

Abstract
Many virulence factors secreted from pathogenic Gram-negative bacteria are autotransporter proteins. The final step of autotransporter secretion is C → N-terminal threading of the passenger domain through the outer membrane (OM), mediated by a cotranslated C-terminal porin domain. The native structure is formed only after this final secretion step, which requires neither ATP nor a proton gradient. Sequence analysis reveals that, despite size, sequence, and functional diversity among autotransporter passenger domains, >97% are predicted to form parallel β-helices, indicating this structural topology may be important for secretion. We report the folding behavior of pertactin, an autotransporter passenger domain from Bordetella pertussis. The pertactin β-helix folds reversibly in isolation, but folding is much slower than expected based on size and native-state topology. Surprisingly, pertactin is not prone to aggregation during folding, even though folding is extremely slow. Interestingly, equilibrium denaturation results in the formation of a partially folded structure, a stable core comprising the C-terminal half of the protein. Examination of the pertactin crystal structure does not reveal any obvious reason for the enhanced stability of the C terminus. In vivo, slow folding would prevent premature folding of the passenger domain in the periplasm, before OM secretion. Moreover, the extra stability of the C-terminal rungs of the β-helix might serve as a template for the formation of native protein during OM secretion; hence, vectorial folding of the β-helix could contribute to the energy-independent translocation mechanism. Coupled with the sequence analysis, the results presented here suggest a general mechanism for autotransporter secretion.
Keywords: parallel β-sheet, contact order, outer membrane protein, protein structure prediction, virulence factor
The autotransporter (AT) (also known as Type Va) secretion mechanism is deceptively simple (1). Each AT has a signal sequence that directs secretion across the cytoplasmic membrane, a nonhomologous passenger domain that forms the mature virulence factor, and a conserved C-terminal ≈30-kDa domain that forms a β-barrel pore in the outer membrane (OM) (2). OM secretion of the passenger occurs from the C to N terminus, presumably through the porin domain (1) (Fig. 1A). Alternatively, the porin domain might facilitate OM secretion of the passenger through a larger, multimeric pore (3) or an unidentified translocase (4). Regardless, many studies have indicated that the passenger domain must maintain or adopt a largely unfolded conformation to efficiently pass through the OM (5–7). Intriguingly, transport of the passenger domain in an unfolded conformation would require the passenger domain to remain in or adopt an unfolded conformation during the time required to translate, translocate, and assemble the C-terminal porin in the OM (Fig. 1A). Yet because OM secretion is ATP-independent and proton gradient-independent (1, 8), the driving force for efficient translocation is unknown. Here, we predict the structural properties of all identified AT proteins. We also examined the in vitro folding properties of the AT passenger domain of pertactin, from Bordetella pertussis, to determine how the folding of this passenger domain might be connected to maturation of the virulence factor in vivo.
Fig. 1.

Pertactin biogenesis and native structure. (A) Current models for AT secretion include passage through the IM via an N-terminal signal sequence (pink), followed by insertion of the 30-kDa C-terminal porin sequence (blue) into the OM; the passenger domain (black) then passes through the central pore of the porin [or a cluster of porins (3)] before folding to the native structure outside the cell. (B) Ribbon diagrams of the pertactin crystal structure (10). Each rung of the parallel β-helix consists of three parallel β-strands arranged in a roughly V-shaped cross section, connected by loops of varying length, producing a long parallel β-sheet on each of the three faces of the β-helix. The locations of the seven Trp residues are represented as red space-filled atoms; Ala-335 is shown in blue. (C) A cross-section view through the seven central rungs of the β-helix (residues 140–357), showing the processively wound stacking of the parallel β-strands and the variable lengths of the connecting loops. The longest connecting loop (35 residues) contains the RGD integrin binding sequence.
Pertactin is an extracellular integrin binding protein that mediates the attachment of B. pertussis to the ciliated cells of the upper respiratory system and is a major virulence factor for whooping cough (9). Passenger domain structures for pertactin (10) (Fig. 1B) and an unrelated AT, hemoglobin protease (11), have been solved and represent the two longest right-handed parallel β-helix structures solved to date. The β-helix is a growing family of all-parallel-β-sheet structures. Seventeen β-helix structures have been solved, including the structures of the secretion domain of filamentous hemagglutinin (12).
In this study, we used sequence analysis to model the structure of passenger domains from all identified ATs (>500). Despite the absence of sequence homology and/or functional similarity between many of the passenger domains, >97% are predicted to adopt a right-handed parallel β-helix structure, suggesting this structure may be important for AT biogenesis. In vitro, pertactin folds extremely slowly, perhaps because the native structure has no elements of truly local structure (i.e., α-helices). Interestingly, the equilibrium unfolding behavior of pertactin reveals that the β-helix contains a well defined, stable core at its C terminus. This partially folded equilibrium intermediate, coupled with the extremely slow folding kinetics, suggests pertactin folding after secretion across the OM may be initiated by formation of C-terminal β-helix structure, which serves as a scaffold for the folding of the remainder of the passenger domain β-helix. Given the prevalence of predicted β-helical structure in AT passenger domains, these results suggest a conserved mechanism for AT biogenesis.
Results
AT Passenger Domains Are Predicted to Form β-Helices.
AT proteins vary greatly in length, sequence, and function. Moreover, sequence features in the C-terminal porin domains resemble porins found in members of the two-partner secretion (TPS; Type Vb) protein family and YadA-like trimeric secreted proteins (Type Vc) (1, 13, 14). Despite the sequence diversity and possibly distinct secretion mechanisms, the few structures available for various Type V proteins reveal a preponderance of precessively coiled, parallel β-sheet structures, including the Type Va structures of pertactin (10) and hemoglobin protease (11), Type Vb filamentous hemagglutinin (12), and the Type Vc collagen-binding adhesin YadA (15). As a result, these proteins are sometimes lumped together as ATs, although the details of the various OM secretion mechanisms might be quite distinct. A recent search for the word “autotransporter” in the National Center for Biotechnology Information (NCBI) RefSeq nonredundant protein sequence database returned 866 proteins, of which 507 appear as bona fide Type Va ATs (see Materials and Methods). There is no sequence homology or functional similarity across the passenger domains of the AT family (1), and as expected, the 507 AT sequences varied greatly in both length (from 510 to 4,248 aa; Fig. 2) and sequence composition (209 of the sequences have <40% identity to one another).
Fig. 2.

Length distribution of AT proteins, including those predicted to form a right-handed parallel β-helix structure (filled bars) and those predicted not to form a β-helix (open bars).
To identify other Type Va ATs that may adopt a β-helical fold, the 507 identified AT sequences were analyzed by using the β-helix structure prediction program betawrappro (16). This program scores pairwise residue probabilities between adjacent β-strands in a sequence profile, allowing β-sheet signal to be found in absence of a particular local sequence repeat. By using the betawrappro bootstrap approach described in Materials and Methods, 496 of the 507 AT sequences (>97%; Fig. 2) were found to have highly significant sequence similarity to predicted β-helical passenger domains, with none of the sequences in the negative set detected. These computational results strongly suggest that all AT passenger domains adopt a right-handed parallel β-helix structure.
Elimination of the 59-aa C-Terminal Disordered Sequence Does Not Affect Pertactin Folding/Unfolding.
Most of the measurements described in this work were performed by using both the full-length pertactin passenger domain and a truncated version lacking the C-terminal 59 residues that are disordered in the pertactin crystal structure (10). These measurements confirmed that this C-terminal segment does not participate in the pertactin folding/unfolding process. For example, the full-length and truncated proteins had almost indistinguishable far-UV CD spectra and thermal denaturation behavior (see Fig. 6, which is published as supporting information on the PNAS web site).
Tryptophan (Trp) Fluorescence Emission Spectra and Chemical Denaturation Features.
The pertactin β-helix includes seven Trp residues, located in various chemical and structural environments (Fig. 1B). The change in Trp fluorescence emission intensity was used to monitor chemical denaturation with guanidine hydrochloride (Gdn·HCl) (Fig. 3A). The maximum fluorescence emission of native pertactin occurs at 335 nm. In 3 M Gdn·HCl, the pertactin emission decreases, and the maximum shifts to 350 nm, characteristic of the emission spectrum of free Trp in aqueous solution. Equivalent results were obtained for denaturation using urea (data not shown).
Fig. 3.

Pertactin Trp fluorescence. (A) Emission spectra of native (black), partly folded (blue; in 1.5 M Gdn·HCl), and denatured (red; in 3 M Gdn·HCl) pertactin, and refolded pertactin in 0.5 M Gdn·HCl after unfolding at 4 M Gdn·HCl (green). (B) Pertactin steady-state fluorescence emission at various Gdn·HCl concentrations. Unfolding samples (filled symbols) were incubated for the times shown; refolding samples (open symbols) were first unfolded in 4 M Gdn·HCl for at least 12 h before dilution to the indicated denaturant concentrations. Each time point represents a fresh aliquot from a large sample volume. (C) Time course of pertactin refolding. Pertactin was unfolded for 30 min in 4 M Gdn·HCl and diluted to 0.5 M Gdn·HCl. a.u., arbitrary units.
Pertactin equilibrium denaturation occurred as two distinct transitions, for both Gdn·HCl-induced (Fig. 3B) and thermal denaturation (Fig. 6). Three-state unfolding behavior also was observed for chemical denaturation monitored by far-UV CD (data not shown). Interestingly, after 30 min (Fig. 3B) or 2 days (data not shown) of Gdn·HCl-induced unfolding/refolding, the titration curves did not overlay at all, indicating the samples had not yet reached equilibrium. The most significant differences were seen in the region from 1.0 to 2.0 M Gdn·HCl: formation of the partially folded structure, which has an emission maximum at 340 nm, was extremely slow. Likewise, formation of native pertactin in 0.5 M Gdn·HCl was extremely slow (Fig. 3C), with equilibrium reached only after 9 days of refolding (Fig. 3B). Dissolution of pertactin structure in higher concentrations of Gdn·HCl (2.0–2.7 M) was also very slow, suggesting the residual structure in the partially folded state is extremely stable and resistant to denaturation.
Upon equilibration, unfolding and folding titrations did overlay, and emission spectra collected for native and refolded pertactin were indistinguishable (Fig. 3 A and B), confirming that the unfolding/refolding process is fully reversible. Likewise, no evidence was found for aggregation or other intermolecular associations: High-speed centrifugation of titration samples revealed no pelletable material, and static light-scattering measurements confirmed the molecular mass of the partially folded structure is ≈60 kDa (M.C.F., A. F. Palmer, and P.L.C., unpublished results), as expected for monomeric pertactin.
Characterization of the Partially Folded State with Limited Proteolytic Digestion.
The fluorescence emission intensity of the partially folded state was roughly halfway between the intensities of native and denatured pertactin (Fig. 3B). This observation suggests two extreme models for the residual structure present in the partially folded state: (i) a high level of residual structure concentrated within a subset of rungs; i.e., half of the protein was folded, or (ii) residual structure evenly distributed along the β-helix; i.e., the entire protein was half-folded. Other conformations, incorporating elements from both of these models, and/or including nonnative structure, also would be consistent with the equilibrium denaturation results. Limited proteolytic digestion was used to distinguish between these models and highlight the location of ordered structure within the partially folded state.
Proteinase K (proK) digestion of native pertactin was slow; after 20 min, a significant amount of full-length, undigested pertactin remained (Fig. 4A). At later time points, the band corresponding to full-length pertactin disappeared and was replaced with two bands with apparent molecular masses of 27 and 28 kDa; no bands corresponding to smaller fragments were detected. Together, these two fragments add up to 55 kDa, equivalent to the size of full-length pertactin. Conversely, digestion of the partially folded state populated at 1.5 M Gdn·HCl resulted in the relatively rapid disappearance of full-length pertactin and the appearance of a stable fragment of ≈21 kDa; this fragment remained detectable for at least 24 h (Fig. 4B). During the digestion of the partially folded state, several bands corresponding to fragments of discrete sizes between 21 and 55 kDa also appeared, but no fragments <21 kDa were detected. The protease resistance of the 21-kDa fragment result strongly supports the retention of significant structure within approximately one-half of the pertactin sequence (model i).
Fig. 4.

Characterization of the pertactin partially folded state. ProK digestion patterns of native pertactin (A) and pertactin equilibrated in 1.5 M Gdn·HCl (B), resolved by SDS/PAGE and silver staining. In both experiments, the pertactin:proK ratio was 2,000:1. Digestion was stopped by boiling for 10 min. As a control for autoproteolysis, proK alone was incubated for 50 min (lane proK). Molecular mass standards are indicated in kDa.
Location of the Partially Folded Stable Structure Within the Pertactin Sequence.
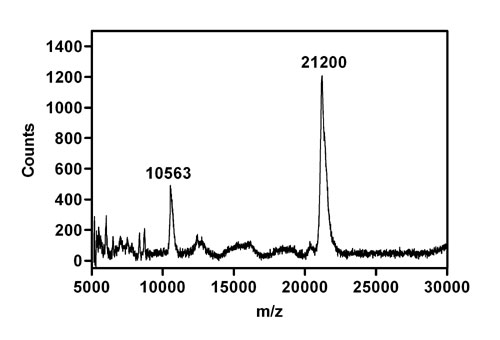
To identify the boundaries of the ordered structure in the partially folded state, the proK-resistant fragment (Fig. 4B) was subjected to further analysis. The fragment was first eluted from the polyacrylamide gel and subjected to mass analysis, confirming the preservation of an intact fragment with a mass of 21.2 kDa (see Fig. 7, which is published as supporting information on the PNAS web site). Given that proK is a nonspecific protease, the width of the mass envelope is surprisingly narrow.
To map the sequence of the 21-kDa fragment onto the sequence of full-length pertactin, the recovered fragment also was subjected to complete digestion with trypsin; this process yielded numerous smaller fragments of defined sizes. Mass analysis of the tryptic peptides (Fig. 5A) revealed seven unambiguous assignments to the pertactin sequence, spanning a 16.4-kDa segment within the C-terminal half of the β-helix (Fig. 5B).
Fig. 5.

Mass analysis of the pertactin 21-kDa stable core. (A) MALDI-TOF mass analysis of tryptic peptides from the pertactin 21-kDa stable core fragment. Peaks corresponding to identified tryptic peptides are labeled. No fragments >4,000 m/z were detected. (B) Locations of the tryptic peptides in the full-length pertactin sequence. All labeled peaks deviate less than ±2 Da from the calculated size. Residue numbering is taken from numbering in the PDB file (PDB ID code 1DAB). The upper bar shows the complete preprotein sequence from B. pertussis, including the N-terminal signal sequence, the flexible proline-rich repeat (PRR) at the C terminus of β-helix, and the C-terminal β-porin domain. The middle bar represents the mature β-helix domain used in this study, with labeled tryptic peptides magnified in the lower bar. Symbols correspond to labeled peaks in A.
To determine the precise N- and C-terminal boundaries of the stable core, the 21-kDa proK-resistant fragment also was subjected to N-terminal Edman degradation and cleavage by cyanogen bromide (CNBr) followed by mass spectrometry. N-terminal sequencing yielded in a sequence of eight residues, A-Q-G-K-A-L-L-Y, corresponding to the pertactin sequence from Ala-335 to Tyr-342. CNBr cleavage produced four fragments of the expected sizes, given the three Met residues within the 21-kDa region from Ala-335 to Pro-539 (see Fig. 8, which is published as supporting information on the PNAS web site). Results from the mass analyses and Edman degradation are consistent with the assignment of the pertactin stable structural core as the residues from the C-terminal six rungs of the native β-helix structure.
Discussion
The AT secretion mechanism represents the most common mechanism for the secretion of virulence factors from pathogenic Gram-negative bacteria (1). The secretion requirements, all contained within a single polypeptide chain, are astonishingly simple, in direct contrast to other mechanisms for OM secretion, which can require as many as 14 accessory proteins (7). Yet despite this simplicity, much remains unknown regarding the details of AT secretion.
Three mechanisms have been proposed for the transport of an AT passenger domain across the OM (1, 7). (i) The first is threading of the unfolded passenger through a pore formed by a single C-terminal porin domain. In support of this mechanism, the recent structure determination of an AT porin domain revealed the dimensions of the pore (1.0–1.3 nm) would require the passenger maintain or adopt a largely unfolded conformation before passage through the OM. In addition, the N terminus of the crystallized AT porin (immediately C-terminal to the passenger) is inserted into the center of the porin, perhaps serving to plug the central pore after secretion of the passenger (2). (ii) Alternatively, a larger pore might be created by clustering several (8–10) C-terminal porin domains, as has been observed in vitro for the IgA protease porin domain (3). This cluster could produce a larger (≈2 nm) pore, yet one still too small to allow passage of natively folded β-helical structures [for example, native pertactin has a diameter of 3–4 nm (10)]. In both mechanisms i and ii, it is predicted that secretion of the passenger proceeds from C to N terminus, consistent with studies using truncated passenger domains (6, 17). (iii) OM secretion could alternatively proceed from N to C terminus, using an as-yet-unidentified transporter, but this mechanism is inconsistent with passenger domain truncation studies.
Intriguingly, all three AT OM secretion mechanisms require the passenger domain to maintain or adopt a largely unfolded conformation in the periplasm. Periplasmic chaperones have been implicated in the proper assembly of porins in the OM (18, 19), and the protease DegP is involved in the proper localization of the AT IcsA to the cell poles (20), but no evidence exists for chaperone-mediated unfolding in the periplasm, and the scarcity of ATP outside the inner membrane (IM) makes it unclear how a canonical ATPase-driven molecular chaperone or unfoldase (akin to Clp/Hsp100) would work in this environment. Alternatively, a subset of ATs have unusually long N-terminal signal sequences (1), and a recent report suggested the additional length may play a role in preventing misfolding of this passenger domain in the periplasm, while it awaits secretion across the OM (21). Yet the extremely slow folding of the isolated pertactin passenger domain suggests that, because the β-helix folds very slowly, it might not require additional cellular factors to remain unfolded in the periplasm during secretion and might explain why only a subset of ATs have unusually long signal sequences.
The extremely slow folding of the pertactin β-helix stands in stark contrast to the microsecond-to-second folding rates reported for the majority of protein-folding studies. However, most folding studies have focused on the folding of small (often <100 aa), α-helix-rich single-domain proteins. Fast folding can play a crucial role in suppressing off-pathway aggregation by minimizing the accumulation of partially folded, aggregation-prone folding intermediates (22, 23). Yet some proteins that are rich in β-structure fold orders of magnitude more slowly than typical folding models (24, 25). In the case of pertactin (and by extension, other β-helical AT proteins), slow folding may confer a functional advantage, allowing the protein to remain in a secretion-competent conformation during IM secretion and OM assembly of the C-terminal porin. Nevertheless, it remains to be determined how pertactin avoids aggregation during β-sheet folding, particularly given the slow folding rate.
Although the results reported here do not include detailed kinetic measurements, it is clear that the pertactin in vitro refolding rate is much slower than the doubling time for Gram-negative bacteria. Of course, a purified refolding system does not include all components present in vivo; moreover, another major difference in vivo is the vectorial appearance of the chain into various folding environments. During IM and OM secretion, the pertactin polypeptide chain appears first (for IM secretion) from N → C terminus and, finally (for OM secretion), from C → N terminus. In general, vectorial folding allows a polypeptide chain to explore conformational space in a fundamentally different way than when the entire chain folds at once upon dilution from denaturant (26). In addition, IM secretion (from N → C) is fundamentally different from OM secretion (from C → N) and could result in dramatically different folding kinetics. During OM secretion, the C terminus of the β-helix emerges first into the extracellular space, and equilibrium denaturation of the pertactin β-helix demonstrated that the C-terminal rungs are considerably more stable than the N-terminal rungs. From the pertactin crystal structure, it is not at all clear what stabilizes the C-terminal rungs of the β-helix vs. the N-terminal rungs. Yet a functional role is suggested: the C-terminal rungs might form a template for the efficient formation of the native β-helix. At the moment, however, it is unknown whether the stable structure formed by the C-terminal rungs is indeed β-helical; alternatively, nonnative could be formed, which might serve as a folding template.
Regardless of the structure of the C-terminal rungs, an additional consideration for any proposed OM secretion mechanism is the driving force for efficient secretion. In the absence of external energy sources, our results suggest that the free energy released by the C- to N-terminal vectorial folding of the passenger domain after OM secretion might be sufficient to drive secretion or at least to prevent “backsliding” of the passenger domain into the periplasm.
Previous sequence comparisons and deletion studies have identified a conserved junction sequence in the extreme C terminus of some AT passenger domains that is important for efficient maturation (i.e., secretion and/or folding) of some passenger domains (6, 17, 27). In pertactin, this sequence corresponds to the C-terminal two rungs of the β-helix (6). Results presented here suggest that the C-terminal passenger sequence that is important for pertactin folding extends further into the β-helix, beyond the observable sequence conservation, and may therefore be present in other ATs that lack a detectable junction sequence.
It is important to note that a few studies have shown that replacement of an endogenous passenger domain sequence with a heterologous sequence (i.e., a native structure that is not β-helical) does not abolish AT secretion across the OM (28, 29). The effects of premature folding of a heterologous passenger in the periplasm are conflicting (4, 28, 30–32), but in at least one case, secretion of a heterologous passenger domain was not impaired by disulfide bond formation in the periplasm (28). Nevertheless, it is important to note that the heterologous passenger chosen for this study consisted of small (≈2-nm diameter), relatively low-contact-order Ig domains. From the results presented here, we propose that replacement of the β-helical passenger domain with a similarly sized, higher-contact-order structure will result in less efficient secretion than the wild-type β-helical passenger, as will disulfide bonds within the β-helix.
Intriguingly, the finding that most AT proteins are β-helical offers a solution to the conundrum of how to achieve a variety of functions while preserving a common topology important for efficient secretion and folding. Mutational (33, 34) and sequence analyses (35) have shown that the β-helix topology is highly tolerant of sequence variations. In addition, because the β-helix is a processive fold, larger proteins (or larger active sites) can be accommodated simply by increasing the number of rungs or extending one or more loops to form a functional domain [as seen for hemoglobin protease (11)], while still preserving the β-helix core.
In conclusion, we propose that the conserved β-helical structure of the AT passenger domains may represent a fold that is conserved, not for reasons related to the ultimate function of these proteins, but rather to preserve a folding pathway required for specific, efficient OM secretion and folding. This mechanism also may extend to Type Vb and Vc secretion mechanisms; the Type Vb protein filamentous hemagglutinin is known to contain β-helical structure (12), and OM secretion of this protein requires only the expression of a separately expressed 30-kDa β-porin (36).
Materials and Methods
Sequence Analysis and Structure Prediction.
A search for the term “autotransporter” in the National Center for Biotechnology Information (NCBI) RefSeq database of nonredundant protein products (www.ncbi.nlm.nih.gov/projects/RefSeq) returned 866 sequences (August 12, 2005). These sequences were identified primarily by conserved features in the C-terminal porin domain; hence, there was no initial bias toward selecting sequences with a specific passenger domain feature. Of the 866 sequences, 507 met the criteria for Type Va ATs, i.e., included a porin domain [pfam (37) classification Autotransporter; E < 0.0001] no more than 100 residues from the C terminus and at least 250 aa from the N terminus (to accommodate a signal peptide and a minimal passenger domain) and did not include other related protein classifications such as YadA-like (15) trimeric left-handed β-helices/rolls (pfam classification Hep_Hag).
A bootstrap approach using betawrappro (16) and psi-blast (38) was used to confidently detect AT proteins with a β-helical passenger domain. betawrappro uses pairwise residue correlations between adjacent β-sheets in the Protein Data Bank (PDB) to detect the β-helix fold in a sequence profile, even in the absence of detectable sequence similarity to known structures. The test set was comprised of the 507 AT sequences, plus 507 non-β-helical sequences randomly selected from a nonredundant version of the PDB (August 4, 2005) (negative set). betawrappro was run on this test set, and the sequences scoring better than the solved β-helical AT structures (P < 0.0003; 31 sequences) were used to create an initial database of putative β-helical passenger domains. An iterative method then was applied: in each round, psi-blast was run for four iterations to align the remaining test set sequences against the putative positive database. The test sequences that aligned with a predicted β-helix region in the putative positive database with a psi-blast bit score of at least 200 were added to the putative positive database. These psi-blast rounds were repeated until no new sequences were found (four rounds total). By querying the remaining test sequences against the database of putative β-helices, this bootstrap approach in effect searches for a match between each test sequence and a profile from the database, leading to higher sensitivity for sequences with several matches to the putative β-helices in the set.
Because the initial putative positive database was created with betawrappro predictions, it contains only sequences with a very high probability of forming the β-helix fold but that also represent a wide range of sequence diversity. From this starting point, psi-blast found other sequences with similarity to the initial predictions. In each round, the positive database was expanded, which allows more distant matches to be found.
Molecular Biology.
Escherichia coli expression plasmid pPERtac36, encoding P.69 pertactin, was a generous gift from N. Isaacs (University of Glasgow, Glasgow, Scotland). Upon sequencing the pertactin gene, two point mutations were discovered: D228E and P298S. The P298S mutation appears in almost all clinical isolates (for example, ref. 39); the conservative D228E mutation is in the RGD integrin binding sequence and has been used to modulate pertactin function (40). Neither mutation significantly affects pertactin folding or structural properties (N. Isaacs, personal communication; data not shown). Pertactin expression plasmid pPERPLC01, incorporating a stop codon after residue 539 (numbering from PDB ID code 1DAB), eliminated the C-terminal disordered residues (10) and was constructed by using pPERtac36 and the QuikChange site-directed mutagenesis procedure (Stratagene).
Protein Expression and Purification.
Pertactin was overexpressed in E. coli and purified from insoluble inclusion bodies by using standard methods. A detailed description is available in Supporting Text, which is published as supporting information on the PNAS web site.
Fluorescence Spectroscopy.
A QM-6 fluorimeter (PTI, Birmingham, NJ) was used for all fluorescence measurements. Samples were measured at 20°C, by using 0.5 μM pertactin in 50 mM Tris (pH 8.8) (plus various concentrations of Gdn·HCl) in a 10-mm cuvette. All data were collected at an excitation wavelength of 280 nm, with an integration time of 1 s. For equilibrium time course experiments, lamp-related degradation of the Trp chromophores was avoided by measuring, at each time point, the fluorescence intensity of a fresh aliquot from a large sample volume.
Proteolytic Digestion.
Pertactin was digested with proK (2,000:1 pertactin:protease) at room temperature in 50 mM Tris (pH 8.8) with or without 1.5 M Gdn·HCl, supplemented with 7.5 mM CaCl2. Proteolysis was stopped by boiling samples for 10 min. Digestion fragments were resolved by SDS/PAGE using 14% polyacrylamide gels. The 21-kDa pertactin fragment was eluted from the gel by using the Insite system (National Diagnostics). Protein bands were stained with a fluorescence dye present in the SDS/PAGE running buffer. Bands were visualized by UV illumination, and the 21-kDa fragment was eluted by incubating excised gel fragments overnight in 25 mM ammonium bicarbonate. Trypsin digestion of the 21-kDa fragment was performed overnight at 37°C.
Mass and Sequencing Analysis of Pertactin Digestion and Cleavage Fragments.
For MALDI-TOF analysis, samples were diluted in acetonitrile, by using α-cyano-4-hydroxycinnamic acid (for sizes < 10 kDa) or 3,5-dimethoxy-4-hydroxycinnamic acid (for larger fragments) as a matrix. MALDI mass spectra were acquired on a Voyager-DE TOF mass spectrometer (PerSeptive Biosystems, Framingham, MA). Typically, 50–150 laser shots were averaged. TOF to mass conversion was achieved by external calibration by using mass spectral peaks detected for purchased standards. Eight rounds of Edman degradation were performed on an Applied Biosystems Procise 494 cLC at the Keck Foundation Biotechnology Resource Laboratory at Yale University.
Supplementary Material
Acknowledgments
We thank Neil Isaacs for providing pPERtac36; Bill Boggess for performing tryptic digests and assistance with the mass spectrometer; Katie O’Sullivan for assistance with fluorescence measurements; and Seth Brown, Brian Baker, Doug Barrick, Jonathan King, Kevin Plaxco, Michael Brudno, and members of the P.L.C. laboratory for critical discussions and reading of this manuscript. This project was supported by National Science Foundation CAREER Award MCB-0237934 and American Heart Association Scientist Development Grant 0330151N. P.L.C. received an award from the Clare Boothe Luce Program of the Henry Luce Foundation. M.J. was supported in part by an award from the Deutscher Akademischer Austauschdienst.
Abbreviations
- AT
autotransporter
- Gdn·HCl
guanidine hydrochloride
- IM
inner membrane
- OM
outer membrane
- PDB
Protein Data Bank
- proK
proteinase K.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Henderson I. R., Navarro-Garcia F., Desvaux M., Fernandez R. C., Ala’Aldeen D. Microbiol. Mol. Biol. Rev. 2004;68:692–744. doi: 10.1128/MMBR.68.4.692-744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oomen C. J., Van Ulsen P., Van Gelder P., Feijen M., Tommassen J., Gros P. EMBO J. 2004;23:1257–1266. doi: 10.1038/sj.emboj.7600148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veiga E., Sugawara E., Nikaido H., de Lorenzo V., Fernandez L. A. EMBO J. 2002;21:2122–2131. doi: 10.1093/emboj/21.9.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skillman K. M., Barnard T. J., Peterson J. H., Ghirlando R., Bernstein H. D. Mol. Microbiol. 2005;58:945–958. doi: 10.1111/j.1365-2958.2005.04885.x. [DOI] [PubMed] [Google Scholar]
- 5.Klauser T., Pohlner J., Meyer T. F. EMBO J. 1990;9:1991–1999. doi: 10.1002/j.1460-2075.1990.tb08327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oliver D. C., Huang G., Nodel E., Pleasance S., Fernandez R. C. Mol. Microbiol. 2003;47:1367–1383. doi: 10.1046/j.1365-2958.2003.03377.x. [DOI] [PubMed] [Google Scholar]
- 7.Henderson I. R., Navarro-Garcia F., Nataro J. P. Trends Microbiol. 1998;6:370–378. doi: 10.1016/s0966-842x(98)01318-3. [DOI] [PubMed] [Google Scholar]
- 8.Thanassi D. G., Stathopoulos C., Karkal A., Li H. Mol. Membr. Biol. 2005;22:63–72. doi: 10.1080/09687860500063290. [DOI] [PubMed] [Google Scholar]
- 9.Smith A. M., Guzman C. A., Walker M. J. FEMS Microbiol. Rev. 2001;25:309–333. doi: 10.1111/j.1574-6976.2001.tb00580.x. [DOI] [PubMed] [Google Scholar]
- 10.Emsley P., Charles I. G., Fairweather N. F., Isaacs N. W. Nature. 1996;381:90–92. doi: 10.1038/381090a0. [DOI] [PubMed] [Google Scholar]
- 11.Otto B. R., Sijbrandi R., Luirink J., Oudega B., Heddle J. G., Mizutani K., Park S. Y., Tame J. R. J. Biol. Chem. 2005;280:17339–17345. doi: 10.1074/jbc.M412885200. [DOI] [PubMed] [Google Scholar]
- 12.Clantin B., Hodak H., Willery E., Locht C., Jacob-Dubuisson F., Villeret V. Proc. Natl. Acad. Sci. USA. 2004;101:6194–6199. doi: 10.1073/pnas.0400291101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yen M. R., Peabody C. R., Partovi S. M., Zhai Y., Tseng Y. H., Saier M. H. Biochim. Biophys. Acta. 2002;1562:6–31. doi: 10.1016/s0005-2736(02)00359-0. [DOI] [PubMed] [Google Scholar]
- 14.Loveless B. J., Saier M. H., Jr. Mol. Membr. Biol. 1997;14:113–123. doi: 10.3109/09687689709048171. [DOI] [PubMed] [Google Scholar]
- 15.Nummelin H., Merckel M. C., Leo J. C., Lankinen H., Skurnik M., Goldman A. EMBO J. 2004;23:701–711. doi: 10.1038/sj.emboj.7600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonnell A. V., Palmer N., Menke M., King J., Cowen L., Berger B. Proteins Struct. Funct. Bioinform. in press. [Google Scholar]
- 17.Velarde J. J., Nataro J. P. J. Biol. Chem. 2004;279:31495–31504. doi: 10.1074/jbc.M404424200. [DOI] [PubMed] [Google Scholar]
- 18.Gentle I., Gabriel K., Beech P., Waller R., Lithgow T. J. Cell Biol. 2004;164:19–24. doi: 10.1083/jcb.200310092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bulieris P. V., Behrens S., Holst O., Kleinschmidt J. H. J. Biol. Chem. 2003;278:9092–9099. doi: 10.1074/jbc.M211177200. [DOI] [PubMed] [Google Scholar]
- 20.Purdy G. E., Hong M., Payne S. M. Infect. Immun. 2002;70:6355–6364. doi: 10.1128/IAI.70.11.6355-6364.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szabady R. L., Peterson J. H., Skillman K. M., Bernstein H. D. Proc. Natl. Acad. Sci. USA. 2005;102:221–226. doi: 10.1073/pnas.0406055102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wetzel R. Cell. 1996;86:699–702. doi: 10.1016/s0092-8674(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 23.Engelhard M., Evans P. A. Protein Sci. 1995;4:1553–1562. doi: 10.1002/pro.5560040813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roumestand C., Boyer M., Guignard L., Barthe P., Royer C. A. J. Mol. Biol. 2001;312:247–259. doi: 10.1006/jmbi.2001.4928. [DOI] [PubMed] [Google Scholar]
- 25.Burns L. L., Ropson I. J. Proteins Struct. Funct. Genet. 2001;43:292–302. doi: 10.1002/prot.1040. [DOI] [PubMed] [Google Scholar]
- 26.Clark P. L. Trends Biochem. Sci. 2004;29:527–534. doi: 10.1016/j.tibs.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 27.Ohnishi Y., Nishiyama M., Horinouchi S., Beppu T. J. Biol. Chem. 1994;269:32800–32806. [PubMed] [Google Scholar]
- 28.Veiga E., de Lorenzo V., Fernandez L. A. Mol. Microbiol. 2004;52:1069–1080. doi: 10.1111/j.1365-2958.2004.04014.x. [DOI] [PubMed] [Google Scholar]
- 29.Klauser T., Pohlner J., Meyer T. F. EMBO J. 1992;11:2327–2335. doi: 10.1002/j.1460-2075.1992.tb05292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brandon L. D., Goldberg M. B. J. Bacteriol. 2001;183:951–958. doi: 10.1128/JB.183.3.951-958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jose J., Kramer J., Klauser T., Pohlner J., Meyer T. F. Gene. 1996;178:107–110. doi: 10.1016/0378-1119(96)00343-5. [DOI] [PubMed] [Google Scholar]
- 32.Veiga E., de Lorenzo V., Fernandez L. A. Mol. Microbiol. 1999;33:1232–1243. doi: 10.1046/j.1365-2958.1999.01571.x. [DOI] [PubMed] [Google Scholar]
- 33.Haase-Pettingell C., King J. J. Mol. Biol. 1997;267:88–102. doi: 10.1006/jmbi.1996.0841. [DOI] [PubMed] [Google Scholar]
- 34.Schuler B., Furst F., Osterroth F., Steinbacher S., Huber R., Seckler R. Proteins Struct. Funct. Genet. 2000;39:89–101. [PubMed] [Google Scholar]
- 35.Kajava A. V., Cheng N., Cleaver R., Kessel M., Simon M. N., Willery E., Jacob-Dubuisson F., Locht C., Steven A. C. Mol. Microbiol. 2001;42:279–292. doi: 10.1046/j.1365-2958.2001.02598.x. [DOI] [PubMed] [Google Scholar]
- 36.Newman C. L., Stathopoulos C. 2004;30:275–286. doi: 10.1080/10408410490499872. [DOI] [PubMed] [Google Scholar]
- 37.Bateman A., Coin L., Durbin R., Finn R. D., Hollich V., Griffiths-Jones S., Khanna A., Marshall M., Moxon S., Sonnhammer E. L., et al. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul S. F., Madden T. L., Schaffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parkhill J., Sebaihia M., Preston A., Murphy L. D., Thomson N., Harris D. E., Holden M. T., Churcher C. M., Bentley S. D., Mungall K. L., et al. Nat. Genet. 2003;35:32–40. doi: 10.1038/ng1227. [DOI] [PubMed] [Google Scholar]
- 40.Everest P., Li J., Douce G., Charles I., De Azavedo J., Chatfield S., Dougan G., Roberts M. Microbiology. 1996;142:3261–3268. doi: 10.1099/13500872-142-11-3261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}