

Abstract
Several major insecticides, including α-endosulfan, lindane, and fipronil, and the botanical picrotoxinin are noncompetitive antagonists (NCAs) for the GABA receptor. We showed earlier that human β3 homopentameric GABAA receptor recognizes all of the important GABAergic insecticides and reproduces the high insecticide sensitivity and structure-activity relationships of the native insect receptor. Despite large structural diversity, the NCAs are proposed to fit a single binding site in the chloride channel lumen lined by five transmembrane 2 segments. This hypothesis is examined with the β3 homopentamer by mutagenesis, pore structure studies, NCA binding, and molecular modeling. The 15 amino acids in the cytoplasmic half of the pore were mutated to cysteine, serine, or other residue for 22 mutants overall. Localization of A-1′C, A2′C, T6′C, and L9′C (index numbers for the transmembrane 2 region) in the channel lumen was established by disulfide cross-linking. Binding of two NCA radioligands [3H]1-(4-ethynylphenyl)-4-n-propyl-2,6,7-trioxabicyclo[2.2.2]octane and [3H] 3,3-bis-trifluoromethyl-bicyclo[2,2,1]heptane-2,2-dicarbonitrile was dramatically reduced with 8 of the 15 mutated positions, focusing attention on A2′, T6′, and L9′ as proposed binding sites, consistent with earlier mutagenesis studies. The cytoplasmic half of the β3 homopentamer pore was modeled as an α-helix. The six NCAs listed above plus t-butylbicyclophosphorothionate fit the 2′ to 9′ pore region forming hydrogen bonds with the T6′ hydroxyl and hydrophobic interactions with A2′, T6′, and L9′ alkyl substituents, thereby blocking the channel. Thus, widely diverse NCA structures fit the same GABA receptor β subunit site with important implications for insecticide cross-resistance and selective toxicity between insects and mammals.
Keywords: β3 homopentamer, transmembrane 2, insecticide, disulfide trapping, receptor model
Pest insect control in the past 60 years was achieved, in part, by application of >3 billion (3 × 109) pounds of polychlorocycloalkane insecticides, including cyclodienes (e.g., α-endosulfan and dieldrin), lindane and its isomers, and others, which are now highly restricted or banned except for endosulfan and some uses of lindane (1–3). One of the replacement compounds is the phenylpyrazole fipronil. All of these insecticides and the botanical picrotoxinin (PTX) have widely diverse chemical structures but appear to act at the same nerve target. It is therefore important to understand how these compounds work in mammals and insects, or how they do not work when resistant insect strains appear.
The GABA-gated chloride channel is the target for the insecticides and toxicants referred to above based on radioligand binding and electrophysiology studies (3–10). Important radioligands in these developments are [3H]dihydroPTX (4, 11), [35S]t-butylbicyclophosphorothionate (TBPS) (5, 12), [3H]1-(4-ethynylphenyl)-4-n-propyl-2,6,7-trioxabicyclo[2.2.2]octane (EBOB) (6), and [3H]3,3-bis-trifluoromethyl-bicyclo[2,2,1]heptane-2,2-dicarbonitrile (BIDN) (8) (Fig. 1A). All of these compounds act in mammals and insects as noncompetitive antagonists (NCAs) to block chloride flux so the target is referred to as the GABA receptor NCA-binding site. Vertebrate GABA receptors consist of α, β, γ, ρ, and other subunits in various combinations, for example, α1β2γ2 as a heteropentamer and ρ1 as a homopentamer (13–15). The molecular localization of the NCA site defined here (Fig. 1B) was first indicated by mutagenesis studies (16) as A2′ (17–20), T6′ (21, 22), and L9′ (23, 24) in the cytoplasmic half of the transmembrane 2 domain of the channel (Fig. 2). Drosophila resistant to dieldrin (RDL) have a mutation conferring GABA receptor insensitivity identified as A2′S (17). The NCA target of the GABAA receptor requires a β subunit, and a β3 homopentamer is sufficient for binding (9, 26). Importantly, the β3 subunit from human brain, when expressed in insect Sf9 cells, assembles to form a receptor sensitive to all of the important GABAergic insecticides (9) and, surprisingly, reproduces the insecticide sensitivity and structure-activity relationships of the native insect receptor (27). Studies of the GABA receptor NCA site are therefore simplified by using this highly sensitive β3 homopentamer, an approach verified by showing here that Cys and Ser or Phe mutations in β3 at each of the 2′, 6′, and 9′ positions greatly reduce or destroy NCA radioligand binding.
Fig. 1.

Structure-activity relationships of seven GABA receptor noncompetitive antagonists. (A) Structures of three important insecticides (lindane, fipronil, and α-endosulfan) and four radioligands (asterisk designates labeling position). The high potencies of each compound with the β3 homopentamer are indicated by the 2.7 nM Kd for [3H]EBOB on direct binding and 0.47–59 nM IC50 values for the other compounds in displacing [3H]EBOB binding (9). (B) Models of four antagonists positioned as in A showing their proposed β3 homopentamer M2 binding sites in the channel lumen. A, L, and T refer to the side chains of the interacting 2′, 6′, and 9′ residues, respectively.
Fig. 2.

Alignment of the cytoplasmic half of the M2 and flanking sequences of various GABA receptor subunits. The species are human or rat for α, β, and γ, rat for ρ, and Drosophila for WT and RDL. Index numbers for positioning in M2 (25) are shown at the top. The β3 homopentamer region studied here is shown in a box with the channel lumen residues defined in the present investigation by disulfide cross-linking in bold type (−1′, 2′, 6′, and 9′). The resistance-associated RDL mutation (A2′S) in Drosophila (17) is underlined.
This study tested the hypothesis that insecticides and convulsants of many chemical types act at the same GABA receptor site in the same way to initiate insecticidal action and mammalian toxicity. The goal was to characterize the GABA receptor–NCA interaction by using the human GABAA receptor recombinant β3 homopentamer as a model. The first step was to prepare Cys and other mutations to scan the cytoplasmic half of M2 and the flanking region (−4′ to 10′), overall 22 mutants involving 15 positions. The mutants were used to identify Cys residues undergoing disulfide cross-linking as a guide to channel pore structure (28). Next, [3H]EBOB and [3H]BIDN were used to identify positions where mutation altered binding (6, 8). Finally, modeling of the NCA-binding domain (29, 30) was applied to the β3 homopentamer to determine whether the wide diversity of NCAs could fit the same site.
Results
Mutagenesis and Protein Expression.
The transfection efficiency of each recombinant baculovirus was examined by PCR analysis. The nonrecombinant virus would give one 839-bp band of its polyhedrin region and the recombinant virus incorporating the 1,425-bp β3 cDNA would appear at 2.3 kb. Each extracted recombinant virus gave only one 2.3-kb band (Fig. 3A), indicating a recombination efficiency for the target gene of nearly 100% for all mutants and the WT. Further, all PCR products from virus extraction were sequenced and confirmed as the right mutations.
Fig. 3.

Baculovirus transfection efficiencies and protein expression levels of WT (S-3′S) and mutant β3 subunits. (A) PCR analysis of recombinant efficiency. (B) SDS/PAGE-Western blotting analysis of protein expression level. VWT refers to membrane transfected with WT baculovirus. Samples were treated with 10 mM DTT in sample buffer.
The expression levels of the WT and mutant β3 subunits were determined by Western blotting. The monoclonal anti-β-chain antibody recognized a very specific band at ≈55 kDa with similar intensity for membrane extracts of the WT and each mutant (Fig. 3B). Equal protein transfer levels were determined by Ponceau S staining. As exceptions, two mutants (L3′C and L3′F) were not expressed.
Disulfide Cross-Linking Profiles.
Oxidation of the Cys mutants with copper sulfate:1,10-phenanthroline (Cu:phen) resulted in four cases of a molecular mass increase from 55 kDa to ≈130 kDa for the monomer and dimer, respectively, as detected by SDS/PAGE and immunoblotting (Fig. 4). Cys substituents at −1′, 2′ (weak), 6′, and 9′ formed disulfide-linked dimers in the presence but not in the absence of Cu:phen. Only trace amounts of the −1′ and 9′ monomers are left with Cu:phen indicating more extensive reaction possibly due to higher flexibility at these positions. Dimers were not detected under the same conditions for Cys substituents at 0′, 1′, 4′, 5′, 7′, 8′, and 10′, although in some cases there were apparent losses in receptor levels on oxidation. Disulfides at −1′, 2′, and 9′ were completely reversed with DTT, but the one at 6′ was only partially reversed.
Fig. 4.

Disulfide cross-linking profiles. Samples are control without Cu:phen or DTT (–/–), oxidized with Cu:phen but not treated with DTT (+/–), or oxidized with Cu:phen then reduced with 10 mM DTT (+/+). Reactions were terminated with 10 mM N-ethylmaleimide before SDS/PAGE-Western blotting analysis.
Effect of Site-Specific Mutations on [3H]EBOB and [3H]BIDN Binding.
Membranes (100 μg of protein) from the WT were assayed with [3H]EBOB (1 nM) or [3H]BIDN (2.5 nM) by using incubations for 90 min at 25°C. Specific binding (n = 10) was 2,458 ± 250 dpm for [3H]EBOB and 1,253 ± 100 dpm for [3H]BIDN with nonspecific binding of 496 ± 45 and 225 ± 16 dpm, respectively, i.e., 83–85% specific relative to total binding. Using the same conditions and amounts of receptors, the mutants were then compared to the WT for both [3H]EBOB and [3H]BIDN binding. The binding activities of A-4′S, T7′C and T10′C were similar to the WT, whereas A-2′S and V1′C gave reduced binding (Fig. 5). All of the rest gave little or no specific binding. It was indeed surprising to find that the low binding for mutants involves the whole segment from A2′ to I5′, in addition to the expected T6′ to L9′, with the two exceptions of L3′ not expressed and T7′ normal. More generally, mutations in the lowest region of the channel have no (−4′ or −3′) or little (−2′) influence on activity, whereas those in the region of −1′ to 10′, except 1′, 7′, and 10′, drastically reduce [3H]EBOB and [3H]BIDN binding. This reduction is not due to interference from oxidation of the Cys moiety because (i) DTT did not restore the activity of the eight low-binding mutants (data not shown), and (ii) the findings are essentially the same with Ser (0′, 2′, and 9′), Leu (5′), Phe (6′), and Ala (4′ and 8′) as well as for the corresponding Cys mutants. It is assumed that the mutants, which do not bind NCAs, form functional channels that are correctly assembled on the cell surface because, on the Western blot, they all have a protein of similar size and presumably maturely glycosylated. Most importantly, on an overall basis, the results are essentially the same with [3H]EBOB and [3H]BIDN.
Fig. 5.

Effect of site-specific mutations (Cys, Ser, Ala, Leu, or Phe) on specific binding of [3H]EBOB and [3H]BIDN. NE, not expressed. Data are percent of WT (S-3′S) ± SD.
Two methanethiosulfonate (MTS) sulfhydryl-modification reagents provided further information on the NCA site by comparing their effect on [3H]EBOB binding for the Cys active mutants 1′, 7′, and 10′ compared with the WT. With both sulfhydryl reagents, there was a site-dependent effect on [3H]EBOB binding with little inhibition for the T7′C mutant, moderate for T10′C, and almost complete for V1′C.
Structural Model for NCA Binding.
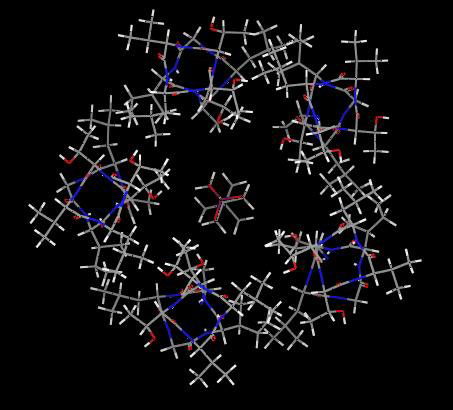
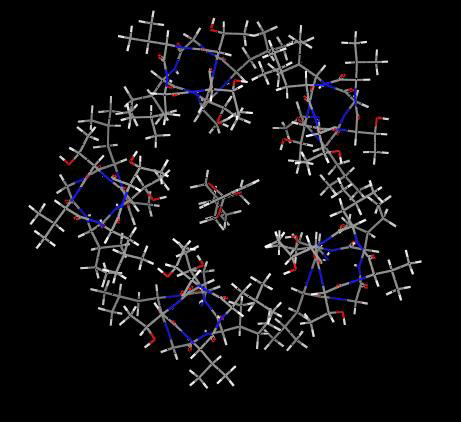
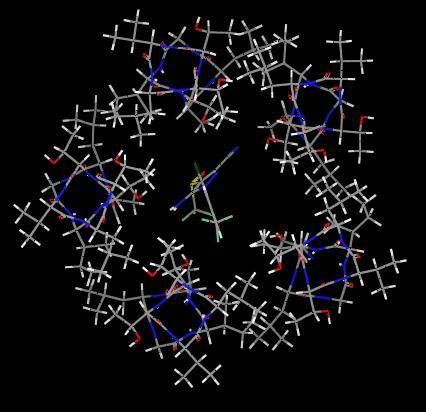
Fig. 6Upper Left shows a model of the channel lumen from the 2′ to 9′ positions with five β3 α-helices and lindane docked into the putative binding site, which it clearly fills to block the pore. Similar models of the six other NCAs also show filling of the pore space.
Fig. 6.

Proposed interactions of seven noncompetitive antagonists at the same GABAA receptor β3 homopentamer binding site. (Upper Left) lindane (space fill, red and blue for partial negative and positive charges of chlorine and carbon, respectively) binds to the GABAA receptor (five β3 α-helices shown in green) to block the channel pore shown as the 2′ to 9′ positions viewed from the top into the pore. Remaining panels: seven ligands (see Fig. 1A) docked at their optimized positions with the perspective chosen for ease of viewing. A, L, and T refer to the side chains of the interacting 2′, 6′, and 9′ residues, respectively. van der Waals contacts are illustrated in green (see text for discussion of hydrogen bonding). The space filling aspects of all of the ligands are most readily evident in supporting information.
Attention was focused on A2′, T6′, and L9′, because these residues are in the channel lumen (based on disulfide trapping) and mutations (Cys versus Ser or Phe in each case) at these sites greatly reduce or abolish binding. The interacting sites are shown in Figs. 1B and 6. Docking of EBOB positions the A2′ methyls interacting with the normal-propyl and two O-methylenes, two T6′ hydroxyls interacting with the oxygens (H---O distance ≈3.1Å), T6′ methyls binding to the phenyl moiety, and, at a slightly longer range, a L9′ methyl also interacting with the ethynyl substituent (evident in Fig. 1B but not Fig. 6). TBPS has numerous favorable A2′ interactions with the tertiary-butyl moiety, and the T6′ methyls and hydroxyls interact with the sulfur and cage oxygens. PTX has A2′ methyl interactions with the isopropenyl methyl and methylene and three T6′ hydroxyl hydrogen bonding interactions to three PTX oxygens. BIDN has multiple contact points with A2′ methyls and T6′ methyls and hydroxyls. A cyano nitrogen and a fluorine each form hydrogen bonds to a T6′ hydroxyl. Lindane bridges A2′ methyls and T6′ hydroxyls and methyls, each interacting with multiple chlorines. α-Endosulfan and fipronil have multiple interaction sites and types, with A2′ methyls and T6′ methyls and hydroxyls for both compounds reinforced by L9′ side chains for fipronil. More complete depictions of the β3 homopentamer model and the docked ligands are given in supporting information, which is published on the PNAS web site.
Discussion
Mutagenesis and Expression.
The cytoplasmic half of the M2 region contains 11 amino acids (0′ to 10′), and this number is extended to 15 (−4′ to 10′) with the flanking region of interest. Site-specific mutagenesis introduced Cys at 12 sites (A-1′C to T10′C), Ser at five sites (A-4′S, A-2′S, R0′S, A2′S, and L9′S), and Phe at two sites (L3′F and T6′F). In addition, three mutations were introduced with little change in polarity, i.e., G4′A, I5′L and V8′A. The 3′-position was an exception because L3′C and L3′F did not show detectable expression by Western blotting either in the β3 homopentamer studied here or the α1β3 heteropentamer (data not shown).
Pore-Lining Residues.
The position of pore-lining residues was determined by disulfide cross-linking, cysteine accessibility, and molecular modeling. Cys sulfhydryl substituents in the pore lining can be oxidized to disulfides resulting in dimerization. Disulfide trapping for Cys mutants in the present study places the sulfhydryl substituents of −1′, 2′, 6′, and 9′ within the channel lumen; disulfide trapping of A-1′C, A2′C, and L9′C was not established before. The tight protein packing in the 2′ position (31) may account for the weak dimer formation by limiting the required flexibility and close proximity for disulfide bond formation. Disulfides are not formed with 0′, 1′, 4′, 5′, 7′, 8′, and 10′, indicating they are probably not in the pore or have low mobility/flexibility. For T6′, similar findings are obtained with the α1T6′Cβ1T6′C receptor but only in the presence of GABA (28), suggesting that the β3 homopentamer of the present investigation assumes the spontaneous open state (32). Homology of the GABA receptor β3 homopentamer with the nicotinic acetylcholine receptor (33) indicates the narrowest gating region of the pore is between 9′ and 14′, suggesting the positioning of L9′C in the pore (24, 31). In the β1 subunit of the α1β1γ2 receptor, A2′C, T6′C, T7′C (slow reaction rate), V8′C, L9′C, and T10′C are all accessible to a sulfhydryl-modification reagent depending on the state of the channel (31). Methanethiosulfonate reagents in the present β3 homopentamer study show that V1′C is transiently available in the channel lumen in contrast to T7′C and T10′C, which are not readily accessible. In addition, reaction with the cationic methanethiosulfonate reagent suggests that the anion-selective filter may be below V1′C. Molecular modeling of the β3 homopentamer as an α-helix (Fig. 6) places −1′, 2′, 6′, and 9′, but not 0′, 1′, 3′, 4′, 5′, 7′, 8′, or 10′, in the channel pore (see supporting information), consistent with the other approaches.
Sites for NCA Interactions.
The interacting residues are considered to be A2′ (or more generally the A2′-I5′ hydrophobic pocket) and T6′ (the highly conserved and most important structural determinant) with a supplemental role for L9′. A biophysical calculation model focused on PTX interactions with A2′ and T6′ of the ρ1 receptor (29). The present study uses site-specific mutations in the β3 homopentamer to determine the importance of 10 other amino acid residues in NCA binding, i.e., the whole cytoplasmic half of the M2 region. A-4′, S-3′, and A-2′ are apparently outside of the binding site. β3 homopentamer mutants A-1′C, R0′C, and R0′S block binding, perhaps because of proximity to A2′. Sulfhydryl modification at V1′C impedes [3H]EBOB binding (this study), possibly by overlapping the sensitive A2′ position. Further, for 2′, the low sensitivity of the Drosophila RDL homomeric receptor to [3H]EBOB with A2′S (or A2′G) (34) suggests this site for binding with confirmation here from A2′C and A2′S mutants in the β3 homopentamer. In addition, with V2′C at the α1 subunit of the α1β1γ2 receptor, PTX protects against sulfhydryl derivatization (18), and a sulfhydryl-reactive fipronil analog [-C(O)CH2Br replaces -S(O)CF3] serves as an irreversible blocker (19). The involvement of 3′, directly or by influencing the neighboring A2′, is shown by L3′F at β3 of the α1β3 receptor almost abolishing TBPS and PTX binding (20). The structurally critical apolar pocket in the β3 homopentamer appears to involve A2′, L3′, G4′, and I5′, i.e., a tightly packed and completely hydrophobic region that may play a role in stabilizing the helical structure (31, 35). Although G4′ is on the backside of the helix, the side chains introduced with the G4′C and G4′A mutants appear to perturb the tightly packed 2′-5′ region of the channel lumen to disturb NCA binding. The T6′C and T6′F mutations in the β3 homopentamer abolish NCA sensitivity, and introducing T6′F in β2 (or α1 or γ2) of α1β2γ2 greatly reduces PTX sensitivity (21). Mutagenesis of the 6′ position of ρ1 and ρ2 receptors from rats showed this site to be important in PTX sensitivity (22). T7′C and V8′C fall outside the pore and, therefore, are not expected to be important binding sites, yet the 8′ mutants block binding, perhaps, by changing the shape of the pore. For L9′, where a mutation can potentially perturb the gating kinetics (24), the L9′C and L9′S mutations for β3 abolish NCA binding here and L9′S reduces PTX sensitivity in each subunit of α1β2γ2 (24). Finally, with ρ1, several mutations at L9′ also reduce PTX sensitivity (23). Lying outside the pore, T10′C does not affect [3H]EBOB or [3H]BIDN binding.
Widely Diverse NCA Structures Fit the Same Site.
The RDL A2′S mutation confers cross-resistance of insects to all classes of commercial NCA insecticides (10, 17), and this cross-resistance also applies to the highly potent model compounds EBOB and BIDN. The effect of all mutations is essentially the same with [3H]EBOB and [3H]BIDN, indicating that they both have the same binding site. More generally, an extremely wide diversity of chemical types, each with configurational specificity, appears to act the same way as GABA receptor NCAs (3, 9, 36, 37). Figs. 1B and 6 illustrate how they, in fact, may all fit the same site by showing the proposed interactions of seven NCAs with the β3 homopentameric receptor. Favorable hydrophobic interactions are observed for the A2′ methyls with the alkyl substituents of EBOB, TBPS, PTX, and BIDN, the trifluoromethylsulfinyl and pyrazole cyano of fipronil, the hexachlorocyclopentenyl moiety of endosulfan, and the hexachlorocyclohexane isomer lindane. The T6′ methyls interact with the ethynylphenyl and trifluoromethylphenyl substituents of EBOB and fipronil, respectively, the trifluoromethyls and cyanos of BIDN, and the exocyclic oxygen of α-endosulfan. The T6′ hydroxyl substituent hydrogen bonds (H-X distance <3.5 Å) to multiple electronegative sites, i.e., the trioxabicyclooctane oxygens of EBOB and TBPS; the exocyclic oxygen of endosulfan; the epoxy, hydroxyl, and lactone exocyclic oxygens of PTX; the pyrazole, amino, and cyano nitrogens of fipronil; and a cyano nitrogen and fluorine of BIDN. In lindane, four chlorines are <3.5 Å from T6′ hydroxyl hydrogens. On calculating the relative energies of the bound ligands by using maestro/macromodel (Schrödinger LLC, Portland, OR), the most potent γ isomer lindane binds in a more stable configuration than the less active α, β, and δ isomer(s) by >30 kJ/mol and the more active α-endosulfan versus the less potent β-endosulfan by 15 kJ/mol. The L9′ side chains associate with the phenyl group of EBOB and fipronil, the ethynyl of EBOB and the aryl trifluoromethyl and chloro substituents of fipronil, enhancing the potency of these long or extended molecules. The more compact NCAs, including TBPS, lindane, and BIDN, require only A2′ and T6′ for fit lengthwise or lying across the pore, and this positioning probably also applies to α-endosulfan and PTX. These docking proposals are consistent with current structure-activity relationships and may help in further ligand optimization.
The NCAs are chloride channel blockers, i.e., their potency in binding to the NCA site is proportional to their effectiveness in inhibiting chloride flux (38, 39). In the proposed binding site model, the NCAs fill up and actually block the pore, although they also may act allosterically by changing the channel conformation. The internuclear distance across the channel pore is on the order of 8.5 Å, which is the same as or only slightly longer than the distance across multiple types of NCAs (6–8 Å).
NCA Potency and Selectivity Conferred by Subunit Specificity.
The β3 homopentamer has higher NCA sensitivity than other vertebrate GABA receptors and any replacement subunits of those tested reduce ligand affinity (9). The β3 homopentamer can form a spontaneously opening ion channel (32), potentially facilitating ligand binding. GABA and other agonist modulators affect NCA binding with native and α1 subunit-containing receptors but not with the β3 homopentamer (9, 12, 40). As with related ligand-gated ion channels the NCA potency profile varies with subunit composition. Selectivity is conferred by these additional subunits as evident by comparing native receptors with α1β2γ2 heteropentameric and β3 homopentameric recombinant receptors (9, 27). NCAs with excellent fit for the β3 homopentamer model may show less favorable docking in the heteropentameric native receptors associated with subunit variation at the 2′ position.
Concluding Remarks.
The human GABAA receptor recombinant β3 homopentamer retains the NCA site in its most sensitive form, equal to the insect site. Both the β3 homopentamer pore and principal radioligand [3H]EBOB are symmetrical, thereby greatly facilitating receptor modeling and ligand positioning. Ligands of widely diverse structures approach similar potency when optimized. The effect of mutations is the same for [3H]EBOB and [3H]BIDN binding and possibly for the other NCAs as well. A model for the GABAA receptor M2 region applied to the β3 homopentamer brings these observations together to propose structural aspects of the NCA site. Further test of this proposal requires direct rather than indirect structural analysis of the homopentameric and heteropentameric GABA receptors.
Materials and Methods
Site-Directed Mutagenensis.
cDNA encoding the human GABAA receptor β3 subunit inserted in the pVL1392 baculovirus transfer vector was described in ref. 9. Point mutations were introduced with the QuikChange Site-Directed Mutagenesis kit (Stratagene). Mutagenic oligonucleotides were prepared by Operon (Huntsville, AL). All mutations were confirmed by double-strand DNA sequencing (DNA Sequencing Facility, University of California, Berkeley).
Cell Culture and Protein Expression.
Insect Sf9 cells (serum-free adapted, derived from ovaries of Spodoptera frugiperda) were maintained by described methods in refs. 9 and 41. Recombinant baculoviruses were constructed by using a Bacfectin-mediated transfection kit (BD Biosciences Clontech). The Invitrogen protocol was used for PCR analysis of recombinant virus. All PCR products were recycled with GelQuick Gel Extraction Kit (Qiagen, Valencia, CA) and then were sequenced as described above. Log phase Sf9 cells were infected with recombinant baculovirus at a multiplicity of infection of 5–8. Cells were harvested at 65 h after infection. They were pelleted at 1,500 × g for 5 min and washed once with PBS (155 mM NaCl/3.0 mM NaH2PO4/1.0 mM K2HPO4, pH 7.4). Cell pellets were stored at −80°C until ready to use.
Membrane Preparation.
The pelleted cells were resuspended in PBS and homogenized in a glass tube with a motor-driven Teflon pestle (9, 41). Cellular debris was removed by centrifugation at 500 × g for 10 min at 4°C. The supernatant was centrifuged at 100,000 × g for 40 min at 4°C, and the resulting pellet was resuspended in PBS and stored at −80°C. Protein concentration was determined with the detergent-compatible Lowry assay (Bio-Rad).
Western Blotting.
Membrane preparations were mixed with Laemmli sample buffer (1.5% SDS/5% glycerol/65 mM Tris·HCl, pH 6.8, with or without 10 mM DTT). After boiling at 100°C for 5 min, samples were analyzed by SDS/PAGE (10% acrylamide) by using a Mini-PROTEAN II apparatus (Bio-Rad). Proteins were transferred onto poly(vinylidene difluoride) membranes for 2 h at 100 V and 4°C by using the Transblot apparatus (Bio-Rad). The membranes were blocked in Tris-buffered saline (Bio-Rad) containing 2% nonfat dry milk with 0.5% Tween 20 for 1 h at room temperature and incubated with the mouse anti-GABAA receptor, β-chain monoclonal antibody (Chemicon International, Temecula, CA), at a dilution of 1:1,000, also for 1 h at room temperature. After three 5-min washings in TBS with 0.5% Tween 20, the blots were incubated with anti-mouse horseradish peroxidase-linked secondary antibodies (Santa Cruz Biotechnology) at a dilution of 1:2,000 for 1 h at room temperature. After extensive washing, immunoreactivity was detected by chemiluminescence kit (PerkinElmer). Finally, the transferred protein was visualized by incubation in Ponceau S solution (Bio-Rad).
Disulfide Cross-Linking and Sulfhydryl Modification.
For disulfide cross-linking, the membrane preparation (100 μg of protein) in PBS (100 μl) was oxidized with Cu:phen (100 μM:400 μM) (28, 42) for 5 min at 25°C. The reaction was terminated by adding 10 mM N-ethylmaleimide and 1 mM EDTA (final concentrations). After 3 min, the membranes were recovered by centrifugation (20,000 × g for 15 min at 4°C), resuspended in PBS, mixed with sample buffer with or without 10 mM DTT, and subjected to SDS/PAGE (10% acrylamide) and Western blot analysis. For sulfhydryl modification, two methanethiosulfonate reagents were used, 2-(trimethylammonium)ethyl methanethiosulfonate bromide and sodium (2-sulfonatoethyl)methanethiosulfonate, under described conditions (18, 43) with analysis for their effect on [3H]EBOB binding.
[3H]EBOB and [3H]BIDN Binding.
Assay mixtures contained 1 nM [3H]EBOB (48 Ci/mmol; 1 Ci = 37 GBq) (PerkinElmer) (6, 9) or 2.5 nM [3H]BIDN (50 Ci/mmol) (8) and the recombinant expressed receptor (100 μg protein) in PBS (500 μl final volume) (6). After incubation for 90 min at 25°C, the samples were filtered through GF/B filters (presoaked in 0.2% polyethyleneimine for 3 h) and rinsed three times with ice-cold saline (0.9% NaCl). Nonspecific binding was determined in the presence of 1 μM α-endosulfan for [3H]EBOB or 5 μM unlabeled BIDN for [3H]BIDN by using 5 μl dimethyl sulfoxide to add the displacing agent immediately before incubation. Each experiment was repeated three or more times with duplicate samples. The binding activity of mutants was expressed as percent (mean ± SD) of that for the WT. Supplemental binding studies were made by using DTT preincubation (10 mM for 5 min at 25°C) in deoxygenated PBS continuously bubbling with argon to rule out any spontaneous disulfide formation.
Modeling Receptor–Ligand Interactions.
Modeling started from the α1β2γ2 GABAA receptor based on the homologous nicotinic acetylcholine receptor and acetylcholine binding protein (30). This α-helical structure was reconstructed here as the β3 homopentamer, i.e., the two β2 subunits were directly replaced by β3, because they have the same M2 sequence, then the two α1 subunits and one γ2 subunit were replaced with β3, by using the original α1 and γ2 backbone atom positions as a guide, to make the homopentameric model. The cytoplasmic side of the M2 region and adjacent residues are considered, i.e., A-4′ to T10′ (Fig. 2), with particular attention to A2′ to L9′.
All modeling was done with MAESTRO 6.5 (Schrödinger LLC). Macromodel atom types were used to assign partial charges (44). van der Waals contacts were defined as C = (distance between atomic centers)/(radius 1st atom + radius 2nd atom) where good, bad, and ugly contacts are defined as C = 1.3, 0.89, and 0.75 Å, respectively. The antagonists were manually docked into the putative binding site to maximize good contacts, and then the ligand geometry and location were allowed to optimize relative to the β3 homopentamer, which was itself constrained. In this optimization, all settings were left at the default values except a water model (generalized Born/surface area) was used instead of a gas phase model. In each case, sufficient optimization steps were performed as necessary to ensure that the convergence criteria were met.
Supplementary Material
Acknowledgments
We thank our department colleagues Jung-Chi Liao, Motohiro Tomizawa, Gary Quistad, Daniel Nomura, and Shannon Liang for helpful advice and Myles Akabas, Gerald Brooks, Richard Olsen, and David Weiss for important suggestions. This work was supported by the William Mureice Hoskins Chair in Chemical and Molecular Entomology (to J.E.C.) and National Science Foundation Grant CHE-0233882 (to K.A.D.). Molecular modeling was performed on workstations purchased with National Science Foundation support matched with an equipment donation from Dell.
Abbreviations
- BIDN
3,3-bis-trifluoromethyl-bicyclo[2,2,1]heptane-2,2-dicarbonitrile
- Cu:phen
copper:phenanthroline
- EBOB
1-(4-ethynylphenyl)-4-n-propyl-2,6,7-trioxabicyclo[2.2.2]octane
- M2
transmembrane 2
- NCA
noncompetitive antagonist
- PTX
picrotoxinin
- RDL
resistant to dieldrin
- TBPS
t-butylbicyclophosphorothionate.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Casida J. E., Quistad G. B. Annu. Rev. Entomol. 1998;43:1–16. doi: 10.1146/annurev.ento.43.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Tomlin C. D. S., editor. The Pesticide Manual. 13th Ed. Alton, Hampshire, U.K: Brit. Crop Prot. Counc; 2003. p. 1344. [Google Scholar]
- 3.Brooks G. T. In: Handbook of Pesticide Toxicology: Agents. 2nd Ed. Krieger R. I., editor. Vol. 2. New York: Academic; 2001. pp. 1131–1156. [Google Scholar]
- 4.Matsumura F., Ghiasuddin S. M. J. Environ. Sci. Health B. 1983;18:1–14. doi: 10.1080/03601238309372355. [DOI] [PubMed] [Google Scholar]
- 5.Lawrence L. J., Casida J. E. Life Sci. 1984;35:171–178. doi: 10.1016/0024-3205(84)90136-x. [DOI] [PubMed] [Google Scholar]
- 6.Cole L. M., Casida J. E. Pestic. Biochem. Physiol. 1992;44:1–8. [Google Scholar]
- 7.Casida J. E. Arch. Insect Biochem. Physiol. 1993;22:13–23. doi: 10.1002/arch.940220104. [DOI] [PubMed] [Google Scholar]
- 8.Rauh J. J., Benner E., Schnee M. E., Cordova D., Holyoke C. W., Howard M. H., Bai D., Buckingham S. D., Hutton M. L., Hamon A., et al. Brit. J. Pharmacol. 1997;121:1496–1505. doi: 10.1038/sj.bjp.0701215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ratra G. S., Kamita S. G., Casida J. E. Toxicol. Appl. Pharmacol. 2001;172:233–240. doi: 10.1006/taap.2001.9154. [DOI] [PubMed] [Google Scholar]
- 10.Bloomquist J. R. In: Biochemical Sites of Insecticide Action and Resistance. Ishaaya I., editor. Berlin: Springer; 2001. pp. 17–41. [Google Scholar]
- 11.Ticku M. K., Ban M., Olsen R. W. Mol. Pharmacol. 1978;14:391–402. [PubMed] [Google Scholar]
- 12.Squires R. F., Casida J. E., Richardson M., Saederup E. Mol. Pharmacol. 1983;23:326–336. [PubMed] [Google Scholar]
- 13.Barnard E. A., Skolnick P., Olsen R. W., Mohler H., Sieghart W., Biggio G., Braestrup C., Bateson A. N., Langer S. Z. Pharmacol. Rev. 1998;50:291–313. [PubMed] [Google Scholar]
- 14.Enna S. J., Bowery N. G., editors. The GABA Receptors. 2nd Ed. Totowa, NJ: Humana; 1997. [Google Scholar]
- 15.Martin D. L., Olsen R. W., editors. GABA in the Nervous System: the View at Fifty Years. Philadelphia: Lippincott; 2000. [Google Scholar]
- 16.Chang Y., Weiss D. S. In: GABA in the Nervous System: the View at Fifty Years. Martin D. L., Olsen R. W., editors. Philadelphia: Lippincott; 2000. pp. 127–139. [Google Scholar]
- 17.ffrench-Constant R. H., Anthony N., Aronstein K., Rocheleau T., Stilwell G. Annu. Rev. Entomol. 2000;48:449–466. doi: 10.1146/annurev.ento.45.1.449. [DOI] [PubMed] [Google Scholar]
- 18.Xu M., Covey D. F., Akabas M. H. Biophys. J. 1995;69:1858–1867. doi: 10.1016/S0006-3495(95)80056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perret P., Sarda X., Wolff M., Wu T.-T., Bushey D., Goeldner M. J. Biol. Chem. 1999;274:25350–25354. doi: 10.1074/jbc.274.36.25350. [DOI] [PubMed] [Google Scholar]
- 20.Buhr A., Wagner C., Fuchs K., Sieghart W., Sigel E. J. Biol. Chem. 2001;276:7775–7781. doi: 10.1074/jbc.M008907200. [DOI] [PubMed] [Google Scholar]
- 21.Gurley D., Amin J., Ross P. C., Weiss D. S., White G. Recept. Channels. 1995;3:13–20. [PubMed] [Google Scholar]
- 22.Zhang D., Pan Z.-H., Zhang X., Brideau A. D., Lipton S. A. Proc. Natl. Acad. Sci. USA. 1995;92:11756–11760. doi: 10.1073/pnas.92.25.11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang Y., Weiss D. S. Mol. Pharmacol. 1998;53:511–523. doi: 10.1124/mol.53.3.511. [DOI] [PubMed] [Google Scholar]
- 24.Chang Y., Weiss D. S. Biophys. J. 1999;77:2542–2551. doi: 10.1016/s0006-3495(99)77089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller C. Neuron. 1989;2:1195–1205. doi: 10.1016/0896-6273(89)90304-8. [DOI] [PubMed] [Google Scholar]
- 26.Slany A., Zezula J., Tretter V., Sieghart W. Mol. Pharmacol. 1995;48:385–391. [PubMed] [Google Scholar]
- 27.Ratra G. S., Casida J. E. Toxicol. Lett. 2001;122:215–222. doi: 10.1016/s0378-4274(01)00366-6. [DOI] [PubMed] [Google Scholar]
- 28.Horenstein J., Wagner D. A., Czajkowski C., Akabas M. H. Nature Neurosci. 2001;4:477–485. doi: 10.1038/87425. [DOI] [PubMed] [Google Scholar]
- 29.Zhorov B. S., Bregestovski P. D. Biophys. J. 2000;78:1786–1803. doi: 10.1016/S0006-3495(00)76729-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Mara M., Cromer B., Parker M., Chung S.-H. Biophys. J. 2005;88:3286–3299. doi: 10.1529/biophysj.104.051664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goren E. N., Reeves D. C., Akabas M. H. J. Biol. Chem. 2004;279:11198–11205. doi: 10.1074/jbc.M314050200. [DOI] [PubMed] [Google Scholar]
- 32.Wooltorton J. R. A., Moss S. J., Smart T. G. Eur. J. Neurosci. 1997;9:2225–2235. doi: 10.1111/j.1460-9568.1997.tb01641.x. [DOI] [PubMed] [Google Scholar]
- 33.Miyazawa A., Fujiyoshi Y., Unwin N. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 34.Cole L. M., Roush R. T., Casida J. E. Life Sci. 1995;56:757–765. doi: 10.1016/0024-3205(95)00006-r. [DOI] [PubMed] [Google Scholar]
- 35.Popot J.-L., Engelman D. M. Annu. Rev. Biochem. 2000;69:881–922. doi: 10.1146/annurev.biochem.69.1.881. [DOI] [PubMed] [Google Scholar]
- 36.Calder J. A., Wyatt J. A., Frenkel D. A., Casida J. E. J. Comput. Aided Mol. Des. 1993;7:45–60. doi: 10.1007/BF00141574. [DOI] [PubMed] [Google Scholar]
- 37.Ozoe Y., Akamatsu M. Pest Manag. Sci. 2001;57:923–931. doi: 10.1002/ps.375. [DOI] [PubMed] [Google Scholar]
- 38.Obata T., Yamamura H. I., Malatynska E., Ikeda M., Laird H., Palmer C. J., Casida J. E. J. Pharmacol. Exp. Ther. 1988;244:802–806. [PubMed] [Google Scholar]
- 39.Huang J., Casida J. E. J. Pharmacol. Exp. Ther. 1996;279:1191–1196. [PubMed] [Google Scholar]
- 40.Korpi E. R., Gründer G., Lüddens H. Prog. Neurobiol. 2002;67:113–159. doi: 10.1016/s0301-0082(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 41.Elster L., Schousboe A., Olsen R. W. J. Neurosci. Res. 2000;61:193–205. doi: 10.1002/1097-4547(20000715)61:2<193::AID-JNR10>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 42.Yu H., Kono M., McKee T. D., Oprian D. D. Biochemistry. 1995;34:14963–14969. doi: 10.1021/bi00046a002. [DOI] [PubMed] [Google Scholar]
- 43.Slotboom D. J., Konings W. N., Lolkema J. S. J. Biol. Chem. 2001;276:10775–10781. doi: 10.1074/jbc.M011064200. [DOI] [PubMed] [Google Scholar]
- 44.Mohamadi F., Richards N. G. J., Guida W. C., Liskamp R., Lipton M., Caufield C., Chang G., Hendrickson T., Still W. C. J. Comput. Chem. 1990;11:440–467. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}