

Abstract
Many questions remain about the process of DNA double strand break (DSB) repair by homologous recombination (HR), particularly concerning the exact function played by individual proteins and the details of specific steps in this process. Some recent studies have shown that RecQ DNA helicases have a function in HR. We studied the role of the RecQ helicase Rqh1 with HR proteins in the repair of a DSB created at a unique site within the Schizosaccharomyces pombe genome. We found that DSBs in rqh1+ cells, are predominantly repaired by interchromosomal gene conversion, with HR between sister chromatids [sister-chromatid conversion (SCC)], occurring less frequently. In Δrqh1 cells, repair by SCC is favored, and gene conversion rates slow significantly. When we limited the potential for SCC in Δrqh1 cells by reducing the length of the G2 phase of the cell cycle, DSB repair continued to be predominated by SCC, whereas it was essentially eliminated in wild-type cells. These data indicate that Rqh1 acts to regulate DSB repair by blocking SCC. Interestingly, we found that this role for Rqh1 is independent of its helicase activity. In the course of these studies, we also found nonhomologous end joining to be largely faithful in S. pombe, contrary to current belief. These findings provide insight into the regulation of DSB repair by RecQ helicases.
Keywords: homologous recombination, nonhomologous end joining, rqh1, gene conversion
Double strand breaks (DSBs) pose a major problem for genomic instability and cell survival, because a single unrepaired DSB is, presumably, sufficient to cause cell death (1). The sources of DSBs can be either endogenous, such as those induced during the reshuffling of DNA in Ig gene diversification, or exogenous, such as those induced by exposure to ionizing radiation (2–4). The cell has two major mechanisms for the repair of DSBs: homologous recombination (HR) and nonhomologous end joining (NHEJ), each used to varying degrees in different organisms (2, 3, 5, 6). HR is characterized as an error-free process using homologous sequences as the template to repair the DSB. Repair by HR begins with the formation of 3′ single strand ends at the break that can then invade homologous duplex DNA. The 3′ end of the invading strand is extended by DNA polymerase. At this point, the DSB can be repaired by either DSB repair, which involves formation of a double Holliday junction (HJ) or synthesis-dependent strand annealing (3, 7).
In NHEJ, the DNA ends are resealed by rejoining the broken ends; however, this process can lead to loss of information at the break and is, thus, referred to as error-prone repair (5, 8). Whereas mammalian cells preferentially use NHEJ over HR, budding yeast, for the most part, use HR over NHEJ. In Saccharomyces cerevisiae, the MRX (MRN in Schizosaccharomyces pombe) complex has been shown to play a role in NHEJ (9, 10). In S. pombe, only pKu70/80 and Ligase IV have been identified as functioning in NHEJ, and MRN does not appear to be active in this process (11).
RecQ DNA helicases are found in virtually every organism from bacteria to humans. First described in Escherichia coli as a suppressor of cell death by thymine starvation, RecQ was later found to be a helicase that unwinds DNA in the 3′-to-5′ direction (12, 13). RecQ was first recognized in eukaryotes in S. cerevisiae as a slow growth suppressor (SGS1) of top3 (14). The fission yeast recQ, originally known as rad12+, was renamed rqh1+ for recQ homologue (15–17). There are five recQ homologues in humans. Mutations in three of them (BLM, which leads to Bloom Syndrome (BS); WRN, which leads to Werner’s Syndrome; and RECQL4, which causes Rothman Thomson Syndrome) all present with genomic instability and predisposition to cancer (18–20). How RecQ helicases function in maintaining genomic stability is only beginning to be understood, but several connections between RecQ function and HR have been identified. RecQ mutants show increased rates of HR (21–25). BS patients show very high levels of sister-chromatid exchanges, whereas, in yeast cells, sgs1 and rqh1 mutants have increased levels of HR, based on studies using various reporter systems (17, 26–28). HR is responsible for the synthetic lethality seen between sgs1/rqh1 and srs2, another DNA helicase gene (29, 30). Loss of HR genes suppresses the synthetic interaction between sgs1 and mus81 (31, 32). When the E. coli HJ resolvase RusA was expressed in Δrqh1 cells, their UV and hydroxyurea (HU) sensitivities were partially suppressed, suggesting that in the absence of Rqh1, HJs accumulate (33). Finally, we recently reported that the UV and HU sensitivities of Δrqh1 are suppressed by loss of a subset of HR genes (34). Together, these findings strongly imply that RecQ helicases function through HR to provide genomic stability during both DNA damage and replication arrest.
In this study, we have used a system in which a DSB is induced at a unique site on a nonessential minichromosome (Ch16), which contains the pericentric regions of ChIII. The unique DSB is created by HO endonucleolytic cleavage of MATa, inserted into Ch16 (35). This system allows us to score the events that occur downstream of DSB formation in different genetic backgrounds, enabling us to assess the contributions of various proteins to this process. By using a partial diploid and generating the DSB in a nonessential chromosome, repair is not required for cell survival. We found that, in wild-type cells, DSBs are repaired preferentially through gene conversion (GC) and that this choice is regulated by the action of Rqh1 by suppressing sister-chromatid conversion (SCC) leading to increased GC. Interestingly, the DNA helicase activity of Rqh1 is apparently not required in the suppression of SCC.
Results
Repair of Site-Specific DSB in the Wild-Type Background.
A unique system was used to analyze the repair of a site-specific DSB at an ectopic MATa site through the expression of the HO endonuclease, under control of the thiamine-repressible promoter nmt (Fig. 1) (35). Strains carrying the minichromosome with the MATa site are referred to as TH805. After a period of HO induction, cells were spread onto yeast extract plates and incubated until colonies grew up. Colonies were analyzed for chromosome loss (ChL), GC, SCC, or NHEJ, as described in Methods and in supporting information, which is published on the PNAS web site. The results from the various strains analyzed are shown in Table 1 and Fig. 2a. In wild-type cells after 48 h of induction, 44 ± 5% of colonies remained Ade+ but became G418-sensitive, indicative of repair by GC, whereas 45 ± 4% of colonies remained both Ade+ and G418R, consistent with repair by SCC or NHEJ. At 72 h postinduction, 60 ± 7% of colonies had repaired the DSB by GC, whereas 17 ± 7% remained Ade+ G418R.
Fig. 1.

A schematic depiction of the Th805 system and the predicted products that can form after repair of a DSB induced at MATa.
Table 1.
DSB repair results
| Strain | Hours | ChL, % | SCC/NHEJ, % | GC, % | Total colonies |
|---|---|---|---|---|---|
| Wild type (Th805) | 48 | 11 ± 2 | 45 ± 4 | 44 ± 5 | 3,615 |
| 72 | 23 ± 8 | 17 ± 7 | 60 ± 7 | 2,358 | |
| K5471 | 48 | 18 ± 4 | 40 ± 4 | 42 ± 7 | 2,425 |
| 72 | 29 ± 3 | 22 ± 1 | 48 ± 2 | 740 | |
| rqh1 | 48 | 9 ± 2 | 78 ± 5 | 13 ± 4 | 4,226 |
| 72 | 27 ± 4 | 45 ± 8 | 26 ± 4 | 570 | |
| ku80 | 48 | 2 ± 2 | 55 ± 4 | 43 ± 2 | 973 |
| 72 | 1 ± 1 | 38 ± 3 | 61 ± 3 | 758 | |
| wee1-50 | 48 | 9 ± 2 | 24 ± 7 | 67 ± 4 | 2,322 |
| 72 | 18 ± 3 | 12 ± 2 | 70 ± 2 | 1,524 | |
| rqh1/ku80 | 48 | 2 ± 1 | 90 ± 7 | 8 ± 6 | 921 |
| 72 | 2 ± 2 | 77 ± 9 | 22 ± 7 | 883 | |
| rqh1/wee1-50 | 48 | 3 ± 4 | 88 ± 4 | 8 ± 3 | 3,412 |
| 72 | 1 ± 2 | 73 ± 6 | 26 ± 5 | 5,127 | |
| wee1-50/ku80 | 48 | 2 ± 2 | 3 ± 4 | 95 ± 2 | 2,178 |
| 72 | 2 ± 2 | 1 ± 1 | 97 ± 2 | 2,460 | |
| rqh1/ku80/wee1-50 | 48 | 0 ± 1 | 91 ± 3 | 9 ± 3 | 4,938 |
| 72 | 4 ± 4 | 71 ± 8 | 25 ± 1 | 3,739 |
These data represent the results of a minimum of three experiments.
Fig. 2.

Bar graphs showing comparisons in GC and SCC/NHEJ frequencies in various genetic backgrounds. Each bar represents an average of a minimum of three individual experiments. (a) Comparison of SCC/NHEJ and GC rates among Th805 (wild type), Th805 Δrqh1 (rqh1), Th805 Δpku80 (pku80), and Th805 Δrqh1 Δpku80 (rqh1 pku80). (b) Comparison of Th805 wee1-50 (wee1-50), Th805 Δrqh1 wee1-50 (rqh1 wee1-50), Th805 Δpku80 wee1-50 (rqh1 pku80), and Th805 Δrqh1 Δpku80 wee1-50 (rqh1 pku80 wee1-50). (c) Comparison of Th805 (wild type), Th805 Δrqh1 (rqh1), and Th805 rqh1-K547I (rqh1 K547I).
A shortcoming of this system is that this marker analysis does not allow us to differentiate among uncut substrate, repair by SCC, or repair by NHEJ, because all three result in the retention of both the Ade6+ and G418R phenotype. Other authors have used Southern blot analysis to observe the efficiency of HO cleavage in this system (35, 36). These experiments showed that the cleavage product begins to appear 20 h after induction, with peak cleavage seen at 24 h. However, only a minor amount of cleavage product was visible, suggesting that repair of the DSB is very efficient and/or that cleavage occurs slowly. Because it is critical to know what portion of cells actually experienced an HO-induced DSB, we measured the number of cells that received a DSB by an indirect method.
The appearance of Rad22-yellow fluorescent protein (YFP) nuclear foci in S. pombe has been correlated with DSB formation (37–39). We created a Th805 strain containing rad22-YFP-KanMX6 and looked for the appearance of Rad22-YFP nuclear foci as a measure of HO cleavage. We induced HO endonuclease in Th805-rad22-YFP, collected cells at 2-h intervals, and examined them by fluorescence microscopy. For the first 12 h, the number of cells containing nuclear foci was <5%. This number began to increase at 14 h, and, thereafter, the percentage of cells with visible nuclear foci increased rapidly (Fig. 3). By 28 h after HO induction, we observed foci in >75% of cells, and the level remained constant for the next 6 h. The persistence of Rad22-YFP foci in this study is consistent with previous studies where Rad22 foci were shown to persist for 8 h or longer after damage (37). It is important to note that the products of DSB repair by SCC or faithful NHEJ are substrates for another round of cutting by HO. This ongoing repair of the HO-induced DSB indicates that the fraction of cells with Rad22 foci is, likely, an underestimation of cells that have experienced a DSB, suggesting that, by 48 h postinduction, essentially every cell has experienced at least one DSB.
Fig. 3.

Demonstration of cleavage by HO endonuclease rad22-YFP was introduced into Th805 in a wild-type background. HO endonuclease was induced by growth in media lacking thiamine and the cells followed for 30 h. Before induction, only a few cells ≈2–4% showed Rad22-YFP foci. By ≈16 h postinduction, foci were visible in many more cells (≈30%). By 28 h, nearly all cells (>77%) contained Rad22-YFP foci, suggesting that essentially every cell had received at least one HO-induced break by this time.
To further characterize the repair events that generate Ade+ G418R colonies, we asked whether these repair events depend on Ku70/Ku80 function. Experimental studies of NHEJ using linearized plasmids showed that repair of DSBs by NHEJ is essentially eliminated in the Δku80 background (11). Although rare, Ku-independent repair was detected, resulting in large deletions at the repair junction (11). If the Ade+ G418R colonies represent repair by NHEJ, they should be greatly reduced in the Δku80 background, whereas, if they represent repair by SCC, they should persist in this background. We found that deletion of Ku80 did not eliminate or reduce the Ade+ G418R colonies recovered after HO induction, suggesting that these colonies represent repair by SCC (Fig. 2a and Table 1). In fact, the level of Ade+ G418R colonies increased compared with wild type, likely because of an overall decrease in chromosome loss.
Repair of Site-Specific DSB in the Δrqh1 Background.
To examine the role or Rqh1 in DSB repair, we created Th805-Δrqh1 by crossing rqh1::ura4+ with Th805 (15, 35). ChL, GC, and SCC/NHEJ frequencies were determined for Th805-Δrqh1 at 48 and 72 h after induction of the HO endonuclease. The results are summarized in Table 1 and Fig. 2a. We found significant differences in the mechanism of DNA repair in the Δrqh1 background compared with wild type. In an rqh1+ background, the majority of cells repaired the DSB by GC (60 ± 7% by 72 h after HO induction). By contrast, GC frequencies at 72 h after HO induction in a Δrqh1 background were only 26 ± 4% (P < 0.001), with high levels of SCC/NHEJ (45 ± 8% (P < 0.001) (Fig. 2a and Table 1). These data indicate that, in the absence of Rqh1, DSBs are less likely to be repaired by GC.
One simple explanation for this result is that the HO endonuclease might cleave inefficiently in Δrqh1 cells, accounting for the high levels of Ade+ G418R colonies seen in this background. We were unable to quantify DSBs by measuring Rad22-YFP foci in the Δrqh1 background because of a high level of spontaneous Rad22-YFP foci observed in Δrqh1 cells (J.C.H. and G.A.F., unpublished data). To verify that the level of DSBs after HO induction in a Δrqh1 background is the same as in wild-type cells, we compared the efficiency of HO cutting in Δrqh1 and rqh1+ by Southern blot analysis. Fig. 4 shows the result of the Southern blot, where the 3.5-kb fragment, indicative of HO cutting, is visible in equivalent intensities in both backgrounds, showing that the HO endonuclease cleaves with similar efficiency in both strains. We conclude that the increased number of Ade+ G418R colonies recovered in the Δrqh1 background represent increased repair by SCC or NHEJ and not a decreased frequency of substrate cleavage.
Fig. 4.

HO cleavage is equivalent in wild-type and Δrqh1 backgrounds. Cultures of Th805 and Th805 Δrqh1 were grown for 20 or 24 h in media lacking thiamine to induce the HO endonuclease to cleave at the MATa locus. EcoRI-digested genomic DNA was isolated and used for Southern blot analysis. The blot was probed with a 32P-labeled kanMX6 probe (black bar shown in diagram). The autoradiograph shows that equivalent levels of the 3.5-kb band generated by a combination of EcoRI and HO endonuclease cutting are seen at equivalent levels in both wild-type (WT) and Δrqh1 backgrounds.
In the Absence of Rqh1, DSB Are Preferentially Repaired by SCC.
Our data show that in a Δrqh1 background, Ade+ G418R colonies predominate, but this could be due to repair by either SCC or NHEJ. If the HO break is repaired by NHEJ, this should rely wholly, or in large part, on pKu70/80 (11). We created a Th805 Δrqh1 Δpku80 strain and examined it in the DSB repair assay. We found that, in the absence of both Rqh1 and pKu80, the number of Ade+ G418R colonies actually increased to 90 ± 7% and 77 ± 9% after 48 or 72 h of induction, respectively (Fig. 2a and Table 1). This increase over either single mutant can be accounted for by the apparent suppression of ChL in Δpk80 cells and increased SCC due to loss of Rqh1 activity. These results support the argument that DSBs in Δrqh1 cells are not repaired by NHEJ, leaving SCC as the likely process of repair.
Suppressing SCC Does Not Increase Repair by GC.
Preferential repair by SCC in a Δrqh1 background can be explained in one of two ways; Rqh1 promotes DSB repair by GC, so, in its absence, GC levels decrease, leading to an increase in SCC or Rqh1 blocks DSB repair by SCC, so, in its absence, SCCs simply rise because they are not blocked from forming. We reasoned that, if we could significantly reduce the length of the G2, we would reduce the availability of the sister chromatid as a repair template, limiting the opportunity for SCC. Under these conditions, if Rqh1 promotes GC, then limiting SCC in the absence of Rqh1 should result in increased levels of DSB repair by NHEJ and/or ChL. On the other hand, if Rqh1 blocks SCC, then, under these conditions, GC levels should return to levels comparable with those of wild-type cells. To test this hypothesis, we crossed the conditional mutant wee1-50 into Th805, Th805-Δpku80, Th805-Δrqh1, and Th805-Δrqh1 Δpku80 cells. Cells containing the wee1-50 allele, grown at a semipermissive temperature (33.5°C), have a very short G2 phase, essentially exiting S phase and directly entering mitosis (40, 41).
We found that, in Th805-wee1-50 cells, the majority of colonies recovered represented repair of the DSBs by GC; 66 ± 7% and 68 ± 10% of cells, at 48 and 72 h postinduction, respectively, similar to Th805-wt (Fig. 2b and Table 1). In the TH805-wee1-50 mutant, fewer colonies resulted from repair by SCC/NHEJ (24 ± 5% and 12 ± 2%, at 48 and 72 h postinduction), compared with 45 ± 4% and 17 ± 7%, at 48 and 72 h postinduction for wild type. If SCC is, indeed, impaired in wee1-50, then these Ade+ G418R colonies should represent NHEJ events. Indeed, we found that, in Th805-pku80 wee1-50 cells, where NHEJ is largely blocked, Ade+ G418R colonies were virtually nonexistent (3 ± 3% and 1 ± 1% at 48 and 72 h after HO induction, respectively) (Fig. 2b and Table 1). Thus, the Ade+ G418R colonies observed in wee1-50 were likely the result of NHEJ, confirming that SCC is extremely rare in a wee1-50 background.
When we analyzed Th805 Δrqh1 wee1-50 cells, we found that the majority of colonies were Ade+ G418R (70 ± 14% and 63 ± 10% at 48 and 72 h after HO induction, respectively) (Fig. 2b and Table 1), and GC events were observed in only 8 ± 3% (48 h) and 26 ± 5% (72 h) of colonies. This degree of repair by GC is very similar to that observed in Δrqh1, indicating that limiting SCC does not restore GCs to wild-type levels.
To test whether the Ade+ G418R colonies resulted from repair of the DSB by NHEJ, we created Th805 Δrqh1 Δpku80 wee1-50, where repair by SCC, NHEJ, and GC should all be limited. Surprisingly, the frequency of Ade+ G418R colonies after 72 h was 71 ± 8% and GC frequencies were only 25 ± 1% (Fig. 2b and Table 1), very similar to the levels seen in Th805 Δrqh1 wee1-50, suggesting that repair in this background is largely accomplished by SCC. One possible explanation for these data is that the loss of Rqh1 increases the length of G2 in a wee1-50 background, increasing the possibility of SCC to occur. Two observations argue against this possibility. First, Th805-Δrqh1-Δwee1-50 cells had the same small cell size as the Th805-wee1-50 cells, indicative of a shortened cell cycle. Second, a longer G2 will lead to an overall increase in the length of the cell cycle. Thus, we compared the length of the cell cycle from G1 to cytokinesis of Th805-wee1-50 with Th805-Δrqh1-wee1-50. To accomplish this, both cell lines were arrested in G1 by overnight incubation in media lacking nitrogen. The cells were released from this block by resuspension into yeast extract adenine media. Cells were collected at 20-min intervals, and their septa were stained with Calcofluor. We found that in both backgrounds the number of septa began to increase after 200 min, indicating that the absence of Rqh1 did not significantly affect the length of the cell cycle and, by extrapolation, the length of G2.
We also tested the formal possibility that the cells were still able to repair by NHEJ in this background. Previous studies have shown that, when DSBs are repaired by end joining in the absence of Ku, large deletions at the junctions are observed (11). We isolated several Ade+ G418R colonies, PCR amplified the MATa site, and separated the products on agarose gels. The results demonstrate that there was no apparent change in the size of the PCR products from repaired junctions compared with uncut substrate (see Supporting Information). Sequence analysis of these PCR products showed the HO cut site to be intact in all clones that were tested. Based on these results, we conclude that the Ade+ G418R colonies formed in a TH805 Δrqh1 Δpku80 wee1-50 background arose through repair by SCC. Our interpretation of these results is that DSB repair in the Δrqh1 background occurs primarily by SCC, suggesting that the normal role of Rqh1 is to block repair by SCC. The high rates of SCC, even in a wee1-50 background, demonstrate how significant a role Rqh1 plays in this process.
The Suppression of SCC by Rqh1 Is Independent of Its Helicase Activity.
Previous studies of helicase-dead mutants of RecQ have generally demonstrated that helicase activity is important for most, but not all, of its associated functions. We had shown that suppression of Δtop3 lethality was suppressed more efficiently when rqh1+ was deleted compared with when its helicase activity was inactivated (42). Ahmad et al. (43) showed that recovery from S-phase arrest by Rqh1 only partially depended on its helicase activity. In S. cerevisiae, the ability of Sgs1 to suppress crossovers and reduce GC tract lengths is reportedly independent of its helicase activity (J. Nickloff, personal communications). We tested whether the helicase activity of Rqh1 was important in its role of suppressing repair by SCC, using a helicase-dead mutant rqh1-K547I (42). We found that the mechanism of repair in the helicase-dead mutant was nearly identical to rqh1+ cells, as seen in Fig. 2c and Table 1; GC rates in Th805-wt and Th805-rqh1-K547I at 48 h after HO induction were 44 ± 5% and 42 ± 7% (P = 0.44), respectively, whereas SCC/NHEJ rates were 45 ± 4% and 40 ± 4% (P = 0.14). At 72 h after HO induction, the levels of GCs in Th805-wt compared with Th805-rqh1-K547I were 60 ± 7% and 48 ± 2% (P = 0.04), whereas SCC/NHEJ rates were 17 ± 7% and 22 ± 1% (P = 0.149), respectively, 48 and 72 h after HO induction (Fig. 2c and Table 1). The P values for two independent sample t tests confirm that cells in a rqh1-K547I background do not repair DSBs differently from cells in a wild-type background. These data suggest that the ability of Rqh1 to suppress SCC is largely independent of its helicase activity.
NHEJ of Linearized Plasmids Differs from Repair of an HO Cleavage in the Chromosome.
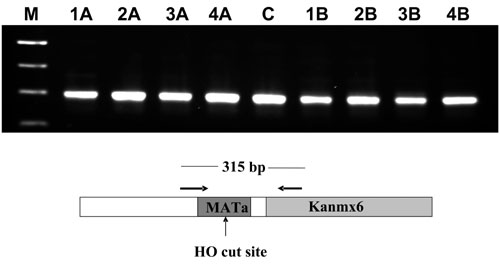
Previous studies of NHEJ events in S. pombe using linearized plasmids found that varying amounts of DNA sequence was usually lost at the repair junction and concluded that NHEJ in S. pombe is generally not faithful (11, 44, 45). We examined the sequences at the cleavage site of MATa in multiple colonies after HO endonuclease induction and looked for evidence of sequence loss consistent with repair by NHEJ. Primers flanking the HO site were used to amplify a 315-nt fragment from 66 Ade+ G418R colonies that formed after a 72-h induction in wild-type (12 colonies), Δrqh1 (34 colonies) and Δpku80 (20 colonies) backgrounds. In every case, 66 of 66 colonies, the MATa locus had been perfectly restored. We initially used these data as evidence that the Ade+ G418R colonies recovered after HO induction represent SCC repair events. However, we analyzed Th805-wee1-50 cells in the DSB repair assay at semipermissive temperatures, where SCC should be largely blocked, a significant number of Ade+ G418R colonies formed, suggestive of repair by NHEJ (Table 1). We confirmed this finding by demonstrating that Ade+ G418R colonies in a wee1-50 background were pKu80-dependent (Fig. 2b and Table 1). We isolated 10 Ade+ G418R Th805-wee1-50 colonies after a 72-h and 96-h HO induction (5 from each), PCR amplified the sequences flanking the HO junction, and analyzed the sequence. In all 10 PCR products, the repair was error-free, suggesting faithful end joining. This finding is in conflict with the conclusion of previous data using linearized plasmid DNA (11, 44, 45).
Discussion
The RecQ helicases are recognized as partners in HR, likely having a late function, possibly in processing HJs (46–48). This is supported by studies showing that sister-chromatid exchanges, which are a visualization of crossovers, increase dramatically in BLM−/− cells (49, 50). Also, biochemical data demonstrated that the BLM protein can catalyze the branch migration of HJs as well as participate in the resolution of a double-HJ-like four-way junction through a process called dissolution (51, 52). In S. pombe, the Δrqh1 phenotype is suppressed significantly when the HJ resolvase RusA is expressed, indicating that Rqh1 functions to prevent the accumulation of HJs (33). We have provided evidence that Rqh1 acts downstream of Rhp55/57 in HR (34). There is also data that support an early role in HR for RecQ helicases. Both BLM and Sgs1 associate with Rad51 and colocalize on ssDNA after exposure to ionizing radiation (53, 54). Similarly, Rqh1 foci form earlier than Rhp51 foci after UV irradiation of S. pombe cells (55). Together, these studies suggest that RecQ helicases act both early and late during HR.
Repair of DSBs by GC Is Reduced in a Δrqh1 Background.
The power of using a specific site-of-damage system is in the ability to follow the products of the repair reaction and determine events that occur downstream of damage in various genetic backgrounds. This particular system has two advantages over those described in S. cerevisiae: first, by placing the DSB on a nonessential chromosome, lack of repair (which would be lethal if the DSB were on an essential chromosome) can be followed; second, the inefficient cleavage by HO endonuclease and/or efficient repair, largely limits DSBs to one sister chromatid at a time, allowing SCC to take place. Our initial observation that repair of the DSB in a Δrqh1 background resulted in reduced GC frequencies as compared with rqh1+ cells seemed to conflict with previous studies that showed RecQ mutants typically have a hyperrecombination phenotype. However, our further studies strongly suggest that this decrease in GC frequencies is actually the result of an increase in the frequency of SCC repair. This conclusion was drawn from several results. First, using Southern blot analysis, we demonstrated that comparable levels of DSBs were being formed in Δrqh1 and rqh1+ cells. Next, we demonstrated that the Ade+ G418R colonies that formed in the Δrqh1 background represented SCC repair events. In Δrqh1 Δpku80 cells, where NHEJ should be largely blocked, the level of Ade+ G418R colonies actually increased over that seen in the Δrqh1 single mutant, indicating that these Ade+ G418R colonies are not the result of NHEJ and, therefore, must arise by SCC. This result also supports previous data suggesting that NHEJ is not commonly used for DSB repair in fission yeast cells (56). Studies of NHEJ in both S. pombe and S. cerevisiae have concluded that, in the absence of Ku proteins, NHEJ is diminished and results in terminal deletions of DNA sequence, often of a substantial length (11, 57). If Δrqh1 Δpku80 or Δpku80 mutants were able to repair DSBs by NHEJ, sequence loss at the HO cut site should be observed. Sequence analysis of the HO junction from multiple Δrqh1 Δpku80 and Δpku80 colonies that were Ade+ G418R failed to reveal any loss of sequence at the junction, supporting the argument that the DSBs were not repaired by pKu80-independent end joining. Together, these data support the argument that it is SCC levels that are elevated in Δrqh1 cells and not NHEJ.
There are two possible explanations for increased levels of SCC in the Δrqh1 background: Rqh1 promotes GCs, and, in the absence of Rqh1, DSB repair shifts to SCC or Rqh1 blocks SCC, so, in its absence, SCC levels increase. We attempted to distinguish between these two possibilities by reducing the length of G2, thus limiting the ability of cells to undergo SCC. The temperature-sensitive wee1-50 mutant was used for the purpose of reducing the length of G2. Our rationale for these studies was that, if Rqh1 blocked SCC, then reducing G2 should greatly limit SCC, returning GC rates to wild-type levels. Alternatively, if Rqh1 promoted GC, then the frequency of GCs should be reduced relative to that of wild-type cells, with concomitant increases in NHEJ, ChL, or both. We first found that in a wee1-50 background, at semipermissive temperatures, the level of Ade+ G418R colonies decreased, whereas the number of colonies that repaired by GC increased, relative to wee1-50+ cells. Furthermore, we showed that the Ade+ G418R colonies that did arise under these conditions resulted from NHEJ, because they were eliminated when pku80+ was deleted. When we measured the level of Ade+ G418R colonies that arose in Th805 Δrqh1 wee1-50 cells after HO induction, they did not decrease but remained at levels comparable with that of Th805 Δrqh1 cells. This result seemed to suggest that repair had switched to NHEJ. However, when we analyzed Th805 Δrqh1 Δpku80 wee1-50 strains in this assay, the level of Ade+ G418R colonies was not diminished, leaving us with two possible explanations: NHEJ occurs in the absence of Ku80, or SCC was still being carried out despite the shortened G2 phase. When we analyzed the HO junctions of multiple Ade+ G418R colonies from this background, all were intact. Thus, repair was almost certainly by SCC. The efficient repair by SCC in the absence of Rqh1, even with a reduced G2, supports the argument that Rqh1 is a potent inhibitor of SCC.
It should be noted that the sequences directly adjacent the HO cut site are not homologous to ChIII, and this could help favor SCC over GC. However, this is true for all backgrounds tested in our study and does not change our interpretation of the data.
The Helicase Activity Defines Two Functions for Rqh1.
Our finding that the GC and SCC frequencies in the helicase-dead rqh1-K547I mutant were very close to wild-type levels supports the argument that the helicase activity is not required in this early role of Rqh1. These results indicate that the helicase activity of Rqh1 activity is not needed for Rqh1 to regulate the pathway of DSB repair. It will be interesting to determine which domains of Rqh1 are responsible for this regulation. Interestingly, we have shown that rqh1-K547I is as sensitive to DNA damage as is Δrqh1 (42). Because rqh1-K547I retains the ability to block SCC, the role of Rqh1 in determining the pathway of DSB repair is not critical for cell viability.
NHEJ in S. pombe Is Faithful.
At the semipermissive temperature, in a wee1-50 background, significant numbers of Ade+ G418R colonies formed that likely arose by NHEJ, yet contained intact sequences at the HO junction. This faithful repair of DSBs by NHEJ differs from DSB repair assays based on linearized plasmids, in which sequence loss was consistently observed (11, 45). We suggest that this difference may be due to the chromosomal context of the DSB in our system, as opposed to the plasmid context of other studies. The HO system more faithfully represents cellular repair of DSBs, where, in the chromosomal context, the DNA is properly packaged into chromatin. We recognize that the universality of faithful repair by NHEJ in fission yeast awaits the study of DSBs with different terminal structures.
Methods
Genetic Manipulations and Creation of Strains.
Standard protocols were used for the creation of strains. A table of strains used in these studies is provided in Supporting Information. The rad22-YFP strain was created by inserting the PCR-amplified YFP-kanMX6 sequences of pDH5 at the 3′ end of rad22+. The resulting strain was shown to have wild-type levels of resistance to ionizing radiation.
HO-Induced DSB Repair Assay.
The protocol followed for HO induction has been described in ref. 35. A detailed description is given in Supporting Information and in Results. Overnight cultures were begun in the presence of thiamine to suppress HO expression. The thiamine was removed and the cells plated at 48 and 72 h onto yeast extract plates and incubated 3–5 days, depending on growth rates. Red and white colonies were counted, and white colonies were picked and analyzed further for G418 sensitivity. Red colonies were scored as ChL.
Analysis of the HO-Recognition Sequence in Cells That Repaired Their HO Cut Site by Either NHEJ or SCC.
Multiple colonies that were Ade+ G418R were isolated after a 72-h induction of HO. Primers adjacent to the HO site (3′ to 5′: CAAGGAGGGTATTCTGGGCC; 5′ to 3′: TCGGTATCTGAGGCCCTTCC) were used to amplify this region. The PCR products were separated on 1.4% agarose gels, and the DNA was isolated and sequenced.
Supplementary Material
Acknowledgments
We thank Tim Humphrey (Medical Research Council Radiation and Genomic Stability Unit, U.K.) for providing us with the Th805 system, John Prudden for his helpful advice in setting up the assay, Ms. Gloria Osorio for technical help, and Dr. Grant Brown for helpful suggestions and critical reading of this manuscript. This work was supported by National Institutes of Health Grant CA072647.
Abbreviations
- ChL
chromosome loss
- DSB
double strand break
- GC
gene conversion
- HJ
Holliday junction
- HR
homologous recombination
- NHEJ
nonhomologous end joining
- SCC
sister-chromatid conversion
- YFP
yellow fluorescent protein.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Bennett C. B., Lewis A. L., Baldwin K. K., Resnick M. A. Proc. Natl. Acad. Sci. USA. 1993;90:5613–5617. doi: 10.1073/pnas.90.12.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson S. P. Carcinogenesis. 2002;23:687–696. doi: 10.1093/carcin/23.5.687. [DOI] [PubMed] [Google Scholar]
- 3.Symington L. S. Microbiol. Mol. Biol. Rev. 2002;66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang C. L., Wabl M. J. Immunol. 2004;172:5815–5821. doi: 10.4049/jimmunol.172.10.5815. [DOI] [PubMed] [Google Scholar]
- 5.Hefferin M. L., Tomkinson A. E. DNA Repair (Amsterdam) 2005;4:639–648. doi: 10.1016/j.dnarep.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Weterings E., van Gent D. C. DNA Repair (Amsterdam) 2004;3:1425–1435. doi: 10.1016/j.dnarep.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 7.Paques F., Haber J. E. Mol. Cell. Biol. 1997;17:6765–6771. doi: 10.1128/mcb.17.11.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Critchlow S. E., Jackson S. P. Trends Biochem. Sci. 1998;23:394–398. doi: 10.1016/s0968-0004(98)01284-5. [DOI] [PubMed] [Google Scholar]
- 9.Lewis L. K., Resnick M. A. Mutat. Res. 2000;451:71–89. doi: 10.1016/s0027-5107(00)00041-5. [DOI] [PubMed] [Google Scholar]
- 10.Teo S. H., Jackson S. P. Curr. Biol. 2000;10:165–168. doi: 10.1016/s0960-9822(00)00317-1. [DOI] [PubMed] [Google Scholar]
- 11.Manolis K. G., Nimmo E. R., Hartsuiker E., Carr A. M., Jeggo P. A., Allshire R. C. EMBO J. 2001;20:210–221. doi: 10.1093/emboj/20.1.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakayama H., Nakayama K., Nakayama R., Irino N., Nakayama Y., Hanawalt P. C. Mol. Gen. Genet. 1984;195:474–480. doi: 10.1007/BF00341449. [DOI] [PubMed] [Google Scholar]
- 13.Umezu K., Nakayama K., Nakayama H. Proc. Natl. Acad. Sci. USA. 1990;87:5363–5367. doi: 10.1073/pnas.87.14.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gangloff S., McDonald J. P., Bendixen C., Arthur L., Rothstein R. Mol. Cell. Biol. 1994;14:8391–8398. doi: 10.1128/mcb.14.12.8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davey S., Han C. S., Ramer S. A., Klassen J. C., Jacobson A., Eisenberger A., Hopkins K. M., Lieberman H. B., Freyer G. A. Mol. Cell. Biol. 1998;18:2721–2728. doi: 10.1128/mcb.18.5.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murray J. M., Lindsay H. D., Munday C. A., Carr A. M. Mol. Cell. Biol. 1997;17:6868–6875. doi: 10.1128/mcb.17.12.6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stewart E., Chapman C. R., Al-Khodairy F., Carr A. M., Enoch T. EMBO J. 1997;16:2682–2692. doi: 10.1093/emboj/16.10.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bachrati C. Z., Hickson I. D. Biochem. J. 2003;374:577–606. doi: 10.1042/BJ20030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakayama H. Oncogene. 2002;21:9008–9021. doi: 10.1038/sj.onc.1205959. [DOI] [PubMed] [Google Scholar]
- 20.Ellis N. A., German J. Hum. Mol. Genet. (5 Spec. No.) 1996:1457–1463. doi: 10.1093/hmg/5.supplement_1.1457. [DOI] [PubMed] [Google Scholar]
- 21.Ajima J., Umezu K., Maki H. Mutat. Res. 2002;504:157–172. doi: 10.1016/s0027-5107(02)00089-1. [DOI] [PubMed] [Google Scholar]
- 22.Harrigan J. A., Bohr V. A. Biochimie. 2003;85:1185–1193. doi: 10.1016/j.biochi.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 23.Hickson I. D. Nat. Rev. Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 24.Myung K., Datta A., Chen C., Kolodner R. D. Nat. Genet. 2001;27:113–116. doi: 10.1038/83673. [DOI] [PubMed] [Google Scholar]
- 25.Yamagata K., Kato J., Shimamoto A., Goto M., Furuichi Y., Ikeda H. Proc. Natl. Acad. Sci. USA. 1998;95:8733–8738. doi: 10.1073/pnas.95.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaganti R. S., Schonberg S., German J. Proc. Natl. Acad. Sci. USA. 1974;71:4508–4512. doi: 10.1073/pnas.71.11.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Onoda F., Seki M., Miyajima A., Enomoto T. Mutat. Res. 2000;459:203–209. doi: 10.1016/s0921-8777(99)00071-3. [DOI] [PubMed] [Google Scholar]
- 28.Watt P. M., Hickson I. D., Borts R. H., Louis E. J. Genetics. 1996;144:935–945. doi: 10.1093/genetics/144.3.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gangloff S., Soustelle C., Fabre F. Nat. Genet. 2000;25:192–194. doi: 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- 30.Maftahi M., Hope J. C., Delgado-Cruzata L., Han C. S., Freyer G. A. Nucleic Acids Res. 2002;30:4781–4792. doi: 10.1093/nar/gkf581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fabre F., Chan A., Heyer W. D., Gangloff S. Proc. Natl. Acad. Sci. USA. 2002;99:16887–16892. doi: 10.1073/pnas.252652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doe C. L., Whitby M. C. Nucleic Acids Res. 2004;32:1480–1491. doi: 10.1093/nar/gkh317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doe C. L., Dixon J., Osman F., Whitby M. C. EMBO J. 2000;19:2751–2762. doi: 10.1093/emboj/19.11.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hope J. C., Maftahi M., Freyer G. A. Genetics. 2005;170:519–531. doi: 10.1534/genetics.104.037598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prudden J., Evans J. S., Hussey S. P., Deans B., O’Neill P., Thacker J., Humphrey T. EMBO J. 2003;22:1419–1430. doi: 10.1093/emboj/cdg119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osman F., Fortunato E. A., Subramani S. Genetics. 1996;142:341–357. doi: 10.1093/genetics/142.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du L. L., Nakamura T. M., Moser B. A., Russell P. Mol. Cell. Biol. 2003;23:6150–6158. doi: 10.1128/MCB.23.17.6150-6158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meister P., Poidevin M., Francesconi S., Tratner I., Zarzov P., Baldacci G. Nucleic Acids Res. 2003;31:5064–5073. doi: 10.1093/nar/gkg719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meister P., Taddei A., Vernis L., Poidevin M., Gasser S. M., Baldacci G. J. Cell Biol. 2005;168:537–544. doi: 10.1083/jcb.200410006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Featherstone C., Russell P. Nature. 1991;349:808–811. doi: 10.1038/349808a0. [DOI] [PubMed] [Google Scholar]
- 41.Russell P., Nurse P. Cell. 1986;45:145–153. doi: 10.1016/0092-8674(86)90546-5. [DOI] [PubMed] [Google Scholar]
- 42.Maftahi M., Han C. S., Langston L. D., Hope J. C., Zigouras N., Freyer G. A. Nucleic Acids Res. 1999;27:4715–4724. doi: 10.1093/nar/27.24.4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmad F., Kaplan C. D., Stewart E. Yeast. 2002;19:1381–1398. doi: 10.1002/yea.917. [DOI] [PubMed] [Google Scholar]
- 44.Baumann P., Cech T. R. Mol. Biol. Cell. 2000;11:3265–3275. doi: 10.1091/mbc.11.10.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson S., Warr N., Taylor D. L., Watts F. Z. Nucleic Acids Res. 1999;27:2655–2661. doi: 10.1093/nar/27.13.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ira G., Malkova A., Liberi G., Foiani M., Haber J. E. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.LeRoy G., Carroll R., Kyin S., Seki M., Cole M. D. Nucleic Acids Res. 2005;33:6251–6257. doi: 10.1093/nar/gki929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu L., Hickson I. D. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 49.Ray J. H., German J. Chromosoma. 1984;90:383–388. doi: 10.1007/BF00294165. [DOI] [PubMed] [Google Scholar]
- 50.Shiraishi Y., Freeman A. I., Sandberg A. A. Cytogenet. Cell Genet. 1976;17:162–173. doi: 10.1159/000130710. [DOI] [PubMed] [Google Scholar]
- 51.Bennett R. J., Keck J. L., Wang J. C. J. Mol. Biol. 1999;289:235–248. doi: 10.1006/jmbi.1999.2739. [DOI] [PubMed] [Google Scholar]
- 52.Karow J. K., Constantinou A., Li J. L., West S. C., Hickson I. D. Proc. Natl. Acad. Sci. USA. 2000;97:6504–6508. doi: 10.1073/pnas.100448097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bischof O., Kim S. H., Irving J., Beresten S., Ellis N. A., Campisi J. J. Cell Biol. 2001;153:367–380. doi: 10.1083/jcb.153.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu L., Davies S. L., Levitt N. C., Hickson I. D. J. Biol. Chem. 2001;276:19375–19381. doi: 10.1074/jbc.M009471200. [DOI] [PubMed] [Google Scholar]
- 55.Laursen L. V., Ampatzidou E., Andersen A. H., Murray J. M. Mol. Cell. Biol. 2003;23:3692–3705. doi: 10.1128/MCB.23.10.3692-3705.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pastwa E., Blasiak J. Acta Biochim. Pol. 2003;50:891–908. [PubMed] [Google Scholar]
- 57.Boulton S. J., Jackson S. P. EMBO J. 1996;15:5093–5103. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}