead directly to loss of telomeric repeat sequences. A critical clue to the mechanism of telomere loss in WS cells comes from a recent study demonstrating the preferential loss of the lagging-strand of telomeres (62), suggesting that telomere loss in WS cells is a result of the requirement for WRN in replication of the G-rich DNA found in the lagging strand. This model is consistent with the involvement of WRN in DNA replication and/or resolution of stalled replication forks (80,98,99), since failure to resolve stalled replication forks can result in DNA DSBs (116,117). The importance of WRN in replication and/or recombination of telomeres is also apparent from its requirement in suppression of sister chromatid exchange (SCE) specifically within telomeric DNA (T-SCE) (118) and activation of the ALT pathway that involves recombination (119), similar to that proposed for the yeast homolog for WRN, Sgs1 (120–122).
NBS1 is another protein that can influence the loss of telomeric repeat sequences. Mutations in NBS1 are responsible for the autosomal recessive disease Nijmegen breakage syndrome (NBS), which displays a wide range of phenotypic abnormalities, including premature aging, increased cancer incidence, chromosomal instability and sensitivity to IR (123,124). NBS1 is part of the MRN complex that also contains the MRE11 and RAD50 proteins (125,126). The MRN complex is a key player in the cellular response to DSBs in that association of the MRN complex with DSBs is required for the localization and activation of ATM (127), which in turn phosphorylates NBS1 (128–130). As a result, similar to cells deficient in ATM, mammalian cells deficient in NBS1 lack the S-phase cell cycle checkpoint (131) and are sensitive to IR (132). In addition to its roles in DNA recombination and repair, the MRN complex also functions in telomere maintenance. Inhibition of NBS1 by RNAi resulted in an increased frequency of telomere association (58). Primary fibroblasts from individuals with NBS have shortened telomeres, which are proposed to play a role in the pathology of this disease (133). In this respect NBS is similar to AT, where accelerated telomere shortening is observed in primary fibroblasts (134), although no difference in telomere length is evident in immortal cells actively maintaining their telomeres (135). In fact, mice with combined knockouts in both ATM and the RNA component of telomerase show accelerated telomere loss and premature aging, leading to the hypothesis that telomere loss is the reason for some of the phenotypic abnormalities observed in AT (136). Like WRN, the function of NBS1 affecting telomere loss is unclear. Similar to WRN, the MRN complex interacts with TRF2 (137), and therefore is likely to function in proper end-cap formation. However, although MRE11 and RAD50 are found at the telomere throughout the cell cycle, NBS1 is associated with the telomere only during S-phase, suggesting that it is involved in telomere replication. Thus, like WRN, a defect in NBS1 may promote telomere loss through problems in DNA replication or the resolution of stalled replication forks in telomeric regions. In fact, WRN associates with MRN (94), and both dominant-negative NBS1 (138) and WRN (111) have been found to cause similar increases in the rate of telomere loss with no change in average telomere length.
MECHANISMS OF GENOMIC INSTABILITY RESULTING FROM TELOMERE LOSS
Selectable marker genes adjacent to telomeres have been used to study the consequences of telomere loss in mammalian cells (139). This approach has the advantage of following the changes in individual chromosomes from the initial event, rather than attempting to reconstruct the sequence of events involved in the generation of complex rearrangements. These marked telomeres contain a Herpes simplex virus thymidine kinase (HSV-tk) selectable-marker gene to select for loss of the telomere, as well as an 18 bp recognition site for the I-SceI endonuclease to introduce DSBs, which has been widely used to study DNA repair and recombination in mammalian cells (75–77). Using this system, the types of chromosome rearrangements resulting from telomere loss have been followed in both mouse ES cells (73) and the EJ-30 human tumor cell line (140–142). In both the mouse ES cells and EJ-30, telomere loss resulted in either a telomere added directly on to the end of the broken chromosome or inverted repeats resulting from sister chromatid fusion. While the addition of a new telomere resulted in stabilization of the marker chromosome, sister chromatid fusion was followed by breakage/fusion/bridge (B/F/B) cycles and amplification of subtelomeric DNA (Figure 3). These B/F/B cycles occur when the chromosome that has lost a telomere is replicated, and the sister chromatids fuse together at their ends. The fused sister chromatids then form a bridge that breaks during anaphase when the two centromeres are pulled in opposite directions. Following DNA replication in the next cell cycle, the sister chromatids fuse once again, and therefore these B/F/B cycles continue until the marker chromosome acquires a new telomere. Because neither direct telomere addition nor sister chromatid fusion have been observed at DSBs generated by I-SceI at interstitial sites (75–77), these results suggest that there is something different about the processing of DSBs occurring near telomeres. Consistent with this conclusion, DSBs generated by I-SceI are poorly repaired by NHEJ near telomeres in yeast, and result in complex chromosome rearrangements (70). Moreover, direct telomere addition on to the ends of broken chromosomes in yeast preferentially occurs near existing telomeric repeat sequences (143).
Figure 3.

B/F/B cycles as a mechanism for chromosome instability resulting from telomere loss. B/F/B cycles are initiated when sister chromatids fuse following the loss of a telomere. Owing to the presence of two centromeres, the fused sister chromatids break when the cell attempts to divide up its sister chromatids at anaphase. Because the break does not occur exactly at the site of the fusion, one daughter cell will receive a copy of the chromosome with an inverted repeat at its end, while the other daughter cell will have a copy of the chromosome with a terminal deletion. Owing to the lack of a telomere, these chromosomes will again undergo sister chromatid fusion after DNA replication, resulting in additional amplification and terminal deletions. The location of telomeres (squares), centromeres (circles) and orientation of the subtelomeric sequences (horizontal arrows) are shown.
One important difference between the mouse ES cells and the EJ-30 human tumor cell line is that while telomeres in mouse ES cells are highly stable (loss of the HSV-tk gene <10−6 events/cell/generation), the telomeres in the human EJ-30 tumor cell line are lost at a relatively high rate (10−4 events/cell/generation). Similar results were observed with other human tumor cell lines (J. P. Murnane, unpublished data). This observation is consistent with other studies demonstrating that cancer cells commonly have telomere instability (144,145). Therefore, although chromosomal rearrangements due to telomere loss in cancer cells are commonly thought to result from the extensive chromosome fusion that occurs during crisis (146,147), a high rate of telomere loss is often observed even in human tumors and tumor cell lines that express telomerase (73,140,144,148,149), suggesting that many tumor cells have a fundamental defect that promotes telomere loss.
Another important difference between the mouse ES cells and human tumor cell lines is that while the direct addition of a telomere at the site of the break is a common event it mouse ES cells (74), telomere loss in human tumor cell lines often results in sister chromatid fusion followed by B/F/B cycles (140). In addition, while B/F/B cycles last only a few generations in mouse ES cells (73,74), in EJ-30 they can last for many cell generations (73,140,141). This inability to terminate B/F/B cycles is likely to contribute to the chromosome instability resulting from telomere loss in human tumor cells.
The prolonged B/F/B cycles in the EJ-30 human tumor cell line results in extensive DNA amplification and terminal deletions of DNA on the end of the marker chromosome that lost its telomere (141). The fused sister chromatids most often break within 1 Mb of the site of fusion, which was confirmed by a subsequent study that found that anaphase bridges formed by sister chromatid fusions most often break near their center, regardless of length (150). As a result, the region amplified by B/F/B cycles without selection is most often relatively small (i.e. <1 Mb). However, some breaks also occur far from the site of fusion, resulting in large duplications and deletions (73,140,141). This type of break is involved in the amplification of selectable marker genes located far from the end of the chromosome, and has been shown to occur at the location of fragile sites (151,152). Thus, the loss of a telomere can result in the amplification of genes anywhere on the arm of a chromosome.
B/F/B cycles end when the chromosome acquires a telomere and again becomes stable. The most common mechanism for telomere acquisition in both the mouse ES cells and the EJ-30 tumor cell line was through translocation of the ends of other chromosomes (139,142). In EJ-30 these translocations were either non-reciprocal (NRT) or involved duplications, as determined by the status of the donor chromosome, i.e. with NRTs, one of the homologs of the donor chromosome is missing part or all of an arm and its telomere, whereas with duplications, both homologs are intact. Both types of events have important consequences for the genome as a whole. The translocations involving duplications commonly involve large portions of the arms of other chromosomes, and therefore generate allelic imbalances involving a large number of genes. On the other hand, NRTs result in the loss of a telomere on the donor chromosome, resulting in its instability and the eventual acquisition of a telomere through translocations from other chromosomes. In fact, in one cell this transfer of instability was found to involve six different chromosomes, demonstrating that the loss of a single telomere can result in instability involving multiple chromosomes. Therefore, in addition to amplification of DNA at the end of a chromosome that has lost a telomere, once initiated, B/F/B cycles can result in other rearrangements, not only involving the chromosome that initially lost its telomere, but other chromosomes as well. In view of the fact that B/F/B cycles can continue for many cell generations, the loss of even a single telomere can generate a wide variety of chromosomal changes in the cell population.
Taken together, the above results demonstrate that loss of telomeric function—whether due to loss of sequence or loss of structure—and the ensuing instability and chromosomal rearrangements, can be expected to have a dramatic impact on the stability of the genome. Therefore, in view of increasing evidence implicating genomic instability in carcinogenesis (153,154), telomere loss is likely to be a significant contributing factor. A role for loss of functional telomeres in the chromosome instability commonly associated with cancer is supported by the large increase in human-like carcinomas in mice deficient in both telomerase and p53, and the presence of chromosome rearrangements typical of B/F/B cycles in these tumors (146,147,155,156). Understanding the mechanisms of telomere maintenance and the various factors that promote telomere instability should therefore provide valuable insights into both human genetic disease and cancer.
Acknowledgments
The work in the laboratory of S.M.B. was supported by NASA grant NNJ04HD83G; additional support from Dr Robert L. Ullrich NIH grant CA43322 and DOE grant DE-FG03-01ER63239 is also gratefully acknowledged. S.M.B. thanks Drs E.H. Goodwin and T. de Lange for cell lines. The work in the laboratory of J.P.M. was supported by National Institute of Environmental Health Science Grant No. RO1 ES008427, and National Cancer Institute Grant No. RO1 CA69044. The Open Access publication charges for this article were waived by Oxford University Press.
Conflict of interest statement. None declared.
REFERENCES
- 1.Muller H.J. The remaking of chromosomes. The collecting net-Woods Hole. 1938;13:181–198. [Google Scholar]
- 2.McClintock B. The stability of broken ends of chromosomes in Zea mays. Genetics. 1941;41:234–282. doi: 10.1093/genetics/26.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cornforth M.N., Meyne J., Littlefield L.G., Bailey S.M., Moyzis R.K. Telomere staining of human chromosomes and the mechanism of radiation-induced dicentric formation. Radiat Res. 1989;120:205–212. [PubMed] [Google Scholar]
- 4.Bailey S.M., Meyne J., Chen D.J., Kurimasa A., Li G.C., Lehnert B.E., Goodwin E.H. DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc. Natl Acad. Sci. USA. 1999;96:14899–14904. doi: 10.1073/pnas.96.26.14899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilley D., Tanaka H., Hande M.P., Kurimasa A., Li G.C., Oshimura M., Chen D.J. DNA-PKcs is critical for telomere capping. Proc. Natl Acad. Sci. USA. 2001;98:15084–15088. doi: 10.1073/pnas.261574698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsu H.-L., Gilley D., Galande S.A., Hande M.P., Allen B., Kim S.-H., Li G.C., Campisi J., Kohwi-Shigematsu T., Chen D.J. Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev. 2000;14:2807–2812. doi: 10.1101/gad.844000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Samper E., Goytisolo F.A., Slijepcevic P., van Buul P.P., Blasco M.A. Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Rep. 2000;1:244–252. doi: 10.1093/embo-reports/kvd051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyne J., Ratliff R.L., Moyzis R.K. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc. Natl Acad. Sci. USA. 1989;86:7049–7053. doi: 10.1073/pnas.86.18.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moyzis R.K., Buckingham J.M., Cram L.S., Dani M., Deaven L.L., Jones M.D., Meyne J., Ratliff R.L., Wu J.-R. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl Acad. Sci. USA. 1988;85:6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blackburn E.H. Structure and function of telomeres. Nature. 1991;350:569–573. doi: 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- 11.Makarov V.L., Hirose Y., Langmore J.P. Long G tails at both ends of human chromosomes suggest a C strand degredation mechanism for telomere shortening. Cell. 1997;88:657–666. doi: 10.1016/s0092-8674(00)81908-x. [DOI] [PubMed] [Google Scholar]
- 12.Wellinger R.J., Sen D. The DNA structures at the ends of eukaryotic chromosomes. Eur. J. Cancer. 1997;33:735–749. doi: 10.1016/S0959-8049(97)00067-1. [DOI] [PubMed] [Google Scholar]
- 13.Huffman K.E., Levene S.D., Tesmer V.M., Shay J.W., Wright W.E. Telomere shortening is proportional to the size of the G-rich telomeric 3′-overhang. J. Biol. Chem. 2000;275:19719–19722. doi: 10.1074/jbc.M002843200. [DOI] [PubMed] [Google Scholar]
- 14.Griffith J.D., Comeau L., Rosenfield S., Stansel R.M., Bianchi A., Moss H., de Lange T. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–514. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 15.Olovnikov A.M. Principle of marginotomy in template synthesis of polynucleotides. Dokl. Akad. Nauk. SSSR. 1971;201:1496–1499. [PubMed] [Google Scholar]
- 16.Watson J.D. The origin of concatemeric T7 DNA. Nature. 1972;239:197–201. doi: 10.1038/newbio239197a0. [DOI] [PubMed] [Google Scholar]
- 17.Lingner J., Cooper J.P., Cech T.R. Telomerase and DNA end replication: no longer a lagging strand problem? Science. 1995;269:1533–1534. doi: 10.1126/science.7545310. [DOI] [PubMed] [Google Scholar]
- 18.Chakhparonian M., Wellinger R.J. Telomere maintenance and DNA replication: how closely are these two connected? Trends Genet. 2003;19:439–446. doi: 10.1016/S0168-9525(03)00135-5. [DOI] [PubMed] [Google Scholar]
- 19.Greider C.W., Blackburn E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 20.Greider C.W., Blackburn E.H. The telomere terminal transferase of tetrahymena is a ribonucleoprotein enzyme with two kinds of specificity. Cell. 1987;51:887–898. doi: 10.1016/0092-8674(87)90576-9. [DOI] [PubMed] [Google Scholar]
- 21.Teixeira M.T., Arneric M., Sperisen P., Lingner J. Telomere length homeostasis is achieved via a switch between telomerase-extendible and -nonextendible states. Cell. 2004;117:323–335. doi: 10.1016/s0092-8674(04)00334-4. [DOI] [PubMed] [Google Scholar]
- 22.Harley C.B., Futcher A.B., Greider C.W. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 23.Bodnar A.G., Ouellette M., Frolkis M., Holt S.E., Chiu C.-P., Morin G.B., Harley C.B., Shay J.W., Lichtsteiner S., Wright W.E. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 24.Jiang X.-R., Jimenez G., Chang E., Frolkis M., Kusler B., Sage M., Beeche M., Bodnar A.G., Wahl G.M., Tlsty T.D., et al. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nature Genet. 1999;21:111–114. doi: 10.1038/5056. [DOI] [PubMed] [Google Scholar]
- 25.Shay J.W., Bacchetti S. A survey of telomerase activity in human cancer. Eur. J. Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 26.Murnane J.P., Sabatier L., Marder B.A., Morgan W.F. Telomere dynamics in an immortal human cell line. EMBO J. 1994;13:4953–4962. doi: 10.1002/j.1460-2075.1994.tb06822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bryan T.M., Englezou A., Gupta J., Bacchetti S., Reddel R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995;14:4240–4248. doi: 10.1002/j.1460-2075.1995.tb00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neumann A.A., Reddel R.R. Telomere maintenance and cancer—look, no telomerase. Nature Rev. Cancer. 2002;2:879–884. doi: 10.1038/nrc929. [DOI] [PubMed] [Google Scholar]
- 29.Dunham M.A., Neumann A.A., Fasching C.L., Reddel R.R. Telomere maintenance by recombination in human cells. Nature Genet. 2000;26:447–450. doi: 10.1038/82586. [DOI] [PubMed] [Google Scholar]
- 30.de Lange T. Protection of mammalian telomeres. Oncogene. 2002;21:532–540. doi: 10.1038/sj.onc.1205080. [DOI] [PubMed] [Google Scholar]
- 31.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 32.Chong L., van Steensel B., Broccoli D., Erdjument-Bromage H., Hanish J., Tempst P., de Lange T. A human telomeric protein. Science. 1995;270:1663–1667. doi: 10.1126/science.270.5242.1663. [DOI] [PubMed] [Google Scholar]
- 33.Zhong Z., Shiue L., Kaplan S., de Lange T. A mammalian factor that binds telomeric TTAGGG repeats in vitro. Mol. Cell. Biol. 1992;12:4834–4843. doi: 10.1128/mcb.12.11.4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bilaud T., Brun C., Ancelin K., Koering C.E., Laroche T., Gilson E. Telomeric localization of TRF2, a novel human telobox protein. Nature Genet. 1997;17:236–239. doi: 10.1038/ng1097-236. [DOI] [PubMed] [Google Scholar]
- 35.Broccoli D., Smogorzewska A., Chong L., de Lange T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nature Genet. 1997;17:231–235. doi: 10.1038/ng1097-231. [DOI] [PubMed] [Google Scholar]
- 36.Loayza D., Parsons H., Donigian J., Hoke K., de Lange T. DNA binding features of human POT1: a nonamer 5′-TAGGGTTAG-3′ minimal binding site, sequence specificity, and internal binding to multimeric sites. J. Biol. Chem. 2004;279:13241–13248. doi: 10.1074/jbc.M312309200. [DOI] [PubMed] [Google Scholar]
- 37.Baumann P., Cech T.R. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292:1171–1175. doi: 10.1126/science.1060036. [DOI] [PubMed] [Google Scholar]
- 38.Kim S.H., Kaminker P., Campisi J. TIN2, a new regulator of telomere length in human cells. Nature Genet. 1999;23:405–412. doi: 10.1038/70508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim S.H., Beausejour C., Davalos A.R., Kaminker P., Heo S.J., Campisi J. TIN2 mediates functions of TRF2 at human telomeres. J. Biol. Chem. 2004;279:43799–43804. doi: 10.1074/jbc.M408650200. [DOI] [PubMed] [Google Scholar]
- 40.Ye J.Z., Donigian J.R., van Overbeek M., Loayza D., Luo Y., Krutchinsky A.N., Chait B.T., de Lange T. TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J. Biol. Chem. 2004;279:47264–47271. doi: 10.1074/jbc.M409047200. [DOI] [PubMed] [Google Scholar]
- 41.Houghtaling B.R., Cuttonaro L., Chang W., Smith S. A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr. Biol. 2004;14:1621–1631. doi: 10.1016/j.cub.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 42.Liu D., Safari A., O'Connor M.S., Chan D.W., Laegeler A., Qin J., Songyang Z. PTOP interacts with POT1 and regulates its localization to telomeres. Nature Cell Biol. 2004;6:673–680. doi: 10.1038/ncb1142. [DOI] [PubMed] [Google Scholar]
- 43.Ye J.Z., Hockemeyer D., Krutchinsky A.N., Loayza D., Hooper S.M., Chait B.T., de Lange T. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18:1649–1654. doi: 10.1101/gad.1215404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li B., Oestreich S., de Lange T. Identification of human Rap1: implications for telomere evolution. Cell. 2000;101:471–483. doi: 10.1016/s0092-8674(00)80858-2. [DOI] [PubMed] [Google Scholar]
- 45.O'Connor M.S., Safari A., Liu D., Qin J., Songyang Z. The human Rap1 protein complex and modulation of telomere length. J. Biol. Chem. 2004;279:28585–28591. doi: 10.1074/jbc.M312913200. [DOI] [PubMed] [Google Scholar]
- 46.Blasco M.A. Telomeres and human disease: ageing, cancer and beyond. Nature Rev. Genet. 2005;6:611–622. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- 47.Karlseder J., Hoke K., Mirzoeva O.K., Bakkenist C., Kastan M.B., Petrini J.H., de Lange T. The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol. 2004;2:E240. doi: 10.1371/journal.pbio.0020240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verdun R.E., Crabbe L., Haggblom C., Karlseder J. Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol. Cell. 2005;20:551–561. doi: 10.1016/j.molcel.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 49.Morin G.B. Telomere integrity and cancer. J. Natl Cancer Inst. 1996;88:1095–1096. doi: 10.1093/jnci/88.16.1095. [DOI] [PubMed] [Google Scholar]
- 50.Harley C.B., Vaziri H., Counter C.M., Allsopp R.C. The telomere hypothesis of cellular aging. Exp. Gerontol. 1992;27:375–382. doi: 10.1016/0531-5565(92)90068-b. [DOI] [PubMed] [Google Scholar]
- 51.Jeggo P.A., Taccioli G.E., Jackson S.P. Menage à trois: double strand break repair, V(D)J recombination and DNA-PK. BioEssays. 1995;17:949–957. doi: 10.1002/bies.950171108. [DOI] [PubMed] [Google Scholar]
- 52.d'Adda di Fagagna F., Hande M.P., Tong W.M., Roth D., Lansdorp P.M., Wang Z.Q., Jackson S.P. Effects of DNA nonhomologous end-joining factors on telomere length and chromosomal stability in mammalian cells. Curr. Biol. 2001;11:1192–1196. doi: 10.1016/s0960-9822(01)00328-1. [DOI] [PubMed] [Google Scholar]
- 53.Hsu H.-L., Gilley D., Blackburn E.H., Chen D.J. Ku is associated with the telomere in mammals. Proc. Natl Acad. Sci. USA. 1999;96:12454–12458. doi: 10.1073/pnas.96.22.12454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bailey S.M., Cornforth M.N., Ullrich R.L., Goodwin E.H. Dysfunctional mammalian telomeres join with DNA double-strand breaks. DNA Repair (Amst) 2004;3:349–357. doi: 10.1016/j.dnarep.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 55.Bailey S.M., Brenneman M.A., Halbrook J., Nickoloff J.A., Ullrich R.L., Goodwin E.H. The kinase activity of DNA-PK is required to protect mammalian telomeres. DNA Repair (Amst) 2004;3:225–233. doi: 10.1016/j.dnarep.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 56.Kurimasa A., Kumano S., Boubnov N.V., Story M.D., Tung C.S., Peterson S.R., Chen D.J. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol. Cell. Biol. 1999;19:3877–3884. doi: 10.1128/mcb.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kilburn A.E., Shea M.J., Sargent R.G., Wilson J.H. Insertion of a telomere repeat sequence into a mammalian gene causes chromosome instability. Mol. Cell. Biol. 2001;21:126–135. doi: 10.1128/MCB.21.1.126-135.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Q., Williams E.S., Askin K.F., Peng Y., Bedford J.S., Liber H.L., Bailey S.M. Suppression of DNA-PK by RNAi has different quantitative effects on telomere dysfunction and mutagenesis in human lymphoblasts treated with gamma rays or HZE particles. Radiat. Res. 2005;164:497–504. doi: 10.1667/rr3366.1. [DOI] [PubMed] [Google Scholar]
- 59.Goodwin E., Meyne J. Strand-specific FISH reveals orientation of chromosome 18 alphoid DNA. Cytogenet. Cell Genet. 1993;63:126–127. doi: 10.1159/000133516. [DOI] [PubMed] [Google Scholar]
- 60.Bailey S.M., Goodwin E.H., Cornforth M.N. Strand-specific fluorescence in situ hybridization: the CO-FISH family. Cytogenet. Genome Res. 2004;107:14–17. doi: 10.1159/000079565. [DOI] [PubMed] [Google Scholar]
- 61.Bailey S.M., Cornforth M.N., Kurimasa A., Chen D.J., Goodwin E.H. Strand-specific postreplicative processing of mammalian telomeres. Science. 2001;293:2462–2465. doi: 10.1126/science.1062560. [DOI] [PubMed] [Google Scholar]
- 62.Crabbe L., Verdun R.E., Haggblom C.I., Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 63.Bilaud T., Koering C.E., Binet-Brasselet E., Ancelin K., Pollice A., Gasser S.M., Gilson E. The telobox, a Myb-related telomeric DNA binding motif found in proteins from yeast, plants and human. Nucleic Acids Res. 1996;24:1294–1303. doi: 10.1093/nar/24.7.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Broccoli D., Chong L., Oelmann S., Fernald A.A., Marziliano N., van Steensel B., Kipling D., Le Beau M.M., de Lange T. Comparison of the human and mouse genes encoding the telomeric pretein, TRF1: chromosomal localization, expression and conserved protein domains. Hum. Mol. Genet. 1997;6:69–76. doi: 10.1093/hmg/6.1.69. [DOI] [PubMed] [Google Scholar]
- 65.van Steensel B., Smogorzewska A., de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 66.Takai H., Smogorzewska A., de Lange T. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 67.Ivessa A.S., Zhou J.-Q., Schulz V.P., Monson E.K., Zakian V.A. Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA. Genes Dev. 2002;16:1383–1396. doi: 10.1101/gad.982902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ivessa A.S., Lenzmeier B.A., Bessler J.B., Goudsouzian L.K., Schnakenberg S.L., Zakian V.A. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein–DNA complexes. Mol. Cell. 2003;12:1525–1536. doi: 10.1016/s1097-2765(03)00456-8. [DOI] [PubMed] [Google Scholar]
- 69.Ohki R., Ishikawa F. Telomere-bound TRF1 and TRF2 stall the replication fork at telomeric repeats. Nucleic Acids Res. 2004;32:1627–1637. doi: 10.1093/nar/gkh309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ricchetti M., Dujon B., Fairhead C. Distance from the chromosome end determines the efficiency of double-strand break repair in subtelomeres of haploid yeast. J. Mol. Biol. 2003;328:847–862. doi: 10.1016/s0022-2836(03)00315-2. [DOI] [PubMed] [Google Scholar]
- 71.Petersen S., Saretzki G., von Zglinicki T. Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp. Cell Res. 1998;239:152–160. doi: 10.1006/excr.1997.3893. [DOI] [PubMed] [Google Scholar]
- 72.Kruk P.A., Rampino N.J., Bohr V.A. DNA damage and repair in telomeres: Relation to aging. Proc. Natl Acad. Sci. USA. 1995;92:258–262. doi: 10.1073/pnas.92.1.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lo A.W.I., Sprung C.N., Fouladi B., Pedram M., Sabatier L., Ricoul M., Reynolds G.E., Murnane J.P. Chromosome instability as a result of double-strand breaks near telomeres in mouse embryonic stem cells. Mol. Cell. Biol. 2002;22:4836–4850. doi: 10.1128/MCB.22.13.4836-4850.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sprung C.N., Reynolds G.E., Jasin M., Murnane J.P. Chromosome healing in mouse embryonic stem cells. Proc. Natl Acad. Sci. USA. 1999;96:6781–6786. doi: 10.1073/pnas.96.12.6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sargent R.G., Brenneman M.A., Wilson J.H. Repair of site-specific double-strand breaks in a mammalian chromosome by homologous and illegitimate recombination. Mol. Cell. Biol. 1997;17:267–277. doi: 10.1128/mcb.17.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Richardson C., Jasin M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature. 2000;405:697–700. doi: 10.1038/35015097. [DOI] [PubMed] [Google Scholar]
- 77.Lin Y., Waldman A.S. Capture of DNA sequences at double-strand breaks in mammalian cells. Genetics. 2001;158:1665–1674. doi: 10.1093/genetics/158.4.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Salk D. Werner's syndrome: a review of recent research with an analysis of connective tissue metablolism, growth control of cultured cells, and chromosome aberrations. Hum. Genet. 1982;62:1–5. doi: 10.1007/BF00295598. [DOI] [PubMed] [Google Scholar]
- 79.Brosh R.M., Bohr V.A. Roles of the Werner syndrome protein in pathways required for maintenance of genome stability. Exp. Gerontol. 2002;37:491–506. doi: 10.1016/s0531-5565(01)00227-3. [DOI] [PubMed] [Google Scholar]
- 80.Shen J.-C., Loeb L.A. The Werner syndrome gene. Trends Genet. 2000;16:213–220. doi: 10.1016/s0168-9525(99)01970-8. [DOI] [PubMed] [Google Scholar]
- 81.Oshima J. The Werner syndrome protein: an update. BioEssays. 2000;22:894–901. doi: 10.1002/1521-1878(200010)22:10<894::AID-BIES4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 82.Martin G.M., Sprague C.A., Epstein C.J. Replicative life-span of cultivated human cells. Effects of donor's age, tissue and genotype. Lab. Invest. 1970;23:86–92. [PubMed] [Google Scholar]
- 83.Fukuchi K., Martin G.M., Monnat R.J.J. Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc. Natl Acad. Sci. USA. 1989;86:5893–5897. doi: 10.1073/pnas.86.15.5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Scappaticci S., Cerimele D., Fraccaro M. Clonal structural chromosomal rearrangements in primary fibroblast cultures and in lymphocytes of patients with Werner's syndromeq. Hum. Genet. 1982;62:16–24. doi: 10.1007/BF00295599. [DOI] [PubMed] [Google Scholar]
- 85.Salk D., Au K., Hoehn H., Martin G.M. Cytogenetics of Werner's syndrome cultured skin fibroblasts: variegated translocation mosaicism. Cytogenet. Cell Genet. 1981;92:92–107. doi: 10.1159/000131596. [DOI] [PubMed] [Google Scholar]
- 86.Gebhart E., Bauer R., Raub U., Schinzel M., Ruprecht K.W., Jonas J.B. Spontaneous and induced chromosomal instability in Werner syndrome. Hum. Genet. 1988;80:135–139. doi: 10.1007/BF00702855. [DOI] [PubMed] [Google Scholar]
- 87.Grigorova M., Balajee A.S., Natarajan A.T. Spontaneous and X-ray-induced chromosomal aberrations in Werner syndrome cells detected by FISH using chromosome-specific painting probes. Mutagenesis. 2000;15:303–310. doi: 10.1093/mutage/15.4.303. [DOI] [PubMed] [Google Scholar]
- 88.Stefanini M., Scappaticci S., Lagomarsini P., Borroni G., Berardesca E., Nuzzo F. Chromosome instability in lymphocytes from a patient with Werner's syndrome is not associated with DNA repair defects. Mutat. Res. 1989;219:179–185. doi: 10.1016/0921-8734(89)90013-1. [DOI] [PubMed] [Google Scholar]
- 89.Shen J.C., Gray M.D., Oshima J., Loeb L.A. Characterization of Werner syndrome protein DNA helicase activity: directionality, substrate dependence, and stimulation by replication protein A. Nucleic Acids Res. 1998;26:2879–2885. doi: 10.1093/nar/26.12.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brosh R.M.J., Orren D.K., Nehlin J.O., Ravn P.H., K,D.M., Machwe A., Bohr V.A. Functional and physical interaction between WRN helicase and human replication protein A. J. Biol. Chem. 1999;274:18341–18350. doi: 10.1074/jbc.274.26.18341. [DOI] [PubMed] [Google Scholar]
- 91.Lebel M., Spillare E.A., Harris C.C., Leder P. The Werner syndrome gene product co-purifies with the DNA replication complex and interacts with PCNA and topoisomerase I. J. Biol. Chem. 1999;274:37795–37799. doi: 10.1074/jbc.274.53.37795. [DOI] [PubMed] [Google Scholar]
- 92.Szekely A.M., Chen Y.H., Zhang C., Oshima J., Weissman S.M. Werner protein recruits DNA polymerase delta to the nucleolus. Proc. Natl Acad. Sci. USA. 2000;97:11365–11370. doi: 10.1073/pnas.97.21.11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sakamoto S., Nishikawa K., Heo S.J., Goto M., Furuichi Y., Shimamoto A. Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes Cells. 2001;6:421–430. doi: 10.1046/j.1365-2443.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- 94.Cheng W.H., von Kobbe C., Opresko P.L., Arthur L.M., Komatsu K., Seidman M.M., Carney J.P., Bohr V.A. Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J. Biol. Chem. 2004;279:21169–21176. doi: 10.1074/jbc.M312770200. [DOI] [PubMed] [Google Scholar]
- 95.Prince P.R., Edmond M.J., Monnat R.J.J. Loss of Werner syndrome protein function promotes mitotic recombination. Genes Dev. 2001;15:933–938. doi: 10.1101/gad.877001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saintigny Y., Makienko K., Swanson C., Emond M.J., Monnat R.J.J. Homologous recombination resolution defect in werner syndrome. Mol. Cell. Biol. 2002;22:6971–6978. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Poot M., Hoehn H., Runger T.M., Martin G.M. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp. Cell Res. 1992;202:267–273. doi: 10.1016/0014-4827(92)90074-i. [DOI] [PubMed] [Google Scholar]
- 98.Constantinou A., Tarsounas M., Karow J.K., Brosh R.M., Bohr V.A., Hickson I.D., West S.C. Werner's syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000;1:80–84. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Franchitto A., Pichierro P. Protecting genomic integrity during DNA replication: correlation between Werner's and Bloom's syndrome gene products and the MRE11 complex. Hum. Mol. Genet. 2002;11:2447–2453. doi: 10.1093/hmg/11.20.2447. [DOI] [PubMed] [Google Scholar]
- 100.Yannone S.M., Roy S., Chan D.W., Murphy M.B., Huang S., Campisi J., Chen D.J. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J. Biol. Chem. 2001;276:38242–38248. doi: 10.1074/jbc.M101913200. [DOI] [PubMed] [Google Scholar]
- 101.Karmakar P., Piotrowski J., Brosh R.M.J., Sommers J.A., Miller S.P., Cheng W.H., Snowden C.M., Ramsden D.A., Bohr V.A. Werner protein is a target of DNA-dependent protein kinase in vivo and in vitro. J. Biol. Chem. 2002;277:18291–18302. doi: 10.1074/jbc.M111523200. [DOI] [PubMed] [Google Scholar]
- 102.Cooper M.P., Machwe A., Orren D.K., Brosh R.M., Ramsden D., Bohr V.A. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;14:907–912. [PMC free article] [PubMed] [Google Scholar]
- 103.Li B., Comai L. Functional interaction between Ku and the Werner syndrome protein in DNA end processing. J. Biol. Chem. 2000;275:28349–28352. doi: 10.1074/jbc.C000289200. [DOI] [PubMed] [Google Scholar]
- 104.Brosh R.M.J., von Kobbe C., Sommers J.A., Darmadar P., Opresko P.L., Piotrowski J., Dianova I., Dianova G.L., Bohr V.A. Werner syndrome protein interacts with human flap endonuclease I and stimulates its cleavage activity. EMBO J. 2001;20:5791–5801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Martin G.M. Genetic modulation of telomeric terminal restriction-fragment length: relevance for clonal aging and late-life disease. Am. J. Hum. Genet. 1994;55:866–869. [PMC free article] [PubMed] [Google Scholar]
- 106.Schulz V.P., Zakian V.A., Ogburn C.E., McKay J., Jarzebowicz A.A., Edland S.D., Martin G.M. Accelerated loss of telomeric repeats may not explain accelerated replicative decline of Werner syndrome cells. Hum. Genet. 1996;97:750–754. doi: 10.1007/BF02346184. [DOI] [PubMed] [Google Scholar]
- 107.Choi D., Whittier P.S., Oshima J., Funk W.D. Telomerase expression prevents replicative senescence but does not fully reset mRNA expression patterns in Werner syndrome cell strains. FASEB J. 2001;15:1014–1020. doi: 10.1096/fj.00-0104com. [DOI] [PubMed] [Google Scholar]
- 108.Ouellette M.M., McDaniel L.D., Wright W.E., Shay J.W., Schultz R.A. The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum. Mol. Genet. 2000;9:403–411. doi: 10.1093/hmg/9.3.403. [DOI] [PubMed] [Google Scholar]
- 109.Wyllie F.S., Jones C.J., Skinner J.W., Haughton M.F., Wallis C., Wynford-Thomas D., Faragher R.G., Kipling D. Telomerase prevents the accelerated ageing of Werner syndrome fibroblasts. Nature Genet. 2000;24:16–17. doi: 10.1038/71630. [DOI] [PubMed] [Google Scholar]
- 110.Chang S., Multani A.S., Cabrera N.G., Naylor M.L., Laud P., Lombard D., Pathak S., Guarente L., DePinho R.A. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nature Genet. 2004;36:877–882. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 111.Bai Y., Murnane J.P. Telomere instability in a human tumor cell line expressing a dominant-negative WRN protein. Hum. Genet. 2003;113:337–347. doi: 10.1007/s00439-003-0972-y. [DOI] [PubMed] [Google Scholar]
- 112.Boulton S.J., Jackson S.P. Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 1998;17:1819–1828. doi: 10.1093/emboj/17.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Espejel S., Franco S., Rodriguez-Perales S., Bouffler S.D., Cigudosa J.C., Blasco M.A. Mammalian Ku86 mediates chromosomal fusions and apoptosis caused by critically short telomeres. EMBO J. 2002;21:2207–2219. doi: 10.1093/emboj/21.9.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Opresko P.L., von Kobbe C., Laine J.-P., Harrigan J., Hickson I.D., Bohr V.A. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J. Biol. Chem. 2002;277:41110–41119. doi: 10.1074/jbc.M205396200. [DOI] [PubMed] [Google Scholar]
- 115.Wang R.C., Smogorzewska A., de Lange T. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell. 2004;119:355–368. doi: 10.1016/j.cell.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 116.Chen C., Kolodner R.D. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nature Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 117.Myung K., Chen C., Kolodner R.D. Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature. 2001;411:1073–1076. doi: 10.1038/35082608. [DOI] [PubMed] [Google Scholar]
- 118.Bailey S.M., Brenneman M.A., Goodwin E.H. Frequent recombination in telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res. 2004;32:3743–3751. doi: 10.1093/nar/gkh691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Laud P.R., Multani A.S., Bailey S.M., Wu L., Ma J., Kingsley C., Lebel M., Pathak S., DePinho R.A., Chang S. Elevated telomere–telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 2005;19:2560–2570. doi: 10.1101/gad.1321305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cohen H., Sinclair D.A. Recombination-mediated lengthening of terminal telomeric repeats requires the Sgs1 DNA helicase. Proc. Natl Acad. Sci. USA. 2001;98:3174–3179. doi: 10.1073/pnas.061579598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huang P.-H., Pryde F.E., Lester D., Maddison R.L., Borts R.H., Hickson I.D., Louis E.J. SGS1 is required for telomere elongation in the absence of telomerase. Current Biol. 2001;11:125–129. doi: 10.1016/s0960-9822(01)00021-5. [DOI] [PubMed] [Google Scholar]
- 122.Johnson F.B., Marciniak R.A., McVey M., Stewart S.A., Hahn W.C., Guarente L. The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. EMBO J. 2001;20:905–913. doi: 10.1093/emboj/20.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Digweed M. Human genetic instability syndromes: single gene defects with increased risk of cancer. Toxicol. Lett. 1993;67:259–281. doi: 10.1016/0378-4274(93)90061-2. [DOI] [PubMed] [Google Scholar]
- 124.Shiloh Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu. Rev. Genet. 1997;31:635–662. doi: 10.1146/annurev.genet.31.1.635. [DOI] [PubMed] [Google Scholar]
- 125.Carney J.P., Maser R.S., Olivares H., Davis E.M., Beau M., Yates J.R., Hays L., Morgan W.F., Petrini J.H. The hMre11/Rad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 126.Veron R., Vissinga C., Platzer M., Cerosaletti K.M., Chrzanowska K.H., Saar K., Beckmann G., Seemanova E., Cooper P.R., Nowak N.J., et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93:467–476. doi: 10.1016/s0092-8674(00)81174-5. [DOI] [PubMed] [Google Scholar]
- 127.Lee J.H., Paull T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- 128.Zhao S., Weng Y.-C., Yuan S.-S.F., Lin Y.-T., Hsu H.-C., Lin S.-C.J., Gerbino E., Song M.,-h., Zdizienicka M.Z., Gatti R.A., et al. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature. 2000;405:473–477. doi: 10.1038/35013083. [DOI] [PubMed] [Google Scholar]
- 129.Lim D.S., Kim S.T., Xu B., Maser R.S., Lin J., Petrini J.H., Kastan M.B. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- 130.Wu X., Ranganathan V., Weisman D.S.F.H.W., Ciccone D.N., O'Neill T.B., Crick K.E., Pierce K.A., Lane W.S., Rathbun G., et al. ATM phosporylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature. 2000;405:477–482. doi: 10.1038/35013089. [DOI] [PubMed] [Google Scholar]
- 131.Petrini J.H. The Mre11 complex and ATM: collaborating to navigate S phase. Curr. Opin. Cell. Biol. 2000;12:293–296. doi: 10.1016/s0955-0674(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 132.Taalman R.D., Jaspers N.G., Scheres J.M., de Wit J., Hustinx T.W. Hypersensitivity to ionizing radiation, in vitro, in a new chromosomal breakage disorder, the Nijmegen Breakage Syndrome. Mutat. Res. 1983;112:23–32. doi: 10.1016/0167-8817(83)90021-4. [DOI] [PubMed] [Google Scholar]
- 133.Ranganathan V., Heine W.F., Ciccone D.N., Rudolph K.L., Wu X., Chang S., Hai H., Ahern I.M., Livingston D.M., Resnick I., et al. Rescue of a telomere length defect in Nijmegen breakage syndrome cells requires NBS and the telomerase catalytic subunit. Curr. Biol. 2001;11:962–966. doi: 10.1016/s0960-9822(01)00267-6. [DOI] [PubMed] [Google Scholar]
- 134.Metcalfe J.A., Parkhill J., Campbell L., Stacey M., Biggs P., Byrd P.J., Taylor A.M.R. Accelerated telomere shortening in ataxia telangiectasia. Nature Genet. 1996;13:350–353. doi: 10.1038/ng0796-350. [DOI] [PubMed] [Google Scholar]
- 135.Sprung C.N., Bryan T.M., Reddel R.R., Murnane J.P. Normal telomere maintenance in immortal ataxia telangiectasia cell lines. Mutat. Res. 1997;379:177–184. doi: 10.1016/s0027-5107(97)00119-x. [DOI] [PubMed] [Google Scholar]
- 136.Wong K.K., Maser R.S., Bachoo R.M., Menon J., Carrasco D.R., Gu Y., Alt F.W., DePinho R.A. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 2003;421:643–648. doi: 10.1038/nature01385. [DOI] [PubMed] [Google Scholar]
- 137.Zhu X.-D., Kuster B., Mann M., Petrini J.H.J., de Lange T. Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nature Genet. 2000;25:347–352. doi: 10.1038/77139. [DOI] [PubMed] [Google Scholar]
- 138.Bai Y., Murnane J.P. Telomere instability in a human tumor cell line expressing the NBS1 gene with mutations at sites phosphorylated by the ATM protein. Mol. Cancer Res. 2003;1:1058–1069. [PubMed] [Google Scholar]
- 139.Murnane J.P., Sabatier L. Chromosomal rearrangements resulting from telomere dysfunction and their role in cancer. BioEssays. 2004;26:1164–1174. doi: 10.1002/bies.20125. [DOI] [PubMed] [Google Scholar]
- 140.Fouladi B., Miller D., Sabatier L., Murnane J.P. The relationship between spontaneous telomere loss and chromosome instability in a human tumor cell line. Neoplasia. 2000;2:540–554. doi: 10.1038/sj.neo.7900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lo A.W.I., Sabatier L., Fouladi B., Pottier G., Ricoul M., Murnane J.P. DNA amplification by breakage/fusion/bridge cycles initiated by spontaneous telomere loss in a human cancer cell line. Neoplasia. 2002;6:531–538. doi: 10.1038/sj.neo.7900267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sabatier L., Ricoul M., Pottier G., Murnane J.P. The loss of a single telomere can result in genomic instability involving multiple chromosomes in a human tumor cell line. Mol. Cancer Res. 2005;3:139–150. doi: 10.1158/1541-7786.MCR-04-0194. [DOI] [PubMed] [Google Scholar]
- 143.Diede S.J., Gottschling D.E. Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerase α and δ. Cell. 1999;99:723–733. doi: 10.1016/s0092-8674(00)81670-0. [DOI] [PubMed] [Google Scholar]
- 144.Gisselsson D., Jonson T., Petersen A., Strombeck B., Dal Cin P., Hoglund M., Mitelman F., Mertens F., Mandahl N. Telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities in human malignant tumors. Proc. Natl Acad. Sci. USA. 2001;98:12683–12688. doi: 10.1073/pnas.211357798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Meeker A.K., Hicks J.L., Iacoluzio-Donahue C.A., Montgomery E.A., Westra W.H., Chan T.Y., Ronnett B.M., De Marzo A.M. Telomere length abnormalities occur early in the initiation of epithelial carcinogenesis. Clin. Can. Res. 2004;10:3317–3326. doi: 10.1158/1078-0432.CCR-0984-03. [DOI] [PubMed] [Google Scholar]
- 146.Chang S., Khoo C., DePinho R.A. Modeling chromosomal instability and epithelial carcinogenesis in the telomerase-deficient mouse. Cancer Biol. 2001;11:227–238. doi: 10.1006/scbi.2000.0374. [DOI] [PubMed] [Google Scholar]
- 147.Rudolph K.L., Millard M., Bosenberg M.W., DePinho R.A. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nature Genet. 2001;28:155–159. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- 148.Sprung C.N., Afshar G., Chavez E.A., Lansdorp P., Sabatier L., Murnane J.P. Telomere instability in a human cancer cell line. Mutat. Res. 1999;429:209–223. doi: 10.1016/s0027-5107(99)00115-3. [DOI] [PubMed] [Google Scholar]
- 149.Chin K., de Solorzano C.O., Knowles D., Jones A., Chou W., Rodriguez E.G., Kuo W.-L., Ljung B.-M., Chew K., Myambo K., et al. In situ analysis of genome instability in breast cancer. Nature Genet. 2004;16:984–988. doi: 10.1038/ng1409. [DOI] [PubMed] [Google Scholar]
- 150.Shimizu N., Shingaki K., Kaneko-Sasaguri Y., Hashizume T., Kanda T. When, where and how the bridge breaks: anaphase bridge breakage plays a crucial role in gene amplification and HSR generation. Exp. Cell Res. 2005;302:233–243. doi: 10.1016/j.yexcr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 151.Hellman A., Ziotorynski E., Scherer S.W., Cheung J., Vincent J.B., Smith D.I., Trakhtenbrot L., Kerem B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell. 2002;1:89–97. doi: 10.1016/s1535-6108(02)00017-x. [DOI] [PubMed] [Google Scholar]
- 152.Coquelle A., Pipiras E., Toledo F., Buttin G., Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets bounderies to early amplification. Cell. 1997;89:215–225. doi: 10.1016/s0092-8674(00)80201-9. [DOI] [PubMed] [Google Scholar]
- 153.Tlsty T.D. Genomic instability and its role in neoplasia. Curr. Topics Microbiol. Immunol. 1997;221:37–46. doi: 10.1007/978-3-642-60505-5_4. [DOI] [PubMed] [Google Scholar]
- 154.Lengauer C., Kinzler K.W., Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 155.Maser R.S., DePinho R.A. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565–569. doi: 10.1126/science.297.5581.565. [DOI] [PubMed] [Google Scholar]
- 156.Artandi S.E., Chang S., Lee S.-L., Alson S., Gottlieb G.J., Chin L., DePinho R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]