

Abstract
Activation loop tyrosine autophosphorylation is an essential requirement for full kinase activation of receptor tyrosine kinases (RTKs). However, mechanisms involved are not fully understood. In general, kinase domains of RTKs are folded into two main lobes, NH2- and COOH-terminal lobes. The COOH-terminal lobe of vascular endothelial growth factor receptor-2 (VEGFR-2) is folded into seven α-helices (αD–αI). In the studies presented here we demonstrate that leucine residues of helix I (αI) regulate tyrosine autophosphorylation and phosphotransferase activity of VEGFR-2. The presence of leucines 1158, 1161, and 1162 are essential for tyrosine autophosphorylation and kinase activation of VEGFR-2 and are involved in helix-helix packing via hydrophobic interactions. The presence of leucine 1158 is critical for kinase activation of VEGFR-2 and appears to interact with αE, αF, αH, and β7. The analogous residue, leucine 957 on platelet-derived growth factor receptor-β and leucine 910 on colony stimulating factor-1R are also found to be critical for tyrosine autophosphorylation of these receptors. Leucines 1161 and 1162 are also involved in helix-helix packing but they play a less critical role in VEGFR-2 activation. Thus, we conclude that leucine motif-mediated helix-helix interactions are critical for kinase regulation of type III RTKs. This mechanism is likely to be shared with other kinases and might provide a basis for the design of a novel class of tyrosine kinase inhibitors.
Receptor tyrosine kinases (RTKs)2 are a large family of enzymes, many of which mediate vital cellular functions of living organisms. The fine-tuning of RTKs function is essential for their normal physiological roles and their aberrant function contributes to human diseases ranging from cancer to diabetes (1-4). RTKs consist of an extracellular region that serves a ligand binding site, a transmembrane domain, and a cytoplasmic region, which possesses intrinsic tyrosine kinase activity. In the inactive state, the activation loop is thought to occupy the active site preventing substrate access and ATP binding (5, 6). Ligand-mediated RTK activation leads to RTK dimerization. Dimerization is believed to facilitate transphosphorylation of one or two tyrosines within the activation loop. Based on the crystal structure of the insulin receptor (7), fibroblast growth factor receptor (8), it is proposed that activation loop tyrosine autophosphorylation removes the activation loop away from the active site and creates appropriate conformation for optimal substrate and ATP binding (6-8). The catalytic kinase domain of RTKs ranges from 250 to 300 amino acid residues and contains highly conserved amino acid sequences. The typical kinase domain of RTKs is folded into two main lobes, NH2- and COOH-terminal lobes. Catalysis occurs in a cleft between the two domains. Residues in the NH2-terminal lobe are mainly in β-sheets and are involved in ATP binding. Residues in the COOH-terminal lobe, however, are primarily in α-helical conformation and are important for catalysis and protein substrate binding (6, 9).
Vascular endothelial growth factor receptor-2 (VEGFR-2) is a type III RTK and its activation is critical for normal vasculogenesis, pathological angiogenesis, and neural development (10-14). Activation of VEGFR-2 stimulates a number of key signal transduction pathways such as the phosphoinositide 3-OH kinase, which is involved in endothelial cell survival and proliferation (phosphatidylinositol 3-kinase) (15, 16), phospholipase Cγ1, which stimulates endothelial cells tubulogenesis (17), Src kinases (18, 19), and Cbl-E3 ligase (20). VEGFR-2 also associates with a number of adaptor proteins, including VRAP (21) and Shb (22). The crystal structure analysis of VEGFR-2 has revealed that the COOH-terminal loop in the kinase domain folds into 7 α-helices (αD–αI) and two anti-parallel β-sheets (23). Although the key structural features of the kinase domain have been deduced from the crystallographic structures, the biochemical data corroborating those observations is largely unavailable. Only the roles of key motifs such as GXGXXG, HRDLA, DFG, and activation loop autophosphorylation sites are fairly well characterized (6). Furthermore, although activation loop tyrosine autophosphorylation is the most common mechanism used by RTKs for activation, there are a number of other mechanisms for regulation of kinase activation of protein kinases. For instance, the COOH-terminal Src kinase and endothelial growth factor receptor do not require activation loop tyrosine autophosphorylation. Instead, COOH-terminal Src kinase activity is regulated by its SH2 and SH3 domains (24), and epidermal growth factor receptor activity is regulated by intermolecular interactions between the kinase domain and its carboxyl tail (25).
We have recently observed that a large deletion from the carboxyl terminus of VEGFR-2, which also removes several amino acids from the helix-I (αI) region and prevents full tyrosine phosphorylation of VEGFR-2 (26). We hypothesized that helix-I may play a global role in RTK activation, substrate recognition, and phosphorylation. In this study we show that leucines 1158, 1161, and 1162 of helix-I via hydro-phobic interactions with αE, αF, and αH participate in maintaining an appropriate conformation of VEGFR-2 necessary for its tyrosine autophosphorylation and kinase activation. Analogous leucine residues in PDGFR-β and CSF-1R were also found important for their activation. We propose that a highly conserved leucine motif plays a critical role in which the activation loop segment is held into the active conformation in type III RTKs. Because, the leucine motif is conserved in both receptor tyrosine kinases and non-receptor tyrosine kinases we propose that this mechanism is likely to be shared with other kinases and might provide a basis for design of a novel class of tyrosine kinase inhibitors.
MATERIALS AND METHODS
Reagents and Antibodies
Recombinant mouse PDGF-BB and recombinant human macrophage colony stimulating factor (CSF-1) were purchased from BIOSOURCE International (Camarillo, CA) and R&D Systems (Minneapolis, MN), respectively. Mouse monoclonal anti-phosphotyrosine antibodies, 4G10 and PY-20, were purchased from Upstate Biotechnology (Lake Placid, NY) and Transduction Laboratories (Lexington, KY), respectively. Monoclonal mouse anti-VEGFR-2/FLK-1 was purchased from Chemicon International (Temecula, CA). The following antibodies were purchased from Santa Cruz Biotechnology, Inc.: pre-adsorbed goat anti-mouse IgG-horseradish peroxidase, pre-adsorbed goat anti-rabbit IgG-horseradish peroxidase, rabbit polyclonal IgG anti-c-Fms/CSF-1R(C-20), and rabbit polyclonal IgG anti-PDGFR-β (958). Rabbit polyclonal anti-VEGFR-2 sera were raised against either a glutathione S-transferase-VEGFR-2 kinase insert domain fusion protein (1410) or a glutathione S-transferase-VEGFR-2 carboxyl terminus fusion protein (1412) as described elsewhere (29). ATP and poly(Glu:Tyr) (4:1) were purchased from Sigma. Anti-phospho-VEGFR-2 (pY1054/pY1059) was purchased from Cell Signaling Technology (Beverly, MA). rProtein A-Sepharose Fast Flow was purchased from Amersham Biosciences.
Site-directed Mutagenesis
The cDNAs for mouse VEGFR-2, mouse PDGFR-β, and human CSF-1R were used as template to generate the mutant receptors. The creation of the VEGFR-2 chimera (CKR) in which its extracellular domain is replaced with that of human CSF-1R are described elsewhere (29). The mutations were made using the PCR-based site-directed mutagenesis method as previously described (17, 19). The reactions were carried out using Accuprime Pfx DNA Polymerase (Invitrogen). The resultant mutations were verified by sequencing and were subsequently cloned into pLXSN2 or pLNCX2 retroviral vectors by NotI and SalI sites.
In Vitro Kinase Assay and Substrate Phosphorylation
Equal numbers of PAE cells (porcine aortic endothelial cells) expressing CKR or mutant CKRs were serum-starved overnight. Cells without stimulation were lysed. Proteins were immunoprecipitated with anti-VEGFR-2 antibody. The immunoprecipitated proteins were washed once with cold lysis buffer, pH 7.4 (1% Triton X-100, 10 mm Tris-HCl, 5 mm EDTA, 50 mm NaCl, 50 mm NaF, 2 mm sodium orthovanadate, 1 μm leupeptin, 1 mm phenylmethylsulfonyl fluoride, and 20 μg/ml aprotinin), and three times with cold PAN buffer (10 mm PIPES, pH 7.0, 100 mm NaCl, 20 mg/ml aprotinin). In vitro kinase activity was performed by incubating immunoprecipitated proteins with 20 μl of kinase buffer (10 mm MgCl2, 1mm dithiothreitol, 100 mm NaCl, 20 mm Tris-HCl, pH 7.4) containing 0.1 to 1 mm ATP for 15 min at 30 °C. The reaction was stopped by the addition of an equal volume of SDS sample buffer. The samples were denatured and resolved on 7.5% SDS-PAGE and subjected to Western blot analysis using anti-phosphotyrosine antibody. To measure the ability of mutant VEGFR-2s to phosphorylate a substrate, poly(Glu) peptide was used as described (26). Briefly, cells were stimulated with ligand for 10 min, lysed, and immunoprecipitated with anti-VEGFR-2 antibody (1410 or 1412). Substrate phosphorylation was measured as described (27). Briefly, immunoprecipitated proteins were incubated in 10 μCi of [γ-32P]ATP for 15 min at 30 °C in the presence of substrate (5 μg/reaction). The reaction was stopped and samples were spotted on the p81 paper and after extensive washing, the p81 papers were subjected to scintillation counter and their DPM value were measured.
Immunoprecipitation and Western Blot Analysis
PAE cells expressing CKR or mutant CKRs were grown in sparse conditions in 10% fetal bovine serum, and serum starved overnight in Dulbecco's modified Eagle's medium. Cells were left either resting or stimulated with 40 ng/ml CSF-1 at 37 °C for appropriate times as indicated in figure legends. Cells were washed twice with H/S buffer (25 mm HEPES, pH 7.4, 150 mm NaCl, 2 mm Na3VO4) and lysed in lysis (EB) buffer (10 mm Tris-HCl, pH 7.4, 5 mm EDTA, 50 mm NaCl, 50 mm NaF, 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride, 2 mm Na3VO4, and 20 μg/ml aprotinin). VEGFR-2 proteins were immunoprecipitated with anti-VEGFR-2 antibody and immune complexes were bound to Protein A-Sepharose. Immunoprecipitates were resolved on a 7.5% SDS-PAGE gel, and the proteins were transferred to Immobilon membrane. For anti-phosphotyrosine Western blot analysis, the membranes were then incubated in Block buffer containing 10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10 mg/ml bovine serum albumin, 10 mg/ml ovalbumin, 0.05% Tween 20, 0.005% NaN3, and then incubated with primary antibody diluted in Block. The membranes were then washed and incubated with horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit antibodies. Finally, the membranes were washed, and developed with ECL (PerkinElmer Life Sciences). In some occasions, the membranes were stripped by incubating them in a buffer containing 6.25 mm Tris-HCl, pH 6.8, 2% SDS, and 100 mm β-mercaptoethanol in 50 °C for 30 min and re-probed.
Molecular Modeling
The three-dimensional model of VEGFR-2 was built using the homology modeling technique with the Mol-Mol program (28) based on the 2.4-Å resolution x-ray structure of human VEGFR-2. All the atom coordinates were from human VEGFR-2 (23). The side chain interactions were calculated by the Xtal contact of the CCP4 program.
RESULTS
Regulation of Ligand-dependent Tyrosine Phosphorylation of VEGFR-2 by Leucine Residues
The amino acid residues in the COOH-terminal loop of the kinase domain of VEGFR-2 folds into 2 β-sheets and 7 α-helices. The αI consists of 12 amino acid residues (1152–1166 in human VEGFR-2) (23). These residues in mouse VEGFR-2 correspond to residues 1150–1164 (Fig. 1). Deletion of the carboxyl terminus of VEGFR-2 from leucine 1158 abrogates the ligand-dependent autophosphorylation of VEGFR-2. However, deletion of the carboxyl terminus after residue 1211 preserves its ligand-dependent tyrosine autophosphorylation (26). This suggests that autophosphorylation of VEGFR-2 is regulated by the presence of 53 amino acid residues (1158–1211) within this region. To address the mechanism involved in tyrosine autophosphorylation of VEGFR-2, we have created a panel of mutant and truncated mouse VEGFR-2s (Fig. 1) and expressed them in PAE cells. To avoid cross-talk with endogenous VEGF receptors we have utilized a chimeric VEGFR-2 system in which the extracellular domain of VEGFR-2 is replaced with that of human CSF-1R, herein called CKR (29). As shown in Fig. 2A, deletion of the carboxyl terminus of VEGFR-2 from amino acid residue 1172 (ΔCKR/194) preserves ligand-dependent tyrosine autophosphorylation as judged by its ability to undergo tyrosine phosphorylation, using a general anti-phosphotyrosine antibody and a specific anti-phospho-1052/1057 VEGFR-2 antibody (Fig. 2B). Tyrosines 1052 and 1057 are activation loop tyrosines and are involved in VEGFR-2 activation (23, 30, 31). To test which amino acid among the 14 amino acid residues is involved in regulation of autophosphorylation of VEGFR-2, we initially mutated Asn-1160, Gln-1163, Asn-1165, Gln-1167, and Gln-1168 to alanine (A). This mutant receptor herein is referred to as 5A/CKR. The mutant receptor was expressed in PAE cells and its ability to become autophosphorylated in response to ligand stimulation was measured. Fig. 2, D and E, shows that 5A/CKR is able to undergo tyrosine autophosphorylation in response to ligand stimulation, suggesting that amino acids including, Asn-1160, Gln-1163, Asn-1165, Gln-1167, and Gln-1168 are not involved in the regulation of ligand-dependent tyrosine phosphorylation of VEGFR-2.
FIGURE 1.
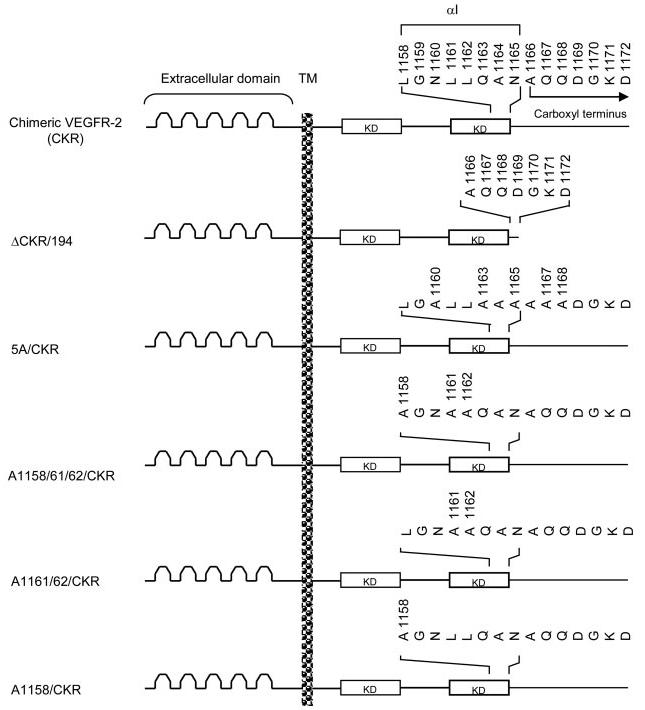
Schematic presentation of truncated and leucine mutant VEGFR-2s. 15 amino acid residues within the boundary of the kinase domain and carboxyl terminus of mouse VEGFR-2 is shown. Based on the crystal structure, Asn-1165 possibly corresponds to the last residue of the kinase domain. Amino acids (1158–1165) are folded in the α-helix conformation and correspond to helix I (23). Alanine 1166 (A1166) corresponds to the first residue of the carboxyl terminus of VEGFR-2. The ΔCKR/194 construct was created by deleting 194 amino acids from the carboxyl-terminal of mouse VEGFR-2 retaining only seven amino acids in the carboxyl-terminal (amino acids 1166–1172 in mouse VEGFR-2). The 5A/CKR construct was created by mutating Asn-1160, Gln-1163, Asn-1165, Gln-1167, and Gln-1168 to Ala. The A1158/1161/1162/CKR construct was created by mutating Leu-1158, Leu-1161, and Leu-1162 to Ala. The A1161/1162/CKR construct was created by mutating Leu-1161 and Leu-1162 to Ala. The A1158/CKR construct was made by mutating Leu-1158 to Ala. The extracellular domain, the transmembrane domain (TM), and the kinase domain (KD) also are shown.
FIGURE 2.
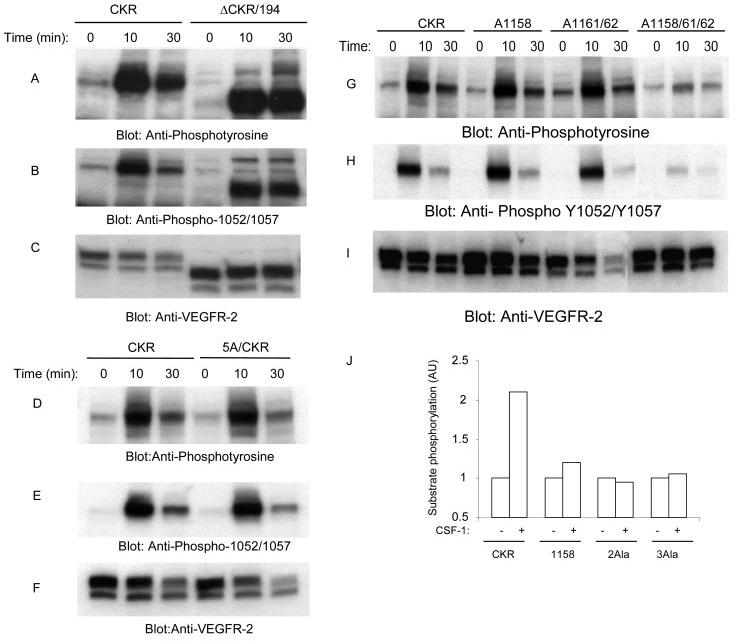
Effect of mutation of leucines 1158, 1161, and 1162 to ligand-dependent tyrosine autophosphorylation of VEGFR-2 and its ability to phosphorylate exogenous substrate. An equal number of serum-starved PAE cells expressing wild type chimeric VEGFR-2 (CKR), carboxyl-terminal deleted, ΔCKR/194, and 5A/CKR were either not stimulated or stimulated for 10 or 30 min with CSF-1 (40 ng/ml). Cells were lysed and total cell lysates were subjected to Western blot analysis using anti-phosphotyrosine antibody (A and D) or a phospho-specific 1052/1057 VEGFR-2 antibody (B and E). The same cell lysates were subjected to Western blot analysis using anti-VEGFR-2 for protein loading as a control (C and F). In a similar manner, an equal number of serum-starved PAE cells expressing wild type chimeric VEGFR-2 (CKR), A1158/CKR, A1161/1162/CKR, and A1158/1161/1162 were either not stimulated or stimulated for 10 or 30 min with CSF-1. Cells were lysed and total cell lysates were subjected to Western blot analysis using anti-phosphotyrosine antibody (G) or a phospho-specific 1052/1057 VEGFR-2 antibody (H). The same cell lysates were subjected to Western blot analysis using anti-VEGFR-2 for protein loading as a control (I). Serum-starved PAE cells expressing wild type chimeric VEGFR-2 (CKR), A1158/CKR (1158), A1161/1162/CKR (2Ala), and A1158/1161/1162/CKR (3Ala) were either not stimulated or stimulated for 10 min with CSF-1. Cells were lysed, and proteins were immunoprecipitated with anti-VEGFR-2 antibody. The immunoprecipitated proteins were extensively washed and subjected to an in vitro kinase assay as described under “Materials and Methods,” and phosphorylation of exogenous substrate was measured (J).
To test whether leucine residues 1158, 1161, and 1162 are associated with the regulation of tyrosine autophosphorylation of VEGFR-2, we generated three additional constructs: including a single leucine 1158 mutation (A1158/CKR), a double mutation leucine 1161 and leucine 1162 (A1162/62/CKR), and a triple leucine mutation leucine 1158, leucine 1161, leucine 1162 (A1158/61/62/CKR) and expressed them in PAE cells. Replacement of single leucine 1158 to alanine and replacement of two leucine residues 1161 and 1162 to alanine appeared to have no effect on tyrosine phosphorylation of CKR (Fig. 2D). However, a triple mutation of leucines 1158, 1161, and 1162 significantly reduced the ability of VEGFR-2 to undergo ligand-dependent tyrosine autophosphorylation. These mutations also substantially reduced the ligand-dependent phosphorylation of activation loop tyrosines 1052 and 1057 (Fig. 2, G and H). This suggests that the presence of three leucine residues collectively contribute to ligand-dependent activation loop tyrosine autophosphorylation of VEGFR-2. To test the ability of leucine mutant VEGFR2s to phosphorylate an exogenous substrate, we assessed their capacity to phosphorylate a poly(Glu) peptide in vitro. Fig. 2J shows that the wild type CKR is able to phosphorylate exogenous substrate in a ligand-dependent manner. However, the ability of leucine mutant VEGFR-2s, including A1158/CKR, A1161/1162/CKR, and A1158/1161/1162/CKR, to phosphorylate the poly(Glu) peptide were severely compromised. This suggests that the presence of leucines 1158, 1161, and 1162 are critical for substrate recognition, phosphotransferase capability, or both of these functions by VEGFR-2.
Leucines 1158, 1161, and 1162 Differentially Contribute to Tyrosine Phosphorylation and Kinase Activation of VEGFR-2
To understand better how the lack of leucines 1158, 1161, and 1162 contribute to tyrosine phosphorylation of VEGFR-2, we tested the ability of wild type and leucine mutant VEGFR-2s to undergo autophosphorylation in the in vitro kinase assay using varying concentrations of ATP. For this purpose, PAE cells expressing wild type and mutant VEGFR-2s were serum-starved and lysed without ligand stimulation. Cell lysates were immunoprecipitated with an anti-VEGFR-2 antibody and subjected to an in vitro kinase assay using different concentrations of ATP as indicated. The ability of these receptors to become autophosphorylated in response to ATP was measured by immunoblotting with an anti-phosphotyrosine antibody. As shown in Fig. 3A, ATP promoted a dose-dependent autophosphorylation of wild type VEGFR-2. The triple leucine mutant receptor, A1158/1161/1162/CKR, however, displayed a highly compromised tyrosine autophosphorylation in response to ATP stimulation (Fig. 3A), suggesting that these residues contribute to both ligand- (Fig. 2G) and ATP-dependent tyrosine phosphorylation of VEGFR-2. ATP-dependent autophosphorylation of the double leucine mutant VEGFR-2 (A1161/1162/CKR) was partially reduced compared with that of the wild type receptor (Fig. 3C). Furthermore, A1158/CKR, which displayed almost normal tyrosine autophosphorylation in response to ligand stimulation (Fig. 2G), also failed to undergo ATP-induced tyrosine phosphorylation (Fig. 3C). In contrast, ATP-induced tyrosine phosphorylation of 5A/CKR, where five residues including Asn-1160, Gln-1163, Asn-1165, Gln-1167, and Gln-1168, were replaced with alanine showed no apparent decrease in tyrosine phosphorylation (Fig. 3E). These findings demonstrate that leucine 1158 plays a critical role in kinase activation of VEGFR-2. Leucines 1161 and 1162 appear to play less critical but a more important role in kinase activation of VEGFR-2. Altogether, the data demonstrate that the presence of leucines 1158, 1161, and 1162 are critically important for kinase activation and for its ability to recognize and phosphorylate substrate.
FIGURE 3.
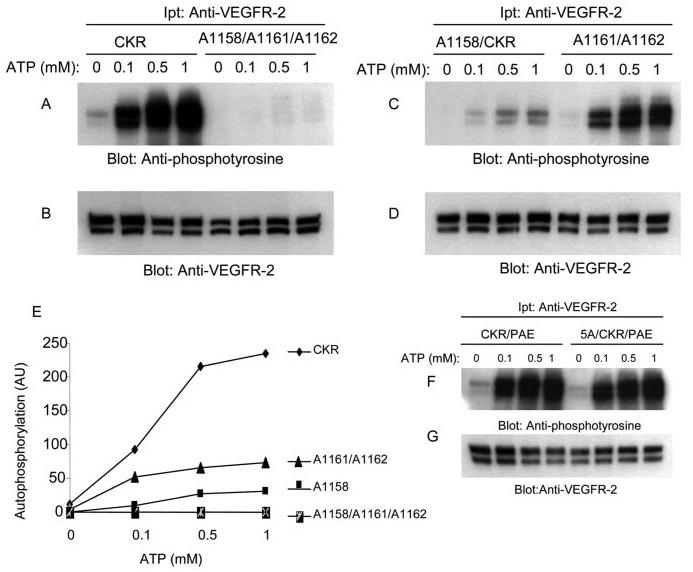
Effect of mutation of leucines 1158, 1161, and 1162 on ATP-induced tyrosine autophosphorylation of VEGFR-2. An equal number of serum-starved PAE cells expressing wild type chimeric VEGFR-2 (CKR), A1158/1161/1162, A1158/CKR, and A1161/1162/CKR were lysed without stimulation. CKR proteins were immunoprecipitated (Ipt) with anti-VEGFR-2 antibody and divided into two groups. One group was subjected to the in vitro kinase assay using ATP (A, C, and E), the second group was subjected to Western blot analysis using anti-VEGFR-2 antibody as a control for protein levels (B, D, and F). In vitro kinase assay was performed as described under “Materials and Methods.” Briefly, the immunoprecipitated proteins were incubated with different concentrations of ATP. After a 15-min incubation, the reaction was terminated by adding 2× sample buffer. The samples were boiled and subjected to Western blot analysis using anti-phosphotyrosine antibody (A, C, and E).
Trans-phosphorylation of Leucine Mutant 1158 VEGFR-2 by the Wild Type VEGFR-2
Because leucine 1158 mutation to alanine compromised the ability of VEGFR-2 to undergo tyrosine phosphorylation in vitro kinase assay, whether the lack of this residue prevents VEGFR-2 to undergo trans-phosphorylation was tested. For this purpose we tested whether A1158/CKR could be tyrosine phosphorylated by wild type VEGFR-2. For this purpose we immunoprecipitated A1158/CKR from serum-starved and non-stimulated PAE cells and incubated with PAE cell lysates expressing no CKR or expressing CKR and stimulated the cells with ligand. The result showed that A1158/CKR could be phosphorylated by the wild type VEGFR-2 (Fig. 4A). Cell lysates from PAE cells expressing no CKR did not phosphorylate A1158/CKR (Fig. 4A). Altogether, in vivo tyrosine phosphorylation of A1158/CKR (Fig. 2G) coupled with the data presented in this figure suggests that leucine 1158 selectively participates in phosphotransferase activity of VEGFR-2, perhaps by interacting with the nucleotide binding loop or activation loop segment of VEGFR-2.
FIGURE 4.
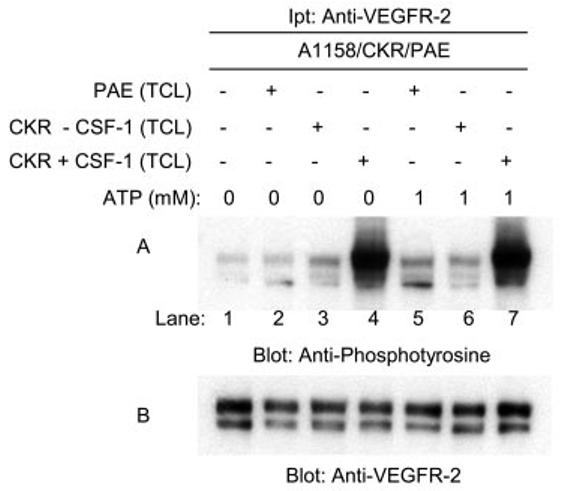
Tyrosine phosphorylation of A1158/CKR by wild type CKR. Serum-starved PAE cells expressing A1158/CKR were lysed without stimulation with ligand, and proteins were immunoprecipitated (Ipt) with anti-VEGFR-2 antibody and the immunoprecipitated 1158/CKR proteins were divided into seven groups (lanes 1–7). Lane 1 was left untreated, lane 2 was incubated with serum-starved PAE cell lysates for 15 min, lane 3 was incubated with lysates from PAE cells expressing CKR without stimulation with CSF-1, lane 4 was incubated with PAE cells expressing CKR stimulated with CSF-1 for 10 min. Lanes 5–7 were preincubated with ATP (1 mm) for 15 min, then incubated either with PAE cell lysates (lane 5), cell lysates from PAE cells expressing CKR (lane 6), or cell lysates from PAE cells expressing CKR stimulated with CSF-1 (lane 7). Reactions were stopped by adding 2× sample buffer, boiled for 5 min, and subjected to Western blot analysis using anti-phosphotyrosine antibody (A). The same membrane was striped and re-blotted with anti-VEGFR-2 antibody for protein level (B).
Mutation of Leucines 1158, 1161, and 1162 Do Not Alter the Ligand-mediated Dimerization of VEGFR-2
Because A1158/CKR failed to undergo kinase activation and its ability to phosphorylate an exogenous substrate was severely compromised, we tested whether this mutation alters the capacity of VEGFR-2 to undergo ligand-induced dimerization. Dimerization is a common mechanism by which ligand-induced activation of RTKs is achieved. Point mutations in the kinase domain of RTKs have been shown to lead to oncogenic activation through receptor dimerization (1). To test whether leucine mutations alters the ability of the VEGFR-2 to undergo dimerization, we analyzed the ligand-stimulated dimerization of the wild type receptor and the leucine mutant receptors. As shown in Fig. 5, VEGFR-2 undergoes ligand-dependent covalent disulfide-linked dimerization. Similarly, A1158/CKR and the triple leucine mutant VEGFR-2 (leucines 1153, 1161, and 1162) were able to undergo ligand-induced disulfide-linked dimerization. Altogether, the data suggest that VEGFR-2 undergoes ligand-stimulated covalent disulfide-linked dimerization and although leucine residues impair the ligand-induced tyrosine autophosphorylation, they have no effect on the ligand-induced dimerization of VEGFR-2.
FIGURE 5.
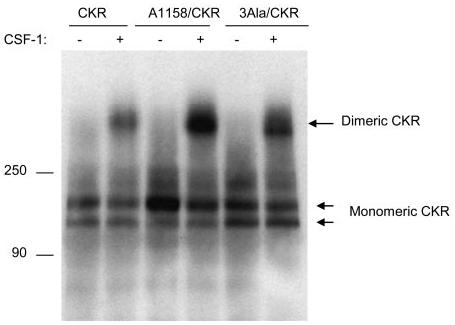
Effect of mutation of leucines 1158, 1161, and 1162 on ligand-induced dimerization of VEGFR-2. An equal number of serum-starved PAE cells expressing wild type chimeric VEGFR-2 (CKR), A1158/CKR, and A1158/1161/1162 (3Ala/CKR) were either not stimulated or stimulated for 10 min with CSF-1 (40 ng/ml). Cells were lysed and total cell lysates were immunoprecipitated with anti-VEGFR-2 antibody. The immunoprecipitated proteins were resolved in a 4–15% gradient non-reducing SDS-PAGE and proteins were transferred to polyvinylidene difluoride membrane and finally subjected to Western blot analysis using an anti-VEGFR-2 antibody. The positions of monomeric and dimeric VEGFR-2s are shown. It is also evident that expression of the mutant VEGFR-2s are slightly higher than the wild type VEGFR-2. This may explain why there is more dimerized mutant of VEGFR-2s compared with that of the wild type VEGFR-2.
Leucines 1158, 1161, and 1162 Are Highly Conserved in Type III RTKs
Amino acid sequence alignment of VEGFR-2 with other type III RTKs as shown in Fig. 6 revealed that leucines, 1158, 1161, and 1162 are highly conserved among these RTKs. Leucine 1158, in particular is conserved in all type III RTKs with the exception of PDGFR-α. Interestingly, PDGFR-α is considered to be a weaker kinase compared with the highly related receptor, PDGFR-β (32). In addition, leucine 1158 is also conserved in non-receptor tyrosine kinases, like Src family kinases (33). The presence of this highly conserved residue among these RTKs is highly suggestive that this residue may play a similar role in the activation of other RTKs. Leucine 1158 of VEGFR-2, in PDGFR-β and CSF-1R corresponds to 957 and 910, respectively. To test whether the corresponding leucine residue in PDGFR-β and CSF-1R are also involved in the regulation of their tyrosine phosphorylation, we mutated leucine 957 in PDGFR-β and leucine 910 in CSF-1R to alanine and expressed them in PAE cells. The wild type receptors and leucine mutant receptors were analyzed for their capacity to undergo tyrosine phosphorylation.
FIGURE 6.

Structure-based amino acid sequence alignment of the kinase domain of VEGFR-2 with the other type III RTKs. The secondary structure of VEGFR-2 indicates that the COOH-terminal lobe of the kinase domain is folded into 7 α-helices and 2 β-sheets (23). Residues from 1150 to 1164 constitute αI. The αI is the last helix and it separates the kinase domain from the carboxyl terminus of VEGFR-2. The position of activation loop tyrosine autophosphorylation sites Tyr-1052 and Tyr-1057 are also shown. Partial sequence alignment of the kinase domain and activation loop regions of human VEGFR-1 and various receptor tyrosine kinases of type III family are shown.
Leucine 957 Regulates Both Ligand and ATP-stimulated Tyrosine Phosphorylation of PDGFR-β
To test the contribution of conserved leucine 957 to tyrosine phosphorylation of PDGFR-β, PAE cells individually expressing PDGFR-β and Ala-957/PDGFR-β were stimulated with PDGF-BB and analyzed for their ability to undergo ligand-dependent tyrosine phosphorylation. As shown in Fig. 7A ligand stimulation of the wild type PDGFR-β resulted in robust tyrosine phosphorylation, Ala-957/PDGFR-β, however, failed to undergo tyrosine phosphorylation. This observation was rather unexpected, because similar mutation in VEGFR-2 impaired only ATP-dependent but not ligand-dependent tyrosine phosphorylation (Figs. 2G and 3C). It also appears that the ability of mutant PDGFR-β to be fully processed and localized in the cell membrane is also altered as seen by the accumulation of the premature form of the Ala-957/PDGFR-β (Fig. 7B). To test whether the leucine 957 mutant can be tyrosine phosphorylated in response to ATP treatment, we immunoprecipitated PDGFR-β and Ala-957/PDGFR-β from serum-starved and non-stimulated PAE cells and subjected them to in vitro kinase assay using different concentrations of ATP as described under “Materials and Methods.” The data shows that the ability of A957/PDGFR-β to become tyrosine phosphorylated in response to ATP is severely compromised (Fig. 7C). Collectively, the data suggest that in PDGFR-β, unlike VEGFR-2, the conserved leucine (1158 in VEGFR-2, 957 in PDGFR-β) is required for both ligand and ATP-stimulated tyrosine phosphorylation of PDGFR-β.
FIGURE 7.
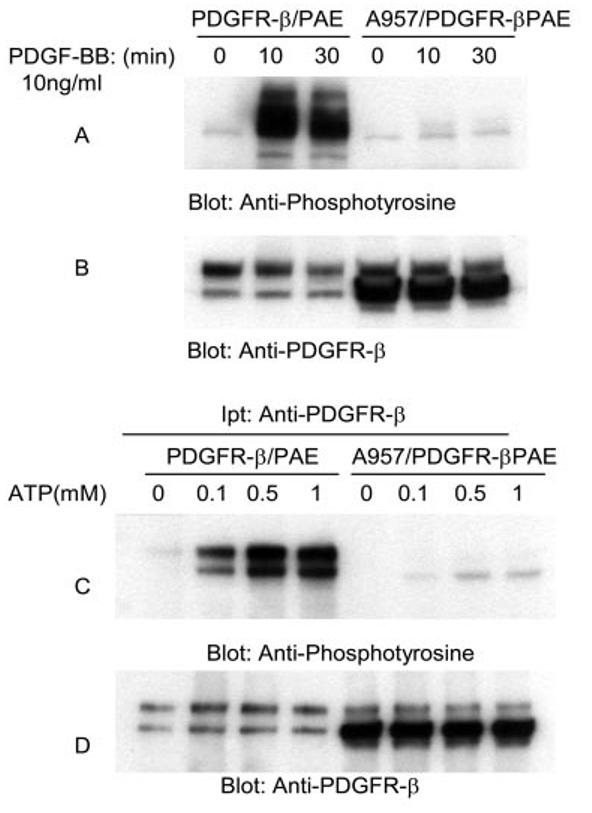
Effect of mutation of leucine 957 on tyrosine phosphorylation of PDGFR-β. Equal numbers of serum-starved PAE cells expressing wild type PDGFR-β and A957/PDGFR-β were either not stimulated (0) or stimulated for 10 or 30 min with PDGF-BB (10 ng/ml). Cells were lysed and total cell lysates were subjected to Western blot analysis using anti-phosphotyrosine antibody (A). The same cell lysates were subjected to Western blot analysis using anti-PDGFR-β antibody for protein loading as a control (B). An equal number of serum-starved PAE cells expressing wild type PDGFR-β and A957/PDGFR-β were lysed without ligand stimulation. CSF-1R proteins were immunoprecipitated (Ipt) with anti-PDGFR-β antibody and divided into two groups. One group was subjected to in vitro kinase assay using ATP (C), the second group was subjected to Western blot analysis using anti-PDGFR-β antibody as a control for protein levels (D). In vitro kinase assay was performed as described in the legend to Fig. 3.
Leucine 910 Regulates Both Ligand and ATP-stimulated Tyrosine Phosphorylation of CSF-1R
In addition to VEGFR-2 and PDGFR-β, we wished to evaluate mutation of conserved leucine to alanine in another type III RTK. To this end, we mutated leucine 910 on human CSF-1R/c-fms to alanine and assessed its impact in tyrosine phosphorylation of CSF-1R. Fig. 8A shows that like PDGFR-β, the ligand-dependent tyrosine phosphorylation of Ala-910/CSF-1R is markedly inhibited. In addition, we evaluated ATP-stimulated tyrosine phosphorylation of CSF-1R. As shown in Fig. 8B, ATP-stimulated tyrosine phosphorylation of Ala-910/CSF-1R is almost totally abolished. In summary, data indicate that the conserved leucine residue in question plays a major role in the activation of RTKs. It also appears that this highly conserved residue plays a differential role in RTKs activation. In VEGFR-2 it alters the ATP-stimulated tyrosine phosphorylation and phosphotransferase activity toward exogenous substrate. In PDGFR and CSF-1R, mutation of this residue impairs tyrosine phosphorylation in response to both ATP and ligand stimulation.
FIGURE 8.
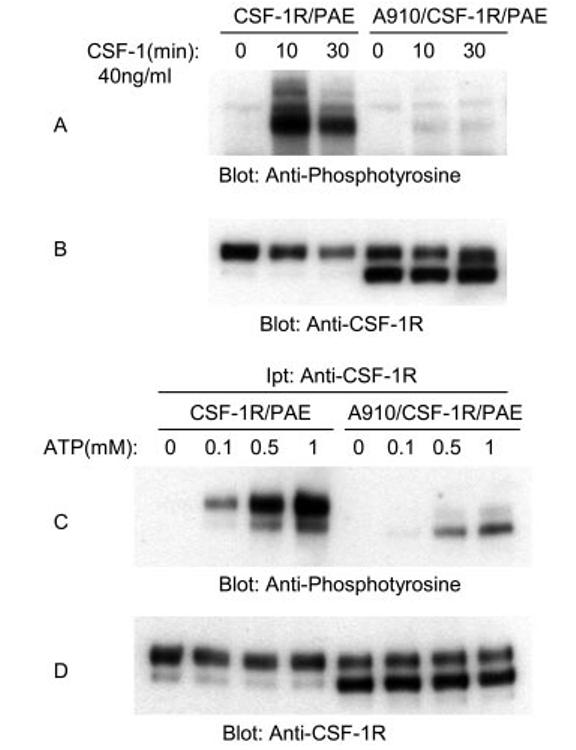
Effect of mutation of leucine 910 on tyrosine phosphorylation of CSF-1R. An equal number of serum-starved PAE cells expressing wild type CSF-1R and A910/CSF-1R were either not stimulated (0) or stimulated for 10 or 30 min with CSF-1 (40 ng/ml). Cells were lysed and total cell lysates were subjected to Western blot analysis using anti-phosphotyrosine antibody (A). The same cell lysates were subjected to Western blot analysis using anti-CSF-1R for protein loading as a control (B). An equal number of serum-starved PAE cells expressing wild type CSF-1R and A910/CSF-1R were lysed without ligand stimulation. CSF-1R proteins were immunoprecipitated (Ipt) with anti-CSF-1R antibody and divided into two groups. One group was subjected to in vitro kinase assay using ATP (C), the second group was subjected to Western blot analysis using anti-CSF-1R antibody as a control for protein levels (D). In vitro kinase assay was performed as described in the legend to Fig. 3.
Leucines 1158, 1161, and 1162 Are Involved in Helix-Helix Interactions
To understand the role of these leucine residues in VEGFR-2 activation, a three-dimensional model of VEGFR-2 was generated based on the x-ray crystal structure of human VEGFR-2 (23). Alignment of human and mouse VEGFR-2 indicated that the kinase domain is highly conserved and the region of leucine 1158 is invariable between the two species. The model (Fig. 9A) shows that leucines, 1158, 1161, and 1162 are part of the α-helical (helix I) conformation and are buried in the VEGFR-2 structure. Leucines 1158, 1161, and 1162 make hydrophobic interactions with a number of residues and are involved in helix-helix interactions, including αE, αF, and αH (Fig. 9B). Whether these leucine residues also interact with the activation and the catalytic loop is not clear, because in the original crystal structure the activation loop of VEGFR-2 is highly disordered (20). Leucines 1158 and 1161 also make hydrophobic interactions with residues of the β-sheet (β7), which separates the catalytic loop from the activation loop. Leucine 1162 mainly associates with the residues of αE, whereas leucine 1161 mostly associates with the residues of αH. Among all the three leucines, leucine 1158 has the most contacts. It associates with the residues of αE, αF, αH, and β-sheet (β7). This might explain why mutation of this site in VEGFR-2, CSF-1R, and PDGFR-β interferes with autophosphorylation of these receptors. Furthermore, it suggests that mutation of leucine 1158 and analogous sites in CSF-1R and PDGFR-β disrupts hydrophobic interactions between leucine 1158 and amino acid residues of αE, αF, αH, and β7.
FIGURE 9.
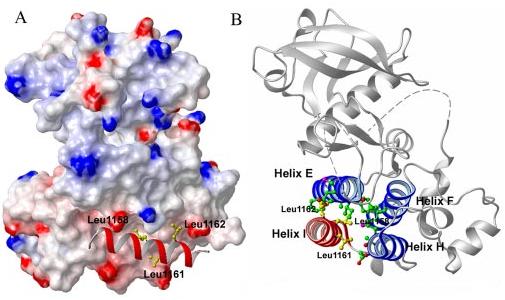
Leucines 1158, 1161, and 1162 are involved in helix-helix interactions. The surface representation of αI of VEGFR-2 is shown. The αI is shown as a red ribbon and the side chains of leucines 1158, 1161, 1162 are shown in yellow (A). The leucine residues are all buried in the structure of VEGFR-2 and are involved in the helix-helix packing via hydrophobic interactions. View of ribbon diagram of VEGFR-2 is shown (B). Helix I (αI) is shown in red, helix-E (αE), helix-F (αF), and helix-H (αH) are shown in blue. The side chains of leucines 1158, 1161, and 1162 are shown in green and the side chain of residues of αE, αF, and αH are shown in yellow. Leucine (Leu1158) associates with amino acid residues of αE, αF, αH, and β7. Leucine (Leu1161) associates largely with αE. Leucine (Leu1162) associates with αE and β7. The figure was drawn using the Mol-Mol program (28) based on the 2.4-Å resolution x-ray structure of human VEGFR-2 (23), but the residue numberings were changed to those of the mouse sequence. The side chain interactions were calculated by XTAL-CONTACT of the CCP4 program.
DISCUSSION
In the present study, we have focused on the molecular mechanisms of VEGFR-2 activation. Our study demonstrates that the highly conserved leucine residues of αI, the last helix of the kinase domain of VEGFR-2, are involved in helix-helix packing and play a critical role in VEGFR-2 activation. Regulation of VEGFR-2 activation plays a key role in the induction of normal and pathological angiogenesis as well as neural development (10-13, 34). Despite its well known physiological importance, the molecular mechanism that governs VEGFR-2 activation is not fully understood. VEGFR-2 is a type III RTK and it is well recognized that activation of RTKs are regulated by tyrosine phosphorylation of activation loop tyrosine sites (7), but it is less clear precisely how tyrosine phosphorylation of the activation loop is regulated. In particular, the influence of the other subdomains on tyrosine phosphorylation and activation of RTKs is not known. Our study demonstrates that leucines 1158, 1161, and 1162 contribute to tyrosine autophosphorylation and kinase activation of VEGFR-2. The lack of leucine 1158 alone impaired the kinase activation of VEGFR-2 as defined by its ability to undergo tyrosine autophosphorylation in an in vitro kinase assay and to phosphorylate exogenous substrate. In vivo ligand-induced tyrosine phosphorylation of VEGFR-2 including phosphorylation of activation loop tyrosines 1052 and 1057, however, were not impaired by the lack of leucine 1158.
The lack of tyrosines 1161 and 1162 also appeared not to impair in vivo ligand-induced tyrosine phosphorylation, but their absence partially impaired kinase activation and diminished phosphorylation of exogenous substrate by VEGFR-2. The data clearly indicate that these leucines participate in maintaining the active conformation of RTKs. The observation that assay conditions (e.g. in vivo versus in vitro) influences tyrosine phosphorylation of these leucine mutant VEGFR-2s, although surprising, may suggest that the lack of these leucine residues predispose RTK to a less kinase active conformation and perhaps the leucine mutant VEGFR-2 proteins are partially misfolded. The analogous leucine sites in PDGFR-β (leucine 957) and CSF-1R (leucine 910) also play a critical role in the tyrosine phosphorylation and activation of these RTKs. The crystal structure of PDGFR-β and CSF-1R are not currently available, the crystal structure of human VEGFR-2 has been resolved (23). It appears that leucines, 1158, 1161, and 1162 are part of the α-helical (helix I) conformation and are buried in the VEGFR-2 structure. Leucines 1158, 1161, and 1162 make hydrophobic interactions with a number of residues and are involved in helix-helix interactions. The interdependence of tyrosine phosphorylation of RTKs and leucines 1158 suggest that leucine 1158 contributes to the stability of the active conformation of the kinase domain and therefore plays a critical role in which the active conformation is held and mutation of this residue may destabilize the active conformation. A number of point mutations in the kinase domain of RTKs including fibroblast growth factor receptor 3, MET, and RET have been identified in which they give rise to gain-of-function in these RTKs (1). The mechanism by which leucine mutations inactivates RTKs is novel, as it appears to abrogate both RTK kinase activation and substrate phosphorylation.
Although in this study we have only evaluated the role of leucine 1158 in VEGFR-2, and the analogous site in PDGFR-β and CSF-1R activation, because this residue is highly conserved in several key tyrosine kinase superfamily including, insulin receptor (35), Src family kinases (36), fibroblast growth factor receptor family, and HGFR family (37), we envision this residue to play a similar role in the tyrosine phosphorylation of these kinases. Understanding the precise mechanisms involved in regulating enzymatic activity of RTKs is important, as it may provide important insights that may facilitate the design of a new class of pharmacological agents to inhibit RTKs activation. In light of the importance of VEGFR-2 activation in normal and pathological angiogenesis, VEGFR-2 provides an attractive target. Understanding the regulation of VEGFR-2 enzymatic activity and the mechanism by which it signals is of great interest not only for the development of new and more effective antagonists, but also because this understanding should provide clues into mechanisms of regulation of angiogenesis.
Footnotes
This work was supported in part by National Institutes of Health Grants EY0137061 and EY012997 (to N. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: RTK, receptor tyrosine kinase; VEGFR-2, vascular endothelial growth factor receptor-2; SH, Src homology domain; CSF, colony stimulating factor; PAE, porcine aortic endothelial; PIPES, 1,4-piperazinediethanesulfonic acid.
REFERENCES
- 1.Robertson SC, Tynan J, Donoghue DJ. Trends Genet. 2000;16:265–271. doi: 10.1016/s0168-9525(00)02021-7. [DOI] [PubMed] [Google Scholar]
- 2.Zwick E, Bange J, Ullrich A. Endocr.-Relat. Cancer. 2001;8:161–173. doi: 10.1677/erc.0.0080161. [DOI] [PubMed] [Google Scholar]
- 3.Blume-Jensen P, Hunter T. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 4.Dibb NJ, Dilworth SM, Mol CD. Nat. Rev. Cancer. 2004;4:718–727. doi: 10.1038/nrc1434. [DOI] [PubMed] [Google Scholar]
- 5.Huse M, Kuriyan J. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 6.Nolen B, Taylor S, Ghosh G. Mol. Cell. 2004;10:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 7.Hubbard SR. EMBO J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohammadi M, Schlessinger J, Hubbard SR. Cell. 1996;86:577–587. doi: 10.1016/s0092-8674(00)80131-2. [DOI] [PubMed] [Google Scholar]
- 9.Hubbard SR, Till JH. Annu. Rev. Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 10.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 11.Carmeliet P. Nat. Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 12.Carmeliet P. Nat. Rev. Genet. 2003;4:710–720. doi: 10.1038/nrg1158. [DOI] [PubMed] [Google Scholar]
- 13.Cleaver O, Melton DA. Nat. Med. 2003;9:661–668. doi: 10.1038/nm0603-661. [DOI] [PubMed] [Google Scholar]
- 14.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 15.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. J. Biol. Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 16.Dayanir V, Meyer RD, Lashkari K, Rahimi N. J. Biol. Chem. 2001;276:17686–17692. doi: 10.1074/jbc.M009128200. [DOI] [PubMed] [Google Scholar]
- 17.Meyer RD, Latz C, Rahimi N. J. Biol. Chem. 2003;278:16347–16355. doi: 10.1074/jbc.M300259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Mol. Cell. 1999;4:915–924. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 19.Meyer RD, Dayanir V, Majnoun F, Rahimi N. J. Biol. Chem. 2002;277:27081–27087. doi: 10.1074/jbc.M110544200. [DOI] [PubMed] [Google Scholar]
- 20.Singh AJ, Meyer RD, Band H, Rahimi N. Mol. Biol. Cell. 2005;16:2106–2118. doi: 10.1091/mbc.E04-08-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumoto T, Bohman S, Dixelius J, Berge T, Dimberg A, Magnusson P, Wang L, Wikner C, Qi JH, Wernstedt C, Wu J, Bruheim S, Mugishima H, Mukhopadhyay D, Spurkland A, Claesson-Welsh L. EMBO J. 2005;24:2342–2353. doi: 10.1038/sj.emboj.7600709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmqvist K, Cross MJ, Rolny C, Hagerkvist R, Rahimi N, Matsumoto T, Claesson-Welsh L, Welshm M. J. Biol. Chem. 2004;279:22267–22275. doi: 10.1074/jbc.M312729200. [DOI] [PubMed] [Google Scholar]
- 23.McTigue MA, Wickersham JA, Pinko C, Showalter RE, Parast CV, Tempczyk-Russell A, Gehring MR, Mroczkowski B, Kan CC, Villafranca JE, Appelt K. Structure Fold Des. 1999;7:319–330. doi: 10.1016/s0969-2126(99)80042-2. [DOI] [PubMed] [Google Scholar]
- 24.Ogawa A, Takayama Y, Sakai H, Chong KT, Takeuchi S, Nakagawa A, Nada S, Okada M, Tsukihara T. J. Biol. Chem. 2002;277:14351–14354. doi: 10.1074/jbc.C200086200. [DOI] [PubMed] [Google Scholar]
- 25.Stamos J, Sliwkowski MX, Eigenbrot C. J. Biol. Chem. 2002;277:46265–46272. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 26.Meyer RD, Singh AJ, Rahimi N. J. Biol. Chem. 2004;279:735–742. doi: 10.1074/jbc.M305575200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rahimi N, Hung W, Tremblay E, Saulnier R, Elliot B. J. Biol. Chem. 1998;273:33714–33721. doi: 10.1074/jbc.273.50.33714. [DOI] [PubMed] [Google Scholar]
- 28.Koradi R, Billeter M, Wuthrich K. J. Mol. Graph. 1996;14:29–32. doi: 10.1016/0263-7855(96)00009-4. 51–55. [DOI] [PubMed] [Google Scholar]
- 29.Rahimi N, Dayanir V, Lashkari K. J. Biol. Chem. 2000;275:16986–16992. doi: 10.1074/jbc.M000528200. [DOI] [PubMed] [Google Scholar]
- 30.Dougher M, Terman BI. Oncogene. 1999;18:1619–1627. doi: 10.1038/sj.onc.1202478. [DOI] [PubMed] [Google Scholar]
- 31.Kendall RL, Rutledge RZ, Mao X, Tebben AJ, Hungate RW, Thomas KA. J. Biol. Chem. 1999;274:6453–6460. doi: 10.1074/jbc.274.10.6453. [DOI] [PubMed] [Google Scholar]
- 32.Rosenkranz S, Kazlauskas A. Growth Factors. 1999;16:201–216. doi: 10.3109/08977199909002130. [DOI] [PubMed] [Google Scholar]
- 33.Hanks SK, Quinn AM, Hunter T. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 34.Jain RK. Nat. Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 35.Hubbard SR, Wei L, Ellis L, Hendrickson WA. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 36.Xu W, Harrison SC, Eck MJ. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- 37.Hanks SK, Quinn AM. Methods Enzymol. 1991;200:38–62. doi: 10.1016/0076-6879(91)00126-h. [DOI] [PubMed] [Google Scholar]