

Abstract
Infectious entry of hepatitis B viruses (HBV) has nonconventional facets. Here we analyzed whether a cell-permeable peptide [translocation motif (TLM)] identified within the surface protein of human HBV is a general feature of all hepadnaviruses and plays a role in the viral life cycle. Surface proteins of all hepadnaviruses contain conserved functional TLMs. Genetic inactivation of the duck HBV TLMs does not interfere with viral morphogenesis; however, these mutants are noninfectious. TLM mutant viruses bind to cells and are taken up into the endosomal compartment, but they cannot escape from endosomes. Processing of surface protein by endosomal proteases induces their exposure on the virus surface. This unmasking of TLMs mediates translocation of viral particles across the endosomal membrane into the cytosol, a prerequisite for productive infection. The ability of unmasked TLMs to translocate processed HBV particles across cellular membranes was shown by confocal immunofluorescence microscopy and by infection of nonpermissive cell lines with HBV processed in vitro with endosomal lysate. Based on these data, we propose an infectious entry mechanism unique for hepadnaviruses that involves virus internalization by receptor-mediated endocytosis followed by processing of surface protein in endosomes. This processing activates the function of TLMs that are essential for viral particle translocation through the endosomal membrane into the cytosol and productive infection.
Keywords: cell permeability, envelope protein, virus entry
Infection with human hepatitis B virus (HBV) can cause acute or chronic inflammation of the liver (1, 2). HBV is the prototype member of the hepadnaviridae family, which encompasses members infecting woodchucks, ground squirrels, and avian viruses isolated from, e.g., pekin ducks, gray herons, and storks.
Duck HBV (DHBV) is a well characterized model system of hepadnaviral infection (3). Cultures of primary duck hepatocytes (PDHs) can be readily established and efficiently infected (3, 4) and therefore provide a suitable tool for analyzing the early steps of hepadnaviral infection on the molecular level. As for HBV (5–7), it is known that DHBV infection is initiated by attachment of the virus particle to the hepatocyte surface via the pre-S domain of the viral surface protein L (8, 9). In DHBV there are two surface proteins embedded in the lipid envelope: The major S protein, a transmembrane protein that encompasses 167 aa, and the L protein, consisting of the S domain N-terminally extended by the 160-aa pre-S domain. Previous work suggested that DHBV enters the cell by receptor-mediated endocytosis (10–13). The mechanism that allows internalized viral particles to escape from the endocytic pathway remained elusive.
Recently, a cell-permeable peptide [translocation motif (TLM)] was identified in the pre-S domain of HBV (14). The TLM is a 12-aa-encompassing domain that forms an amphipathic α-helix. It mediates an energy- and receptor-independent transfer of peptides, nucleic acids, and proteins when fused to them across membranes without affecting their integrity (14–16).
Because the membrane translocation function of the TLM is highly conserved among all hepadnaviridae tested we investigated whether the TLM function is of relevance for the viral life cycle.
Results
The Pre-S Domain of Hepadnaviruses Harbors a Cell Permeability-Mediating Domain.
Detailed analysis revealed that cell permeability mediated by TLMs does not depend on an unique amino acid sequence but on the capacity to form an α-helix with an amphipathic structure. A homology search for potential TLMs in the pre-S domain of the surface proteins of various hepadnaviridae predicted the existence of TLMs in the pre-S domain of all hepadnaviridae (14). In the case of DHBV, the existence of two independent translocation motifs within the pre-S domain is predicted (Fig. 6A, which is published as supporting information on the PNAS web site).
To analyze the potential of these predicted motifs to act as cell-permeable peptides, recombinant fusion proteins with eGFP were engineered. Immunoblot analysis was performed with a GFP-specific antiserum and the cytosolic fraction derived from HepG2 cells grown in medium containing these various purified TLM fusion proteins. The blot revealed comparable amounts of the TLM fusion proteins within the cytosolic fractions, whereas, in the case of cells grown in the presence of WT eGFP, no eGFP-specific protein was detectable (Fig. 6B). Mutation of TLMs prevented translocation of the fusion proteins into the cytosol (Fig. 6C). These data indicate that the predicted TLMs from the hepadnaviridae members analyzed display similar cell permeability when compared with the previously identified HBV-TLM.
Functionality of the TLMs Is Dispensable for DHBV Secretion.
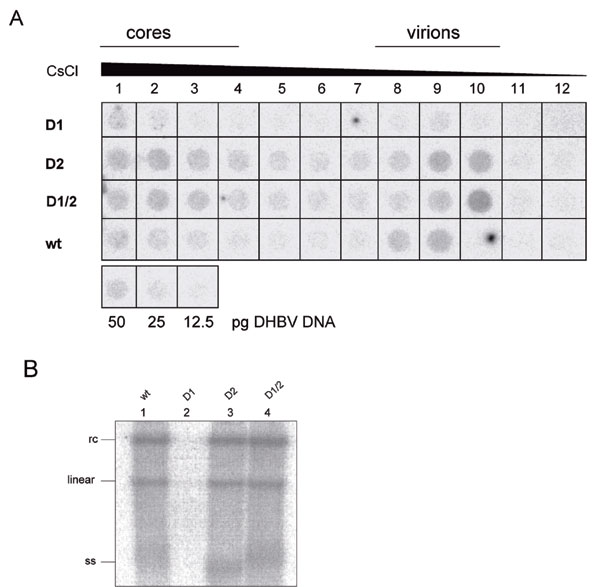
The finding that the membrane translocation function of TLMs is highly conserved throughout hepadnaviridae evolution is suggestive for a crucial role in the viral life cycle. To study this observation, mutated 1.2 DHBV genomes were generated coding for pre-S/S proteins lacking a functional TLM1 (amino acids 20–31) (DHBVD1), a functional TLM2 (amino acids 42–53) (DHBVD2), or both TLMs (DHBVD1/2) without impairing functionality of the polymerase. Transfection of LMH cells with these constructs, followed by cesium chloride centrifugation of the culture supernatants and subsequent quantification of the viral particles by dot blot analysis, revealed reduced amounts of secreted viral particles in the supernatant of DHBVD1-transfected cells as compared with that of WT virus (Fig. 7A, which is published as supporting information on the PNAS web site). In contrast, mutants DHBVD2 and DHBVD1/2 secreted similar amounts of viral particles as the WT virus. Therefore, the observed phenotype of DHBVD1 cannot be due to the TLM deficiency but must have another reason (see below). Comparable amounts of replicative intermediates in LMH cells transfected with WT DHBV, DHBVD2, or DHBVD1/2 and a reduced amount in the case of DHBVD1 detected by Southern blotting (Fig. 7B) corroborates this conclusion. Because none of the TLM mutations introduced alter the viral polymerase protein sequence, an effect of the D1 mutation on a regulatory element in pregenomic RNA encapsidation, in transcription or on the posttranscriptional level, are the most likely explanations for its reduced replication efficiency. Taken together, these results indicate that TLM functionality is dispensable for viral morphogenesis.
Integrity of the TLM Is Crucial for DHBV Infectivity.
To analyze the relevance of the TLMs for the infection process, PDHs were infected at a multiplicity of genome equivalents (MGE) per cell of 100 with WT DHBV and the mutants DHBVD2 or DHBVD1/2. Because of the observed TLM-independent reduction in the replication efficiency of DHBVD1, it was excluded from these assays. The productivity of infection was analyzed 4 days after inoculation. Immunofluorescence staining of the PDHs using surface protein-specific antiserum KpnI (17), which recognizes the mutant proteins (data not shown), revealed de novo synthesis of surface proteins indicative for productive infection only in PDHs infected with WT virus. The TLM-deficient mutants DHBVD2 and DHBVD1/2 failed to infect PDHs (Fig. 1A). This finding was confirmed by core protein-specific immunoblot analysis of cellular lysates from WT-, DHBVD2-, or DHBVD1/2-infected cells (Fig. 1B). In contrast to WT-infected cells, cells infected with DHBD1/2 or DHBVD2 showed no de novo synthesis of core protein. Furthermore, only in cells infected with WT DHBV, but not in cells infected with DHBVD2 or DHBVD1/2, replicative intermediates were found by Southern blot analysis (Fig. 1C). Moreover, covalently closed circular DNA (cccDNA) was detected only in PDHs infected with WT DHBV (Fig. 1D) when analyzed 5 days after infection by cccDNA-selective PCR. These data show that destruction of the TLMs abolishes DHBV infectivity.
Fig. 1.

Destruction of the TLM abolishes infectivity of DHBV. (A) Immunofluorescence microscopy of infected PDH using an L-specific antiserum. Cells were infected with 100 MGE WT DHBV, DHBVD2, or DHBVD1/2 mutant. Cells were fixed 4 days after infection. Hoechst staining was used to visualize nuclei. The photographs were taken at ×200 magnification. (B) Immunoblot analysis of lysates from PDHs infected with WT DHBV or the mutants by using a DHBV core-specific antiserum. Uninfected PDHs served as negative control. (C) PDHs were infected with 100 MGE. Cells were harvested 7 days after infection and analyzed for replicative intermediates by Southern blotting. (D) Analysis of cccDNA by PCR. The cccDNA was isolated 3 days after infection and amplified by PCR by using cccDNA-selective primers. Uninfected PDHs served as negative control.
TLMs Are Dispensable for Attachment and Entry of Viral Particles to and into PDHs.
To control whether the defect in infectivity of TLM mutant viruses is due to reduced binding to PDHs, attachment assays were performed. After inoculation cells were incubated for 2 h at 4°C (viral binding occurs, and internalization is blocked). Then the amount of viral particles attached to the cell was determined by semiquantitative PCR and immunoblotting. Similar amounts of WT DHBV and the mutant viral particles were found to be attached to the cell surface (Fig. 8A Upper Left, which is published as supporting information on the PNAS web site).
A PCR-based analysis of the amount of viral particles that have entered the cells after 3 h revealed similar amounts of intracellular virus in cells inoculated with WT virus and the TLM-deficient viruses (Fig. 8A Lower Left). These data collectively show that the loss of infectivity of TLM mutant viruses is due to neither an impaired attachment nor inhibition of virus entry, but to a post entry block.
TLM Integrity Is Essential for DHBV to Escape from the Endosome.
Recent work suggests that DHBV is internalized by receptor-mediated endocytosis (10–12, 18). Based on these data we hypothesized that TLM deficiency might result in retention of the virus within the endosomal compartment. To investigate this possibility experimentally, we isolated endosomal and cytosolic fractions from infected PDHs 10 h after infection. The subcellular fractions were adjusted to identical protein concentrations, and the amount of DHBV-specific DNA in the endosomal and in the cytosolic compartments was quantified by TaqMan PCR. This study revealed similar amounts of DHBV DNA in the endosomal fraction derived from DHBVD1-, DHBVD2-, and DHBVD1/2-infected cells and a smaller amount in the case of WT DHBV-infected cells. However, in the cytosolic fraction, a significant number of viral genomes was detected only in the case of WT DHBV-infected cells but not in the cytosol from PDHs infected with the mutants (Fig. 2). These results indicate that endosomal escape of internalized viral particles requires proper function of the TLM.
Fig. 2.

TLM-deficient viral particles are trapped in the endosome. PDH were inoculated with WT or mutant DHBV (200 MGE). After 10 h of incubation with WT DHBV, DHBVD1/2, DHBVD2, and DHBVD1, cells were harvested and subfractionated. Cytosolic and endosomal fractions were adjusted to identical protein concentrations, and their purity was controlled by immunoblotting by using grb2 (cytoplasm)- and clathrin HC (endosomes)-specific antisera. For detection of viral DNA in the cytosolic (c) and endosomal (e) fractions, TaqMan PCR was performed. The y axis indicates the number of viral genomes per 25 μl of resuspended subcellular fraction.
Cleavage of Hepatitis B Virus Surface Antigen (HBsAg) by Endosomal Proteases Results in Surface Exposure of the TLM.
The data described above raise the question of why TLMs enable translocation of viral particles across the endosomal membrane but not across the plasma membrane. It can be hypothesized that the TLMs are masked in progeny viral particles, ensuring the hepadnaviral specificity for hepatocytes. After receptor-mediated endocytosis, an unmasking of the TLMs may occur in the endosomal compartment that exposes the TLMs on the surface of the virus particle. Once unmasked, the TLMs allow efficient escape of the viral particles from the endosome.
To verify this hypothesis, accessibility of the TLMs was analyzed by immunoprecipitation of HBV particles with an HBV-TLM-specific antiserum and an HBsAg-specific serum used as a positive control. The precipitates were analyzed for the presence of HBV particles by using a hepatitis B virus core antigen (HBcAg)-specific antiserum. Western blot analysis revealed that native HBV particles could not be precipitated by the TLM-specific serum (Fig. 3, lane 2). However, if HBV particles were incubated with endosomal lysate and then subjected to immunoprecipitation by using the TLM-specific antiserum, a significant amount of virus was precipitated (Fig. 3, lane 8). This result demonstrates that an unmasking step must have occurred exposing the TLM on the viral particle surface and conferring accessibility for the TLM-specific serum. To analyze whether the acidic environment of the endosome is sufficient to induce unmasking of the TLM, the viral particles were incubated for 30 min at pH 5.0 (Fig. 3, lanes 4 and 6). The Western blot of the precipitates shows that acidification alone is not sufficient to unmask the TLM. When a protease inhibitor mixture was added to the endosomal lysate, no subsequent precipitation of viral particles by the TLM-specific antiserum was detected (data not shown). Western blot analysis of viral particles that were incubated with endosomal lysate in vitro confirmed proteolytic processing of viral particles in the endosome (Fig. 9A, which is published as supporting information on the PNAS web site). We concluded from these data that the TLM is not exposed on the surface of mature viral particles. A proteolytic activity in the endosomal compartment is crucial for unmasking the TLM on the viral particle surface.
Fig. 3.

Cleavage of HBsAg by endosomal proteases results in surface exposure of the TLM. Purified HBV particles were subjected to immunoprecipitation with either an HBV-TLM-specific antiserum (lanes 2, 4, 6, and 8), or an HBsAg-specific serum as a positive control (lanes 1, 3, 5, and 7). The precipitated material was immunoblotted by using an HBcAg-specific antiserum. In lanes 1 and 2 precipitates of untreated HBV particles were loaded using an HBsAg-specific serum or a TLM-specific serum. Purified HBV particles were incubated for 30 min at pH 5.0 and immunoprecipitated by using an HBV-TLM-specific antiserum (lane 4) and an HBsAg-specific serum (lane 3). Purified HBV particles were incubated for 30 min at pH 5.0, then by addition of 10× PBS the pH was shifted to ≈7. Afterward, immunoprecipitation was performed by using an HBV-TLM-specific antiserum (lane 6) and an HBsAg-specific serum (lane 5). Purified HBV particles were incubated for 30 min at pH 5.0 in endosomal lysate from HepG2 cells and immunoprecipitated by using an HBV-TLM-specific antiserum (lane 8) and an HBsAg-specific serum (lane 7). Recombinant HBcAg (lane 9) served as positive control.
Processing of HBV Particles by Endosomal Lysate Enables Infection of Nonpermissive Cells.
The resulting question is whether surface-exposed TLMs indeed allow translocation of the processed particle across membranes. HBV particles with unmasked TLMs should be able to translocate across the plasma membrane and to establish infection in Huh7 cells. These cells normally cannot be efficiently infected by HBV.
To test this hypothesis purified HBV particles were pretreated for 60 min with endosomal lysate from HepG2 cells. These processed viral particles were then used to infect Huh7 cells with a MGE of 102 to 103. As a control, cells were infected with a comparable MGE of unprocessed viral particles. Three days after infection de novo synthesis of HBsAg and HBcAg was analyzed by immunofluorescence microscopy by using an HBsAg-specific antiserum (Fig. 4A, red fluorescence) or an HBcAg-specific antiserum (Fig. 4A, blue fluorescence). The immunofluorescence staining revealed no infected cells in cultures incubated with unprocessed virus whereas up to 40% of cells were stained after incubation with processed viral particles. Similar results were obtained when DHBV particles were processed by endosomal lysate from LMH cells and used for successful infection of LMH cells (Fig. 4B). These cells are resistant to infection with authentic DHBV. Furthermore, infectivity was abolished by addition of a protease inhibitor mixture to the endosomal lysate (Fig. 4B). Processing of TLM-deficient mutants DHBVD2 and DHBVD1/2 by endosomal lysate failed to establish an “infection” in LMH cells (Fig. 4B). The results of these immunostaining assays were confirmed and extended by analysis of the cell culture supernatants for viral progeny DNA at different times after inoculation by PCR (Fig. 9B). These data demonstrate the ability of unmasked TLMs to mediate translocation of processed viral particles across membranes.
Fig. 4.

Processing of HBV particles by endosomal lysate enables infection of nonpermissive cells. (A) Confocal microscopy of Huh7 cells infected with unprocessed HBV particles (Left) or with particles incubated with endosomal lysate from HepG2 cells for 30 min before infection (Right). The MGE in both cases was 103. For analysis of the infectivity, HBsAg- and HBcAg-specific antisera were used. Their antigen-specific binding was detected by secondary antibodies visualized by red and blue fluorescence, respectively. Actin filaments were stained by using FITC-conjugated phalloidine. Photographs were taken at ×200 and ×630 magnification. (B) Confocal microscopy of LMH cells infected with unprocessed DHBV particles or with processed WT DHBV, DHBVD1/2, and DHBVD2 particles that had been preincubated with endosomal lysates from LMH cells for 60 min. As a control, processing of WT DHBV was performed in the presence of a protease inhibitor mixture (Roche). The MGE in all cases was 103. For analysis of the infectivity, a surface-specific serum visualized by the blue fluorescence was used. Photographs were taken at ×630 magnification.
Previous experiments had shown that the TLM functions as a cell-permeable and not a fusogenic peptide, which means that peptides or proteins fused to the TLM are translocated across the membrane into the cytoplasm. To analyze whether the TLM enables the translocation of processed viral particles across the plasma membrane into the cytoplasm, 293 cells were incubated with processed WT HBV, processed TLM-deficient HBV, and unprocessed virus. If a fusogenic step is the acting mechanism, then surface proteins should be enriched in the membrane. However, if cell permeability is enhanced because of membrane translocation, then a prominent cytoplasmic staining should be observed. The confocal fluorescence microscopy shows that cells incubated with endosomally processed WT HBV particles exhibit strong cytoplasmic staining for viral envelope protein (Fig. 9C). These data imply that translocation across the membrane indeed occurs and results in delivery of HBsAg to the cytosol. We conclude from these data that after receptor-mediated endocytosis incoming viral particles are processed in the endosome. This processing leads to exposure of the TLMs on the particle surface, a crucial step for endosomal escape across the membrane into the cytosol (Fig. 5).
Fig. 5.

Model of the endosomal processing of hepadnaviral particles. Hepadnaviruses are internalized by receptor-mediated endocytosis. In the endosomal compartment proteolytic cleavage of the surface protein occurs, resulting in a conformational change that exposes the TLMs (shown as red circles) on the surface of the viral particle. The high density of TLMs exposed on the surface of the particle allows endosomal escape into the cytosol to initiate infection.
Discussion
Published work suggested that hepadnaviral infection relies on an endocytic process (10–12, 18), and the presence of enveloped viral particles in purified endosomal fractions at an early stage of infection was very recently visualized by electron microscopy (unpublished data). However, the mechanisms allowing escape of the virus and the viral nucleocapsid from the endocytotic pathway are unknown. Here we show that integrity of the TLM is a crucial prerequisite for infectivity of hepadnaviruses. Functional impairment of the TLM results in a block of the infectious process at a post-entry step. TLM-deficient viral particles are unable to escape from the endosome into the cytosol whereas WT particles can partially and time-dependently escape. Proteolytic processing of the viral particle in the endosome induces the exposure of the TLMs on the surface of the viral particle, enabling translocation across the membrane into the cytoplasm. The unmasking or generation of entry-mediating sequences during a viral infection process is not unprecedented: Influenza virus homotrimers are assembled as hemagglutinin (HA) 0 precursors that display no fusogenic activity. Endolytic cleavage by a furin-like protease results in formation of HA1 and HA2. This cleavage step primes HA for fusion by liberating sequences in HA for fusion-related conformational changes (19, 20). In the case of poliovirus, the interaction with the receptor results in externalization of the N-myristoylated VP4 peptide and exposure of amphipathic sequences of VP1 that can insert into membranes mediating entry in the cytoplasm (21, 22).
In contrast to other enveloped viruses, the TLM-mediated escape from the endosome is not a fusogenic process. The difference from fusogenic mechanisms is that the TLM does not rest in the membrane at the end of the translocation process (14–16). Therefore, it is likely that the cleaved surface protein remains associated with the nucleocapsid during escape from the endocytotic pathway. The surface protein processing in the endosome and the reducing conditions in the cytoplasm, which destroy disulfide bridges in the S-domain, affect the HBsAg–nucleocapsid interaction and allow removal of the envelope from the nucleocapsid.
Endosomal processing of HBV particles and subsequent infection of nonpermissive hepatoma cell lines demonstrate the potential of surface-exposed TLMs to translocate particles across membranes. Moreover, this experimental procedure can be used to investigate post-entry steps in HBV infection in immortalized hepatoma cell lines.
A previous report describing that HepG2 cells can be infected with very low efficiency by HBV particles after preincubation with V8 proteases (23) has to be reconsidered. It is conceivable that the specificity of V8 protease used in the study is not suitable to induce proper processing and unmasking of the TLMs. Unspecific or contaminating protease activity might have processed a small fraction of viral particles and generated unmasked TLMs.
Our data identify TLMs as conserved motifs of HBVs that are essential for infectivity and suggest an entry mechanism for these and possibly other enveloped viruses, as summarized in the following model (Fig. 5): after receptor-mediated endocytosis of the viral particle and endocytic entry, proteolytic processing of surface proteins occurs, which results in unmasking of TLMs. The endosomal processing generates a modified viral particle that is decorated on its surface with TLMs. The high density of TLMs exposed on the particle surface allows exit from the endosome and subsequent establishment of infection.
Materials and Methods
Generation of TLM-Deficient Genomes.
Generation of mutant DHBV genomes lacking a functional TLM was performed based on the subcloned DHBV genome pDHBV16/1.1 (24) by site-directed mutagenesis using the QuikChange site-directed mutagenesis kit from Stratagene. The sequence nucleotides 858–893 encoding the TLM1 was mutated from CTGTTAAACCAACTTGCCGGAAGGATGATCCCAAAA to CTGTCAAACCAACGTGCCGGAAGGACGACCCCAAAA, resulting in conversion of the corresponding amino acid sequence from LLNQLAGRMIPK to LSNQRAGRTTPK. The sequence nucleotides 924–959 encoding TLM2 was mutated from ACACTAGATCACGTGTTAGACCATGTGCAAACAATG to ACACCAGATCACGAGTCAGACCATGCGCAAACAATG, resulting in conversion of TLDHVLDHVQTM to TPDHESDHAQTM.
Generation of mutant HBV genomes lacking a functional TLM was performed based on the subcloned HBV genome pSPT1.2HBV as described above. The TLM-encoding sequence was mutated from CCCATATCGTCAATCTCCTCGAGGATTGGGGACCCT to CCCACATCGTCAACCTTCTCGACGATTGGGGACCCT, resulting in conversion of PISSISSRTGDP to PTSSTFSTTGDP. Mutated nucleotides are highlighted in bold.
Protein Analysis.
SDS/PAGE was performed according to Laemmli (25). Before loading, samples were adjusted to identical protein concentrations. Gels were loaded with 20 μg of total protein per lane.
For immunoblot analysis, a DHBV-pre-S-specific serum, a DHBV-core-specific serum, and a HBV-core-specific serum generated by immunization of rabbits with the respective purified protein were instrumental. Hexokinase (cytoplasm)-specific, histone H1 (nucleus)-specific, cathepsin B (lysosome)-specific, grb2 (cytoplasm)-specific, clathrin HC (endosome)-specific, and TNF-RI (microsomes)-specific sera (Santa Cruz Biotechnology) were used as controls for equal loading and purity control of subcellular fractions. For detection of eGFP, a polyclonal rabbit serum (Invitrogen) was used. Immunoprecipitations were performed as described recently (26) by using a rabbit-derived TLM-specific serum or a goat-derived HBsAg-specific serum (DAKO). The HBV-TLM-specific antiserum was generated by immunization of rabbit with crosslinked peptide.
Indirect immunofluorescence labeling was performed as described in refs. 17 and 26. cccDNA isolation and detection, isolation of replicative intermediates, and Southern blot analysis were performed as described in refs. 13 and 17.
Cell Culture, Transfection, and Subcellular Fractionation.
Fetal PDHs were prepared and cultivated as described (27). PDHs were seeded into 12-well plates at a density of ≈5 × 105 liver cells per well.
Huh7, HepG2, 293, and LMH cells were cultured in DMEM supplemented with 10% FCS. For production of DHBV particles, 20 6-cm dishes with 0.8 × 106 LMH cells were transfected with 2.5 μg of pDHBV16.1.1 or the respective mutants using N-[1-(2,3-dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate (Roche). Cell culture supernatants were harvested daily between days 3 and 7 after transfection, clarified by centrifugation, and stored at 4°C. Aliquots were polyethylene glycol-precipitated as previously described and resuspended in culture medium. Subcellular fractionation was performed as described (14, 16).
TaqMan PCR.
To quantify virus production after transfection experiments, transfected DNA was first eliminated by DNase I treatment. DNase was then destroyed by heating to 94°C, and viral DNA was purified by using the High Pure viral nucleic acid kit (Roche). For quantification of viral particles in subcellular compartments, proteins were removed by purification of the viral nucleic acid by using the same kit. To quantify virus-specific DNA, primers ntDHBV 1311–1331 and ntDHBV 1398–1377 were instrumental. The probe corresponded to ntDHBV 1343–1376 and was synthesized by IBA (Goettingen, Germany). The assay was calibrated in a range corresponding to 102 to 109 DHBV genomes.
In Vitro Processing of Viral Particles.
HBV- or DHBV-positive sera were subjected to gel filtration on a Superose 6 column (Amersham Pharmacia) on an Aekta purifier system (Amersham Pharmacia). The HBV- or DHBV-positive fractions (nearly identical with the exclusion volume) were pooled, and the amount of viral genomes was quantified by TaqMan PCR. A total of 105 genome equivalents were incubated with endosomal lysate isolated from 2 × 105 HepG2 or LMH cells for 30–60 min. Endosomes were prepared as described (14, 28). Briefly, endosomal lysate was obtained by sonication of the endosomal fraction and resuspended in 20 mM Hepes (pH 5.8)/200 mM NaCl. Protease activity was inhibited with a commercial protease inhibitor mixture (Roche) (see Supporting Materials and Methods, which is published as supporting information on the PNAS web site).
Supplementary Material
Acknowledgments
We thank Dr. P. H. Hofschneider for many helpful and stimulating discussions and for support. We thank Sarah Kinkley for critical reading of the manuscript. The Heinrich Pette Institute is supported by the Freie und Hansestadt Hamburg and the Bundesministerium für Gesundheit und Soziale Sicherung. This work was supported by grants from the Deutsche Forschungsgemeinschaft and by the German Competence Network for Viral Hepatitis funded by German Ministry of Education and Research Grant TP13.1.
Abbreviations
- HBV
hepatitis B virus
- MGE
multiplicity of genome equivalents
- TLM
translocation motif
- PDH
primary duck hepatocyte
- DHBV
duck HBV
- cccDNA
covalently closed circular DNA
- HBsAg
hepatitis B virus surface antigen
- HBcAg
hepatitis B virus core antigen
- HA
hemagglutinin.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Beasley R. P., Hwang L. Y., Lin C. C., Chien C. S. Lancet. 1981;2:1129–1133. doi: 10.1016/s0140-6736(81)90585-7. [DOI] [PubMed] [Google Scholar]
- 2.Buendia M. A. Semin. Cancer Biol. 2000;10:185–200. doi: 10.1006/scbi.2000.0319. [DOI] [PubMed] [Google Scholar]
- 3.Tuttleman J. S., Pugh J. C., Summers J. W. J. Virol. 1986;58:17–25. doi: 10.1128/jvi.58.1.17-25.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pugh J. C., Summers J. W. Virology. 1989;172:564–572. doi: 10.1016/0042-6822(89)90199-2. [DOI] [PubMed] [Google Scholar]
- 5.Glebe D., Aliakbari M., Krass P., Knoop E. V., Valerius K. P., Gerlich W. H. J. Virol. 2003;77:9511–9521. doi: 10.1128/JVI.77.17.9511-9521.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neurath A. R., Kent S. B., Strick N., Parker K. Cell. 1986;46:429–436. doi: 10.1016/0092-8674(86)90663-x. [DOI] [PubMed] [Google Scholar]
- 7.Paran N., Geiger B., Shaul Y. EMBO J. 2001;20:4443–4453. doi: 10.1093/emboj/20.16.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klingmuller U., Schaller H. J. Virol. 1993;67:7414–7422. doi: 10.1128/jvi.67.12.7414-7422.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urban S., Breiner K. M., Fehler F., Klingmuller U., Schaller H. J. Virol. 1998;72:8089–8097. doi: 10.1128/jvi.72.10.8089-8097.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kock J., Borst E. M., Schlicht H. J. J. Virol. 1996;70:5827–5831. doi: 10.1128/jvi.70.9.5827-5831.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breiner K. M., Urban S., Schaller H. J. Virol. 1998;72:8098–8104. doi: 10.1128/jvi.72.10.8098-8104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Breiner K. M., Schaller H. J. Virol. 2000;74:2203–2209. doi: 10.1128/jvi.74.5.2203-2209.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Funk A., Mhamdi M., Lin L., Will H., Sirma H. J. Virol. 2004;78:8289–8300. doi: 10.1128/JVI.78.15.8289-8300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oess S., Hildt E. Gene Ther. 2000;7:750–758. doi: 10.1038/sj.gt.3301154. [DOI] [PubMed] [Google Scholar]
- 15.Saher G., Hildt E. J. Biol. Chem. 1999;274:27651–27657. doi: 10.1074/jbc.274.39.27651. [DOI] [PubMed] [Google Scholar]
- 16.Hafner A., Brandenburg B., Hildt E. EMBO Rep. 2003;4:767–773. doi: 10.1038/sj.embor.embor903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Funk A., Hohenberg H., Mhamdi M., Will H., Sirma H. J. Virol. 2004;78:3977–3983. doi: 10.1128/JVI.78.8.3977-3983.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breiner K. M., Schaller H., Knolle P. A. Hepatology. 2001;34:803–808. doi: 10.1053/jhep.2001.27810. [DOI] [PubMed] [Google Scholar]
- 19.Klenk H. D., Rott R., Orlich M., Blodorn J. Virology. 1975;68:426–439. doi: 10.1016/0042-6822(75)90284-6. [DOI] [PubMed] [Google Scholar]
- 20.Skehel J. J., Wiley D. C. Annu. Rev. Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 21.Belnap D. M., Filman D. J., Trus B. L., Cheng N., Booy F. P., Conway J. F., Curry S., Hiremath C. N., Tsang S. K., Steven A. C., Hogle J. M. J. Virol. 2000;74:1342–1354. doi: 10.1128/jvi.74.3.1342-1354.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fricks C. E., Hogle J. M. J. Virol. 1990;64:1934–1945. doi: 10.1128/jvi.64.5.1934-1945.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu X., Block T. M., Gerlich W. H. J. Virol. 1996;70:2277–2285. doi: 10.1128/jvi.70.4.2277-2285.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sprengel R., Kuhn C., Will H., Schaller H. J. Med. Virol. 1985;15:323–333. doi: 10.1002/jmv.1890150402. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli U. K. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Hildt E., Munz B., Saher G., Reifenberg K., Hofschneider P. H. EMBO J. 2002;21:525–535. doi: 10.1093/emboj/21.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pult I., Abbott N., Zhang Y. Y., Summers J. J. Virol. 2001;75:9623–9632. doi: 10.1128/JVI.75.20.9623-9632.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campbell C., Squicciarini J., Shia M., Pilch P. F., Fine R. E. Biochemistry. 1984;23:4420–4426. doi: 10.1021/bi00314a028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}