

Abstract
Chromatin remodeling is required for efficient transcription of eukaryotic genes. In a genetic selection for budding yeast mutants that were defective in induction of the phosphate-responsive PHO5 gene, we identified mutations in ARG82/IPK2, which encodes a nuclear inositol polyphosphate kinase. In arg82 mutant strains, remodeling of PHO5 promoter chromatin is impaired, and the adenosine triphosphate—dependent chromatin-remodeling complexes SWI/SNF and INO80 are not efficiently recruited to phosphate-responsive promoters. These results suggest a role for the small molecule inositol polyphosphate in the regulation of chromatin remodeling and transcription.
DNA in the eukaryotic nucleus is packaged into chromatin, which forms a repressive structure that tends to limit the access of DNA-binding proteins to DNA. Cellular activities have been identified that function to counteract chromatin-mediated repression through acetylation, methylation, or phosphorylation of histones (1). Additionally, complexes such as SWI/SNF alter the association of histones with DNA by using the energy from adenosine triphosphate (ATP) hydrolysis (2). Though many chromatin-modifying activities have been characterized mechanistically, little is known about their regulation.
The budding yeast PHO5 promoter and gene compose a useful system to investigate the relation between chromatin structure and gene expression. Transcription of PHO5 is regulated in response to phosphate availability by the transcription factors Pho4 and Pho2 (3). When yeast cells are grown in a phosphate-rich medium, Pho4 is phosphorylated by the cyclin-CDK (cyclin-dependent kinase) complex Pho80-Pho85 (4) and inactivated (5). In addition, four positioned nucleosomes reside over the PHO5 promoter, and PHO5 transcription is repressed (6). Upon phosphate starvation, Pho4 is unphosphorylated and active (5), the positioned nucleosomes are no longer detectable (6), and PHO5 is induced. Remodeling of PHO5 chromatin structure requires Pho4 and Pho2 (7) and is facilitated by the histone acetyltransferase Gcn5, which acetylates histones in the promoter region (8, 9).
To identify additional factors important for remodeling chromatin at the PHO5 promoter, we designed a genetic selection to identify mutants defective in PHO5 transcription [Supporting Online Material (SOM) Text]. This selection identified mutations in PSE1, which encodes the import receptor for Pho4 (10), and a mutation in ARG82/IPK2 (denoted arg82-153) (SOM Text). Under inducing conditions, PHO5 transcription and chromatin remodeling are reduced in the arg82-153 mutant (fig. S1).
Arg82 functions in at least two different cellular processes: (i) It regulates transcription of arginine-responsive genes (11), and (ii) with Plc1 and Ipk1, Arg82 functions in a pathway leading to the production of soluble inositol polyphosphates in the nucleus (12, 13) (Fig. 1A). Arg82 inositol trisphosphate (IP3) kinase activity and the production of inositol hexakisphosphate (IP6) are required for efficient export of mRNA from the nucleus (14, 15). The relation between the role of Arg82 in transcriptional control and its IP3 kinase activity is unclear, because Arg82 kinase activity is dispensable for transcriptional regulation of some arginine-responsive genes (16).
Fig. 1.
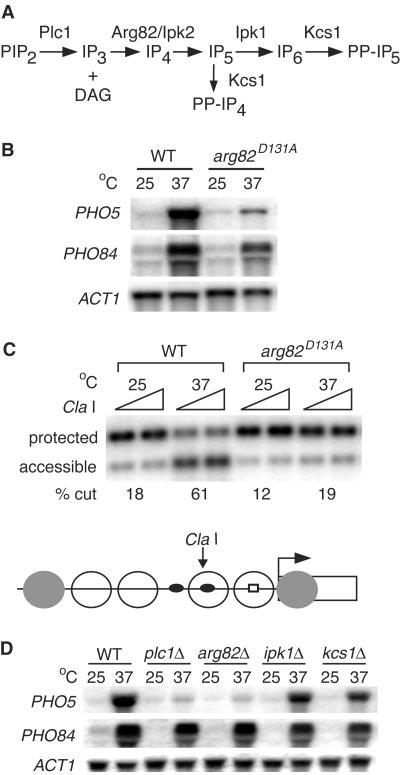
Arg82 kinase activity and IP4 and/or IP5 are important for PHO5 transcription and chromatin remodeling. (A) Pathway for the synthesis of soluble inositol polyphosphates (13, 14). (B) Northern analysis or (C) Cla I restriction enzyme accessibility assay of pho81δ pho80tswild-type (WT) and arg82D131A pho81δ pho80ts strains grown under repressing (25°C) or shifted to inducing (37°C) conditions for 1 hour. PHO5 and PHO84 mRNA amounts in the arg82D131A strain were reduced fivefold and 1.6-fold under inducing conditions. % cut indicates the amount of accessible fragment divided by the sum of accessible and protected fragments. A schematic model of the PHO5 promoter region is shown (bottom), indicating the nucleosomes that are perturbed during induction as open circles, the Pho4 and Pho2 binding sites as dark ovals, the TATA box as an open square, and the Cla I site. (D) Northern analysis of pho81δ pho80ts strains with the relevant genotypes indicated. PHO5 and PHO84 mRNA amounts in the plc1, arg82, ipk1, and kcs1 mutants were 6% and 77%, 4% and 76%, 61% and 64%, and 45% and 50%, respectively, of the wild-type amount under inducing conditions. See fig. S1 for methods.
To determine whether PHO5 induction requires Arg82 kinase activity, we examined PHO5 transcription and chromatin remodeling in a strain expressing the point mutation Asp131 → Ala131 in Arg82 (Arg82D131A), an inositol polyphosphate kinase-deficient mutant (12). The amount of PHO5 mRNA in the arg82D131A strain was reduced under inducing conditions (Fig. 1B) as compared to that in the wild type. We assayed chromatin remodeling in the arg82D131A strain by monitoring a Cla I restriction site in the PHO5 promoter that undergoes an increase in accessibility when wild-type cells are shifted to inducing conditions (Fig. 1C) (6). Little change in Cla I accessibility was observed in the arg82D131A strain (Fig. 1C) as compared with that of the wild-type strain, suggesting that the IP3 kinase activity of Arg82 is important for PHO5 transcription and chromatin remodeling (see also fig. S2).
To better define the role of nuclear inositol polyphosphates in PHO5 transcription, we examined PHO5 induction in plc1, ipk1, and kcs1 mutants. Under inducing conditions, amounts of PHO5 mRNA decreased dramatically in the plc1△ and arg82△ strains but not in the ipk1△ and kcs1△ strains (Fig. 1D) (SOM Text). Therefore, the defect in PHO5 transcription in the arg82△ strain is not likely to result from a lack of IP6 production or a defect in mRNA export, because all these strains are impaired for the production of IP6 and mRNA export (14), whereas only the plc1△ and arg82△ strains are severely defective for PHO5 induction. Also, PP-IP4 is unlikely to play an important role in PHO5 transcription, because kcs1△ strains have only a modest defect in PHO5 activation. Because plc1△ cells do not produce IP3, it is likely the lack of IP4 and/or IP5 production rather than the increased production of IP3 in arg82△ cells that affects PHO5 transcription.
To better understand the requirements for PHO5 promoter chromatin remodeling and transcription, we assayed PHO5 induction in strains with mutations in the following chromatin-remodeling complex components: SNF6, encoding a component of the SWI/SNF complex (SOM Text) (17); ARP8, encoding a component of the INO80 complex (SOM Text) (18); ISW1 and ISW2 (19); and CHD1 (20). For these and subsequent experiments, we induced PHO5 transcription with the use of a strain in which the wild-type CDK Pho85 is replaced with a mutant [Pho85 with the mutation Phe82 → Gly82(Pho85F82G)] that can be selectively inhibited by addition of a cell-permeable kinase inhibitor, 1-NaPP1 (SOM Text). In activating conditions, PHO5 mRNA (and, to a lesser extent, PHO84) was reduced in the snf6△ and arp8△ strains but not in the isw1△, isw2△, or chd1△ strains (Fig. 2A) as compared to the wild-type strain. PHO5 chromatin remodeling was also defective in the snf6△ and arp8△ strains (Fig. 2B), suggesting that SWI/SNF and INO80 are required for efficient remodeling of PHO5 promoter chromatin structure.
Fig. 2.
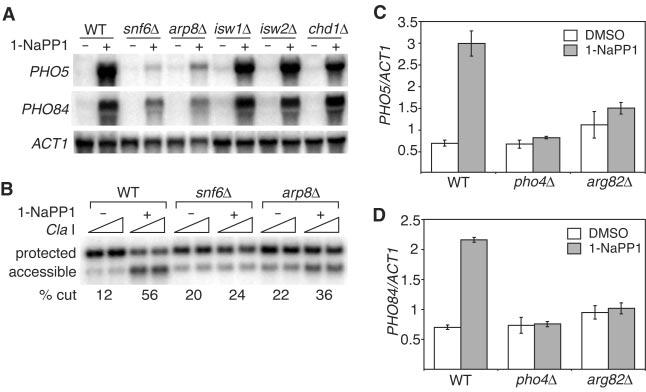
The functions of INO80 and SWI/SNF at the PHO5 and PHO84 promoters are regulated by Arg82. (A) Northern analysis of Pho85F82G-expressing strains with the relevant genotypes indicated. Strains were grown under repressing conditions (-) or shifted to inducing conditions (+) by adding 10 μM 1-NaPP1 for one hour. (B) Cla I restriction enzyme accessibility assay for the PHO85F82G, arp8δ PHO85F82G, and snf6δ PHO85F82G strains, grown under repressing conditions (-) or shifted to inducing conditions (+) by adding 10 m μM 1-NaPP1 for 30 min. % cut, as in Fig. 1C. ChIP using a monoclonal antibody to (C) Ino80-HA or (D) Snf2-Myc in Pho85F82G-expressing strains with the relevant genotypes indicated. Immunoprecipitate efficiency for PHO5 or PHO84 DNA relative to ACT1 DNA is represented by (immunoprecipitated DNA/input DNA)/(ACT1 immunoprecipitated DNA/ACT1 input DNA). Values are the averages of three independent experiments; error bars represent standard error of the mean. DMSO, dimethyl sulfoxide. See fig. S3 for ChIP method.
To determine whether SWI/SNF and INO80 act directly on the PHO5 and PHO84 promoters, we performed chromatin immunoprecipitation (ChIP) experiments. Upon a shift to activating conditions, we observed increased association of Ino80 with PHO5 (Fig. 2C) and PHO84 (fig. S4A) promoters. Recruitment of Ino80 to both promoters was reduced in a pho4△ strain. For Snf2, we observed Pho4-dependent recruitment to the PHO84 promoter under inducing conditions but were unable to detect substantial (> twofold) recruitment to the PHO5 promoter (Fig. 2D). The requirement of Pho4 to efficiently recruit INO80 and SWI/SNF is in agreement with earlier studies demonstrating activator-dependent recruitment of SWI/ SNF to promoters on which it acts (21, 22).
To investigate the connection between inositol polyphosphate metabolism and PHO5 promoter chromatin remodeling, we asked whether the functions of Snf2- or Ino80-containing complexes were influenced by mutations in ARG82. Deletion of ARG82 decreased recruitment of Ino80 to PHO5 (Fig. 2C) and PHO84 (fig. S4A) promoters and Snf2 to the PHO84 promoter (Fig. 2D), suggesting that the function of INO80 and SWI/SNF may be regulated by the production of inositol polyphosphates IP4 and/or IP5.
To determine if Pho4 is still able to bind to phosphate-responsive promoters in arg82△ cells, we characterized Pho4 binding with the use of ChIP. In activating conditions, Pho4 is bound to PHO5 and PHO84 promoters in wild-type and arg82△ strains, though less Pho4 was bound in the arg82△ strain (Fig. 3A and fig. S4B). This result is consistent with the observation that when PHO5 promoter chromatin cannot be remodeled, Pho4 can bind the accessible site in the upstream activation sequence 1 in the PHO5 promoter (UASp1) but not UASp2, which is incorporated into a nucleosome (23). Indeed, in arp8△ and snf6△ strains where chromatin remodeling is defective, the amount of Pho4 bound at the PHO5 promoter was less than that in the wild-type strain and similar to that in the arg82△ strain (Fig. 3B).
Fig. 3.
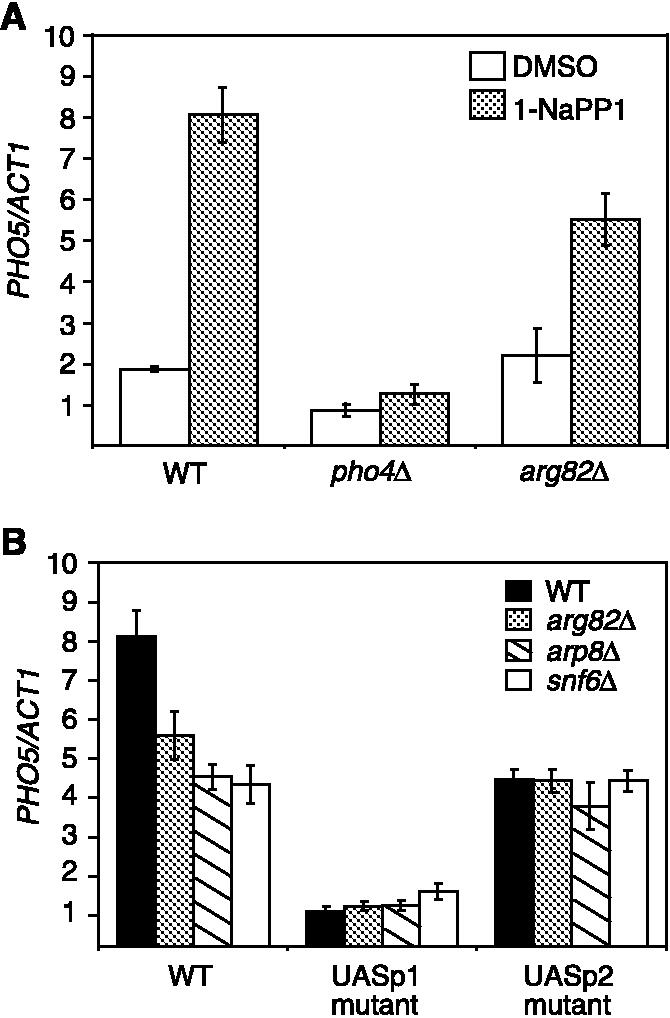
Pho4 binds to UASp1 in mutants impaired for PHO5 chromatin remodeling. PHO5/ACT1, as in Fig. 2C. (A) ChIP using affinity-purified polyclonal antibodies to Pho4 in Pho85F82G-expressing strains with the relevant genotypes indicated. DMSO, as in Fig. 2. (B) ChIP of Pho4 in PHO85F82G, arg82δ PHO85F82G, arp8δ PHO85F82G, and snf6δ PHO85F82G strains carrying substitutions in the Pho4-binding sites of UASp1 and UASp2 in the PHO5 promoter grown under inducing conditions.
To test the hypothesis that Pho4 binds primarily to UASp1 in strains defective for chromatin remodeling, we performed ChIP in strains carrying substitutions in the Pho4-binding sites in UASp1 and UASp2 (fig. S5). The mutation of UASp1 severely impaired binding of Pho4 to the PHO5 promoter in all strain backgrounds (Fig. 3B). In contrast, mutation of UASp2 resulted in ∼50% reduction of Pho4 binding in the wild-type strain but had no significant effect on binding of Pho4 in arg82δ, snf6δ, and arp8δ strains (Fig. 3B). Thus, when Pho4 is nuclear but PHO5 promoter chromatin is not efficiently remodeled, most if not all of the Pho4 binding to the PHO5 promoter occurs at UASp1. We conclude that the defect in recruitment of SWI/SNF and INO80 in the arg82δ strain occurs at a step after Pho4 binding (fig. S6).
We have identified a role for inositol polyphosphates in the regulation of chromatin remodeling and transcription. Our findings suggest that the production of IP4 and/or IP5 modulates the ability of the SWI/SNF and INO80 chromatin remodeling complexes to induce transcription of some phosphate-responsive genes, perhaps by affecting the ability of these complexes to interact with Pho4, Pho2, and/or chromatin. These observations are consistent with results of Wu and colleagues, who demonstrated that the nucleosome-sliding activity of SWI/SNF is stimulated by IP4 and IP5 in vitro and that INO1 transcription is influenced by inositol polyphosphate amounts in vivo (24). It is possible that the amounts or ratios of inositol polyphosphates are altered under certain physiological conditions and that this change may be used by the cell as a signal for global regulation of mRNA export and transcription.
Supplementary Material
Footnotes
Supporting Online Material www.sciencemag.org/cgi/content/full/1078062/DC1 SOM Text Figs. S1 to S6 References and Notes
References and Notes
- 1.Jenuwein T, Allis CD. Science. 2001;293:1074. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 2.Narlikar GJ, Fan HY, Kingston RE. Cell. 2002;108:475. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 3.Oshima Y. Genes. Genet. Syst. 1997;72:323. doi: 10.1266/ggs.72.323. [DOI] [PubMed] [Google Scholar]
- 4.Kaffman A, Herskowitz I, Tjian R, O'Shea EK. Science. 1994;263:1153. doi: 10.1126/science.8108735. [DOI] [PubMed] [Google Scholar]
- 5.Komeili A, O'Shea EK. Science. 1999;284:977. doi: 10.1126/science.284.5416.977. [DOI] [PubMed] [Google Scholar]
- 6.Almer A, Rudolph H, Hinnen A, Hörz W. EMBO J. 1986;5:2689. doi: 10.1002/j.1460-2075.1986.tb04552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fascher KD, Schmitz J, Hörz W. EMBO J. 1990;9:2523. doi: 10.1002/j.1460-2075.1990.tb07432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogelauer M, Wu J, Suka N, Grunstein M. Nature. 2000;408:495. doi: 10.1038/35044127. [DOI] [PubMed] [Google Scholar]
- 9.Barbaric S, Walker J, Schmid A, Svejstrup JQ, Hörz W. EMBO J. 2001;20:4944. doi: 10.1093/emboj/20.17.4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaffman A, Rank NM, O'Shea EK. Genes Dev. 1998;12:2673. doi: 10.1101/gad.12.17.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El Bakkoury M, Dubois E, Messenguy F. Mol. Microbiol. 2000;35:15. doi: 10.1046/j.1365-2958.2000.01665.x. [DOI] [PubMed] [Google Scholar]
- 12.Odom AR, Stahlberg A, Wente SR, York JD. Science. 2000;287:2026. doi: 10.1126/science.287.5460.2026. [DOI] [PubMed] [Google Scholar]
- 13.Saiardi A, Caffrey JJ, Snyder SH, Shears SB. J. Biol. Chem. 2000;275:24686. doi: 10.1074/jbc.M002750200. [DOI] [PubMed] [Google Scholar]
- 14.York JD, Odom AR, Murphy R, Ives EB, Wente SR. Science. 1999;285:96. doi: 10.1126/science.285.5424.96. [DOI] [PubMed] [Google Scholar]
- 15.Saiardi A, Caffrey JJ, Snyder SH, Shears SB. FEBS Lett. 2000;468:28. doi: 10.1016/s0014-5793(00)01194-7. [DOI] [PubMed] [Google Scholar]
- 16.Dubois E, Dewaste V, Erneux C, Messenguy F. FEBS Lett. 2000;486:300. doi: 10.1016/s0014-5793(00)02318-8. [DOI] [PubMed] [Google Scholar]
- 17.Peterson CL, Dingwall A, Scott MP. Proc. Natl. Acad. Sci. U.S.A. 1994;91:2905. doi: 10.1073/pnas.91.8.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen X, Mizuguchi G, Hamiche A, Wu C. Nature. 2000;406:541. doi: 10.1038/35020123. [DOI] [PubMed] [Google Scholar]
- 19.Tsukiyama T, Palmer J, Landel CC, Shiloach J, Wu C. Genes Dev. 1999;13:686. doi: 10.1101/gad.13.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran HG, Steger DJ, Iyer VR, Johnson AD. EMBO J. 2000;19:2323. doi: 10.1093/emboj/19.10.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yudkovsky N, Logie C, Hahn S, Peterson CL. Genes Dev. 1999;13:2369. doi: 10.1101/gad.13.18.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neely KE, et al. Mol. Cell. 1999;4:649. doi: 10.1016/s1097-2765(00)80216-6. [DOI] [PubMed] [Google Scholar]
- 23.Svaren J, Schmidtz J, Hörz W. EMBO J. 1994;13:4856. doi: 10.1002/j.1460-2075.1994.tb06812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen X, Xiao H, Ranallo R, Wu W-H, Wu C. Science. 2003;299:112. doi: 10.1126/science.1078068. [DOI] [PubMed] [Google Scholar]
- 25.We thank C. Wu for communicating results before publication, F. Messenguy and J. York for ARG82 plasmids, and members of the ÒShea laboratory and T. Gruber for helpful discussions. Supported by NIH (GM51377 to E.K.O. and GM51219 to S.R.W.), the Steven and Michelle Kirsch Foundation (S.R.W.), the Howard Hughes Medical Institute (E.K.O.), and the David and Lucile Packard Foundation (E.K.O.). D.J.S. is a Leukemia and Lymphoma Society Special Fellow
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.