

Abstract
Neuropeptide Y (NPY) is thought to have a major role in the physiological control of energy homeostasis. Among five NPY receptors described, the NPY Y5 receptor (Y5R) is a prime candidate to mediate some of the effects of NPY on energy homeostasis, although its role in physiologically relevant rodent obesity models remains poorly defined. We examined the effect of a potent and highly selective Y5R antagonist in rodent obesity and dietary models. The Y5R antagonist selectively ameliorated diet-induced obesity (DIO) in rodents by suppressing body weight gain and adiposity while improving the DIO-associated hyperinsulinemia. The compound did not affect the body weight of lean mice fed a regular diet or genetically obese leptin receptor-deficient mice or rats, despite similarly high brain Y5R receptor occupancy. The Y5R antagonist acts in a mechanism-based manner, as the compound did not affect DIO of Y5R-deficient mice. These results indicate that Y5R is involved in the regulation and development of DIO and suggest utility for Y5R antagonists in the treatment of obesity.
Keywords: antiobesity effect, Y5R-deficient mice, receptor occupancy
Neuropeptide Y (NPY), a 36-aa peptide neurotransmitter, is one of the most potent orexigenic substances when injected into the brain. NPY expression is widely distributed in the CNS, including the hypothalamus, a region involved in energy homeostasis (1). NPY content and mRNA levels in the hypothalamus respond to feeding status, including food deprivation and refeeding (2, 3). Chronic central infusion of NPY in rodents results in a syndrome similar to that in some genetic obesity models, characterized by hyperphagia, insulin resistance, hyperinsulinemia, and reduced thermogenic activity in brown adipose tissue (4). NPY is, therefore, thought to have a major role in the physiological control of energy homeostasis, making it a target for the development of antiobesity agents.
Five types of NPY receptors have been characterized (5). Pharmacological data suggest that the NPY Y5 receptor (Y5R) is involved in feeding regulation. For example, functional and binding activities of different peptide agonists at the Y5R in vitro correlated strongly with their efficacy in stimulating food intake (6). Administration of Y5R antagonists suppressed Y5R agonist-induced feeding (7), and mice lacking the Y5R showed a diminished response to exogenously administered Y5R agonists (8). The Y5R is also reported to regulate brown fat thermogenesis and energy expenditure (9). In addition, chronic intracerebroventricular administration of a Y5R-specific agonist, D-Trp-34NPY, produces obesity (10). These findings suggest that the Y5R is involved in the development of obesity. However, the physiological role of the Y5R in obesity models, rather than models that rely on exogenously added Y5R agonists, remains undefined.
We reported that orally administered Y5R antagonist [2-(3,3-dimethyl-1-oxo-4H-1H-xanthen-9-yl)-5,5-dimethyl-cyclohexane-1,3-dione] reduced Y5R agonist-induced feeding and Y5R agonist-induced obesity (7, 10). To investigate the physiological role of the Y5R in the development of obesity, we examined the effects of this Y5R antagonist in mice subjected to diet-induced obesity (DIO), leptin receptor-deficient (Leprdb/db) mice, and Zucker fatty rats. We show that chronic administration of the Y5R antagonist selectively suppressed body weight gain in the mice with DIO without affecting the other models.
Results
A Y5R Antagonist Can Ameliorate Obesity in Mice.
We administered the Y5R antagonist (30 and 100 mg/kg, orally) for 2 wk to mice consuming a moderately high-fat (MHF) diet. The untreated mice gained 2 g of body weight over the 2-wk period. The Y5R antagonist significantly suppressed the diet-induced body weight gain in a dose-dependent manner (Fig. 1A). The mice on an MHF diet ingested 35% more calories during the test period than mice fed a regular diet. The Y5R antagonist reduced the mean overall caloric intake of an MHF diet by 7.6% and 10.0% at 30 and 100 mg/kg, respectively (Fig. 1B), when compared with controls that did not receive the drug. The Y5R antagonist did not affect mean water intake by mice on an MHF diet at any dose tested (data not shown).
Fig. 1.

Oral treatment with the Y5R antagonist inhibited the body weight gain in mice fed an MHF diet. (A) Body weight change. (B) Mean caloric intake. Veh., vehicle. Data are expressed as means ± SE (n = 8–9). ∗, P < 0.05 (compared with vehicle).
At the end of the study we measured fat content and glucose and insulin levels. Mice on an MHF diet gained 3- to 4-fold more fat in the mesenteric adipose tissue than did the lean mice. Treatment with the Y5R antagonist decreased fat pad weight (Fig. 2A). Histological analysis indicated that adipose cell size in the obese mice was twice that of lean mice and that the Y5R antagonist significantly reduced adipose cell size (data not shown). Plasma insulin levels in the obese mice were elevated 4-fold compared with those in lean mice. Treatment with the Y5R antagonist suppressed this elevation in insulin (Fig. 2C). Plasma glucose levels were not significantly different in any of the groups (Fig. 2B).
Fig. 2.

Effect of the Y5R antagonist on fat weight and plasma glucose and insulin levels. (A) Mesenteric fat weight. (B) Plasma glucose levels. (C) Plasma insulin levels. Data are expressed as means ± SE (n = 8–9). ∗, P < 0.05 (compared with vehicle).
DIO in Y5R-Deficient Mice.
To examine whether the antiobesity effects of the Y5R antagonist are mechanism-based, we administered the Y5R antagonist to knockout (KO) mice lacking the Y5R (Y5R KO) in a crossover study with three dosing phases (14–19 days each arm). Y5R KO mice gained more weight when fed an MHF diet (8.3 g) than did wild-type mice (6.8 g). The Y5R antagonist (100 mg/kg) suppressed the body weight gain in the wild-type mice (Fig. 3A and Table 1); this effect was reproducible as observed from the crossover study. By contrast, the Y5R antagonist had no significant effect on body weight gain in Y5R KO mice throughout the experiment (Fig. 3B and Table 1). Similar results were obtained with wild-type and Y5R KO littermates in a separate study (data not shown). We conclude that the effects of the Y5R antagonist on body weight and appetite are Y5R-dependent and, hence, mechanism-based.
Fig. 3.

Effect of the Y5R antagonist in wild-type (A) and Y5R KO (B) mice. Open symbols represent vehicle, Y5R antagonist (100 mg/kg), and vehicle during the first, second, and third phases, respectively. Filled symbols represent Y5R antagonist, vehicle, and Y5R antagonist, respectively. Data are expressed as means ± SE (n = 4–6).
Table 1.
Body weight change during each crossover dosing arm
| Mouse | Body weight change, g | ||
|---|---|---|---|
| First arm (days 1–19) | Second arm (days 20–33) | Third arm (days 34–46) | |
| Wild type | |||
| Vehicle | 3.60 ± 0.31 | 3.18 ± 0.37 | 1.75 ± 0.54 |
| Y5R antagonist | 1.62 ± 0.61* | 0.05 ± 0.32† | −1.06 ± 0.30† |
| Y5R KO | |||
| Vehicle | 3.12 ± 0.49 | 2.22 ± 0.67 | 2.32 ± 0.40 |
| Y5R antagonist | 3.68 ± 0.38 | 1.15 ± 0.40 | 1.42 ± 0.32 |
Data represent mean ± SE of four to six animals.
*, P < 0.05;
†, P < 0.01 (compared with vehicle-treated group; Student's t test).
No Effect of Y5R Antagonist in Lean Mice or Genetic Obesity Models.
Wild-type Sprague–Dawley rats fed an MHF diet are also sensitive to the Y5R antagonist (data not shown). However, the Y5R antagonist (30 and 100 mg/kg) did not affect body weight gain (or food intake; data not shown) in lean mice fed a regular diet (Fig. 4A), obese Leprdb/db mice fed a regular diet (Fig. 4B), or Zucker fatty rats (Fig. 4D). We also tested Leprdb/db mice on an MHF diet; although they gained more weight than mice on the regular diet, the Y5R antagonist had no effect on either diet (Fig. 4C). There were no significant effects on adipose tissue weight or plasma biochemical parameters of these animals (data not shown).
Fig. 4.

Effect of the Y5R antagonist in lean mice and genetic obesity models. (A) Lean mice. (B) Leprdb/db mice. (C) Leprdb/db mice on MHF diet. (D) Zucker fatty rats. Data are expressed as means ± SE (n = 7–9).
Y5R Occupancy and Plasma Drug Levels.
Brain Y5R occupancy and plasma drug levels were measured to examine whether the ineffectiveness of the Y5R antagonist in some models was due to insufficient receptor occupancy. In lean C57BL/6J mice, a dose- and time-dependent Y5R occupancy was observed (Fig. 5A). A similarly high occupancy of brain Y5R was observed for at least 15 h after a single oral administration in mice fed an MHF diet or in genetically obese Leprdb/db mice or Zucker fatty rats (Fig. 5 A and B). Plasma levels of the drug exceeding 10 μM were sustained for up to 15 h in each of the models (Fig. 5C). Plasma drug levels were similar in the Y5R KO and the wild-type mice (Fig. 5D). The lean mice and Leprdb/db mice exposed to a regular diet had a shorter t1/2 of the Y5R antagonist than did mice fed an MHF diet or Leprdb/db mice and Zucker fatty rats; however, this did not affect brain Y5R occupancy.
Fig. 5.
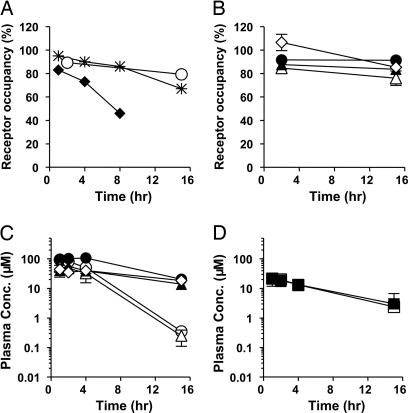
Brain Y5R occupancy and drug exposure levels after oral administration of the Y5R antagonist. (A and B) Brain Y5R occupancy in lean C57BL/6J mice (A) and other obesity models (B). (C and D) Plasma concentration of the Y5R antagonist. In A, filled diamonds, asterisks, and open circles represent 10, 30, and 100 mg/kg, respectively, in lean mice. In B and C, filled circles represent mice with DIO, open circles represent lean mice, open triangles represent Leprdb/db mice, filled triangles represent Leprdb/db mice on an MHF diet, and open diamonds represent Zucker fatty rats. In D, open squares represent wild-type mice, and filled squares represent Y5R KO mice. Data are expressed as means ± SD (n = 3).
Discussion
Previous studies show that Y5R agonists administered into the brain can stimulate food intake and body weight gain (6, 10). Our findings indicate that a highly selective Y5R antagonist can selectively suppress body weight gain in mice and rats exposed to DIO. The weight-controlling effects are accompanied by a reduction of food intake and body fat accumulation. Our unpublished data indicate that both food intake and energy expenditure are impacted by the Y5 receptor (data not shown). Any behavioral abnormalities were not observed during the treatment period. It is possible that the Y5R antagonist changes the dietary preference in normal rodents, because it works only when normal rodents are fed an MHF diet. The Y5R antagonist was ineffective in Y5R KO mice, indicating that the antiobesity effects are attributable to a specific blockade of the Y5R. This assertion is corroborated by the finding that Y5R binding does not occur in Y5R KO brain tissue (data not shown), whereas the Y5R antagonist fully displaces the binding of a distinct Y5R ligand in the wild-type brain. We propose that occupancy of Y5R mediates the development of dietary obesity in rodents, whereas homeostatic control mechanisms intervene in lean rodents or rodents with leptin receptor deficiency. Because similarly high occupancy of Y5R was observed in each model, it is unlikely that the ineffectiveness of the Y5R antagonist in some models is due to differences in drug exposure levels. Thus, the antiobesity effect of the Y5R antagonist appears to be specific to the DIO model.
Several Y5R antagonists have been evaluated in vivo by us and by others (11–16); however, a clear role for Y5R in physiological feeding was not established. Two Y5 antagonists, CGP71683A and GW438014A, were reported to suppress body weight gain in both diet-induced and genetically obese models (11, 12). A recent publication suggests that the CGP71683A effects are probably not mechanism-based, because CGP71683A has potent affinities for muscarinic receptors and the serotonin transporter (13). Moreover, the compound was reported to be equally effective in Y5R KO mice (14). GW438014A has not been tested in Y5R KO mice. Although a benzonitrile derivative compound 1 attenuated Y5R agonist-induced food intake in rats, it did not inhibit overnight food intake, and the compound was not tested in chronic DIO models (15). Although a series of arylimidazoles produced variable responses in acute feeding models, these compounds were not tested in chronic DIO models (16). Another Y5R antagonist, NPY5RA-972, was reported to be ineffective in Wistar rats with DIO (14), whereas the Y5R antagonist used in our study is effective in Sprague–Dawley rats with DIO (A.I., unpublished data). A difference in the pharmacodynamic profiles of each of these Y5R antagonists might account for the differing results. Our unpublished studies indicate that high and sustained Y5R occupancy is required for efficacy in rodents with DIO (data not shown).
Because the Y5R antagonist was ineffective in lean rodents fed regular chow, an involvement of Y5R in feeding regulation and obesity may be absent in normal animals. On the other hand, DIO or diet change to an MHF diet may increase the extent of the contribution of Y5R under these conditions, which renders the Y5R antagonist effective. The DIO-specific effects observed with the Y5R antagonist might be explained by variability in the patterns of NPY expression and its receptors in various rodent models. The density of Y2/Y5-like receptors, which recognize a Y2/Y5-preferring agonist, decreased significantly in a number of hypothalamic and nonhypothalamic brain regions of Zucker fatty rats and Lepob/ob mice, as compared with lean rodents (17, 18), whereas Y2/Y5-like receptors are up-regulated in the hypothalamus of DIO rats (19). Although expression of NPY mRNA in the arcuate nucleus was increased in Zucker fatty rats (20), mice with DIO displayed a profound induction of NPY expression in the dorsomedial hypothalamic and ventromedial hypothalamic nuclei, whereas the level of NPY mRNA in the arcuate nucleus was reduced (21). Therefore, it is possible that differences in NPY and NPY receptor expression among obesity models impact the contribution of NPY to the control of energy homeostasis. The changes in Y5R antagonist sensitivity might occur in a diet-dependent manner, because exposing mice to a different diet impacts Y5R antagonist efficacy (Fig. 1A).
The increased expression levels of NPY mRNA in dorsomedial hypothalamic nuclei (DMH) of mice with DIO might help to explain the selective efficacy of the Y5R antagonist against DIO. It has been reported that DMH-lesioned rats are more sensitive to leptin than intact animals (22) and that chronic exposure to a high-fat diet causes leptin resistance (23, 24). Thus, NPY expression in the DMH may function to diminish the action of leptin by reducing leptin sensitivity. In DIO animals, the tonic inhibitory action of NPY may be enhanced, and the Y5 antagonist may elicit its antiobesity effects by improving leptin sensitivity. This idea is in line with our findings that the Y5 antagonist is ineffective in Zucker fatty rats and Leprdb/db mice in which leptin function is disrupted. Further investigation is needed to elucidate the precise antiobesity mechanism(s) of this Y5R antagonist.
The moderately obese phenotype of Y5R KO mice contrasts the antiobesity effects observed with the Y5R antagonist (ref. 25; S.M., unpublished data). In addition, we have shown that the Y5R KO mice appear more sensitive to developing DIO than their wild-type controls. However, this finding does not negate the hypothesis that the Y5R system is involved in DIO, because the Y5R system is likely not the only CNS pathway involved in the DIO responsiveness. In the case of genetically engineered KO mice, absence of receptors from the early stage of development may cause compensatory mechanisms to be activated that are not observed when compared to treatment with a pharmacological agent in adult rodents. Although details of a discrepancy between genetic and pharmacological ablation are not clear, it may be relevant to consider compensatory effects from other NPY receptors. A selective NPY Y1 receptor (Y1R) antagonist with oral bioavailability has antiobesity effects in Zucker fatty rats (26), suggesting that Y1R is an important contributor to obesity in this model. However, paradoxically, Y1R-deficient mice also develop late-onset obesity (refs. 27 and 28; S.M., unpublished data). An NPY Y2 receptor (Y2R) preferring agonist PYY3–36 (peripherally administered) reduced body weight gain in rats (29), whereas centrally administered Y2R agonists increase food intake. Phenotype of Y2R deletion seems to be controversial; both an increase and a decrease in body weight are reported (30, 31). These observations indicate that the route of administration can affect efficacy of NPY receptor agonists. Perhaps the complete elimination of the Y5R signal during embryonic development results in compensatory mechanisms, which include the activation of the Y1R or the inhibition of the Y2R. Combination studies employing both Y5R and Y1R antagonists along with Y2R agonists could address this hypothesis.
In conclusion, our studies reveal a relevant role for the Y5R only under conditions where diet produces obesity. Both environmental factors (such as unlimited access to high-calorie food and limited physical exercise) and genetic factors that predispose to weight gain contribute to the development of human obesity (32, 33). Because the incidence of monogenic forms of human obesity is low and most human obesity is thought to occur in response to high-calorie diets (34, 35), the DIO model is particularly applicable to human obesity. Our results suggest a potential therapeutic role for Y5R antagonists in the treatment of human obesity.
Methods
Drugs.
The Y5R antagonist [2-(3,3-dimethyl-1-oxo-4H-1H-xanthen-9-yl)-5,5-dimethyl-cyclohexane-1,3-dione] was synthesized by Banyu Pharmaceutical. The specificity of the Y5R antagonist in vitro was reported (10, 36). Briefly, the Y5R antagonist displaced 125I peptide YY binding to human and rat Y5R with Ki values of 26 and 31 nM, respectively, and inhibited the NPY (100 nM)-induced increase in intracellular calcium levels via the human Y5R (IC50 = 210 nM). The Y5R antagonist did not show significant affinity for human NPY receptors at a dose of 10 μM or any significant cross-reactivity with 120 other binding assays and seven enzyme assays, including adrenergic, dopaminergic, GABAergic, histaminergic, and serotonergic receptors, which are considered to be involved in feeding regulation (data not shown). All other chemicals were of the analytical grade.
Animals.
All animals except for Y5R KO mice were purchased from CLEA Japan (Tokyo). Male Y5R KO mice were generated as described (8). Animals were housed individually in plastic cages and kept in temperature- and humidity-controlled rooms at 23 ± 2°C, at 55 ± 15% relative humidity, and on a 12-h light/12-h dark cycle (lights off at 1900 hours). Water and regular diet (CE-2; CLEA Japan) were available ad libitum. All experimental procedures followed the Japanese Pharmacological Society Guideline for Animal Use.
DIO.
Male C57BL/6J mice (22–23 wk old) were divided into three groups to match average values of basal food intake and body weight. Each group was orally administered either vehicle (0.5% methylcellulose in distilled water) or the Y5R antagonist at doses of 30 or 100 mg/kg daily for 2 wk by gavage, and food and water intake and body weight were measured. The diet was changed to an MHF diet (32.6% calories as fat, 4.41 kcal/g; Oriental BioService Kanto, Tsukuba, Japan) 2 days after the beginning of the drug administration (DIO). Another set of mice was divided into three groups, administered the Y5R antagonist or the vehicle similarly, and fed a regular diet throughout the experiment. After the final administration, mice were fasted overnight. Blood samples were collected under ether anesthesia for measurement of plasma parameters.
Y5R KO Mice.
Y5R KO and wild-type mice (12 wk old) were fed an MHF diet for 3–4 wk. Each genotype of mice was divided into two groups (n = 4–6), and they were enrolled in a 45-day crossover study with three 14- to 19-day dosing phases (100 mg/kg once daily).
Leprdb/db Mice.
Effects of 16-day oral administration of the Y5R antagonist (30 or 100 mg/kg once daily; n = 8–9) were examined in male Leprdb/db mice (8 wk old) on an MHF diet or a regular diet under the same protocol as that described for mice with DIO.
Zucker Fatty Rats.
Effect of 2-wk oral administration of the Y5R antagonist (30 or 100 mg/kg once daily) was examined in male Zucker fatty rats fed a regular diet (10 wk old; n = 7–8).
Plasma Measurements.
Plasma glucose levels were measured with commercial kits (Kyowa Medex, Tokyo). Plasma insulin levels were measured with ELISA kits (Morinaga, Kanagawa, Japan).
Brain Y5R Occupancy.
The Y5R antagonist (100 mg/kg) was orally administered by gavage to C57BL/6J and Leprdb/db mice fed a regular or an MHF diet and Zucker fatty rats (n = 3). Two and 15 h after drug administration animals were killed by collecting whole blood from the heart under isoflurane anesthesia. In lean C57BL/6J mice, samples were also taken at 1, 4, 8, and 15 h after administration at 10 and 30 mg/kg. Frozen coronal sections (20 μm) of the striatal region were prepared. Because we have confirmed that equivalent Y5R occupancy was observed in both striatal and posterior hypothalamic regions after oral administration of the Y5R antagonist in the previous studies (data not shown), striatal sections were used for the Y5R occupancy assay. Briefly, brain sections were incubated with a 100 pM concentration of another Y5R antagonist with 35S label (compound A; Banyu Pharmaceutical) in 50 mM Tris·HCl buffer. Compound A is a spiroindoline class Y5R antagonist, binds to Y5R with high affinity (IC50 for human Y5R = 0.99 nM), and is highly selective for Y5R because the binding was displaceable by Y5R-selective compounds and was completely abolished in the Y5R KO mouse brain (data not shown). Nonspecific binding was evaluated by using an adjacent brain slice by the addition of a 10,000-fold excess of nonlabeled Y5R antagonist. The sections were exposed to BAS 5000 imaging plates (Fuji, Kanagawa, Japan), and autoradiographic images were quantified. Ex vivo receptor labeling by 35S compound A in drug-treated animals was calculated and expressed as follows: RO (%) = 100 × [1 − (receptor labeling of drug-treated animals/receptor labeling of vehicle-treated animals)].
Plasma Levels of the Y5R Antagonist.
The Y5R antagonist (100 mg/kg) was orally administered to three animals per strain, and blood samples were collected at 1, 2, 4, and 15 h. Plasma levels of the Y5R antagonist after oral dosing were measured by HPLC.
Statistical Analysis.
Data are expressed as means ± SE or SD. Differences between the values were considered significant at P < 0.05. Body weight changes were analyzed by repeated-measures ANOVA followed by the Bonferroni test. Measurements at single time points were analyzed by a one-tailed Dunnett test. Body weight changes during each crossover dosing arm were analyzed by Student's t test.
Abbreviations
- NPY
neuropeptide Y
- Y1R
NPY Y1 receptor
- Y2R
NPY Y2 receptor
- Y5R
NPY Y5 receptor
- DIO
diet-induced obesity
- MHF
moderately high-fat
- KO
knockout
- Leprdb/db
leptin receptor-deficient.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Stanley B. G. In: The Biology of Neuropeptide Y and Related Peptides. Colmers W. F., Wahlestedt C., editors. Totowa, NJ: Humana; 1993. pp. 457–509. [Google Scholar]
- 2.Beck B., Jhanwar-Uniyal M., Burlet A., Chapleur-Chateau M., Leibowitz S. F., Burlet C. Brain Res. 1990;528:245–249. doi: 10.1016/0006-8993(90)91664-3. [DOI] [PubMed] [Google Scholar]
- 3.Sahu A., White J. D., Kalra P. S., Kalra S. P. Mol. Brain Res. 1992;15:15–18. doi: 10.1016/0169-328x(92)90145-2. [DOI] [PubMed] [Google Scholar]
- 4.Stanley B. G., Kyrkouli S. E., Lampert S., Leibowitz S. F. Peptides. 1986;7:1189–1192. doi: 10.1016/0196-9781(86)90149-x. [DOI] [PubMed] [Google Scholar]
- 5.Blomqvist A. G., Herzog H. Trends Neurosci. 1997;20:294–298. doi: 10.1016/s0166-2236(96)01057-0. [DOI] [PubMed] [Google Scholar]
- 6.Gerald C., Walker M. W., Criscione L., Gustafson E. L., Batzl-Hartmann C., Smith K. E., Vaysse P., Durkin M. M., Laz T. M., Linemeyer D. L., et al. Nature. 1996;382:168–171. doi: 10.1038/382168a0. [DOI] [PubMed] [Google Scholar]
- 7.Sato N., Takahashi T., Shibata T., Haga Y., Sakuraba A., Hirose M., Sato M., Nonoshita K., Koike Y., Kitazawa H., et al. J. Med. Chem. 2003;46:666–669. doi: 10.1021/jm025513q. [DOI] [PubMed] [Google Scholar]
- 8.Kanatani A., Mashiko S., Murai N., Sugimoto N., Ito J., Fukuroda T., Fukami T., Morin N., MacNeil D. J., Van der Ploeg L. H., et al. Endocrinology. 2000;141:1011–1016. doi: 10.1210/endo.141.3.7387. [DOI] [PubMed] [Google Scholar]
- 9.Hwa J. J., Witten M. B., Williams P., Ghibaudi L., Gao J., Salisbury B. G., Mullins D., Hamud F., Strader C. D., Parker E. M. Am. J. Physiol. 1999;277:R1428–R1434. doi: 10.1152/ajpregu.1999.277.5.R1428. [DOI] [PubMed] [Google Scholar]
- 10.Mashiko S., Ishihara A., Iwaasa H., Sano H., Oda Z., Ito J., Yumoto M., Okawa M., Suzuki J., Fukuroda T., et al. Endocrinology. 2003;144:1793–1801. doi: 10.1210/en.2002-0119. [DOI] [PubMed] [Google Scholar]
- 11.Criscione L., Rigollier P., Batzl-Hartmann C., Rüeger H., Stricker-Krongrad A., Wyss P., Brunner L., Whitebread S., Yamaguchi Y., Gerald C., et al. J. Clin. Invest. 1998;102:2136–2145. doi: 10.1172/JCI4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daniels A. J., Grizzle M. K., Wiard R. P., Matthews J. E., Heyer D. Regul. Pept. 2002;106:47–54. doi: 10.1016/s0167-0115(02)00034-4. [DOI] [PubMed] [Google Scholar]
- 13.Della Zuana O., Sadlo M., Germain M., Félétou M., Chamorro S., Tisserand F., de Montrion C., Boivin J. F., Duhault J., Boutin J. A., et al. Int. J. Obes. 2001;25:84–94. doi: 10.1038/sj.ijo.0801472. [DOI] [PubMed] [Google Scholar]
- 14.Turnbull A. V., Ellershaw L., Masters D. J., Birtles S., Boyer S., Carroll D., Clarkson P., Loxham S. J. G., McAulay P., Teague J. L., et al. Diabetes. 2002;51:2441–2449. doi: 10.2337/diabetes.51.8.2441. [DOI] [PubMed] [Google Scholar]
- 15.Elliott R. L., Oliver R. M., Hammond M., Patterson T. A., She L., Hargrove D. M., Martin K. A., Maurer T. S., Kalvass J. C., Morgan B. P., et al. J. Med. Chem. 2003;46:670–673. doi: 10.1021/jm025584p. [DOI] [PubMed] [Google Scholar]
- 16.Elliott R. L., Oliver R. M., LaFlamme J. A., Gillaspy M. L., Hammond M., Hank R. F., Maurer T. S., Baker D. L., DaSilva-Jardine P. A., Stevenson R. W., et al. Bioorg. Med. Chem. Lett. 2003;13:3593–3596. doi: 10.1016/s0960-894x(03)00747-9. [DOI] [PubMed] [Google Scholar]
- 17.Widdowson P. S. Brain Res. 1997;758:17–28. doi: 10.1016/s0006-8993(97)00160-1. [DOI] [PubMed] [Google Scholar]
- 18.Xin X. G., Huang X. F. NeuroReport. 1998;9:737–741. doi: 10.1097/00001756-199803090-00032. [DOI] [PubMed] [Google Scholar]
- 19.Widdowson P. S., Upton R., Henderson L., Buckingham R., Wilson S., Williams G. Brain Res. 1997;774:1–10. doi: 10.1016/s0006-8993(97)81680-0. [DOI] [PubMed] [Google Scholar]
- 20.Sanacora G., Kershaw M., Finkelstein J. A., White J. D. Endocrinology. 1990;127:730–737. doi: 10.1210/endo-127-2-730. [DOI] [PubMed] [Google Scholar]
- 21.Guan X. M., Yu H., Trumbauer M., Frazier E., Van der Ploeg L. H., Chen H. NeuroReport. 1998;9:3415–3419. doi: 10.1097/00001756-199810260-00015. [DOI] [PubMed] [Google Scholar]
- 22.Choi S., Dallman M. F. Endocrinology. 1999;140:4081–4088. doi: 10.1210/endo.140.9.6964. [DOI] [PubMed] [Google Scholar]
- 23.Van Heek M., Compton D. S., France C. F., Tedesco R. P., Fawzi A. B., Graziano M. P., Sybertz E. J., Strader C. D., Davis H. R., Jr J. Clin. Invest. 1997;99:385–390. doi: 10.1172/JCI119171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Widdowson P. S., Upton R., Buckingham R., Arch J., Williams G. Diabetes. 1997;46:1782–1785. doi: 10.2337/diab.46.11.1782. [DOI] [PubMed] [Google Scholar]
- 25.Marsh D. J., Hollopeter G., Kafer K. E., Palmiter R. D. Nat. Med. 1998;4:718–721. doi: 10.1038/nm0698-718. [DOI] [PubMed] [Google Scholar]
- 26.Ishihara A., Kanatani A., Okada M., Hidaka M., Tanaka T., Mashiko S., Gomori A., Kanno T., Hata M., Kanesaka M., et al. Br. J. Pharmacol. 2002;136:341–346. doi: 10.1038/sj.bjp.0704696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedrazzini T., Seydoux J., Künstner P., Aubert J. F., Grouzmann E., Beermann F., Brunner H. R. Nat. Med. 1998;4:722–726. doi: 10.1038/nm0698-722. [DOI] [PubMed] [Google Scholar]
- 28.Kushi A., Sasai H., Koizumi H., Takeda N., Yokoyama M., Nakamura M. Proc. Natl. Acad. Sci. USA. 1998;95:15659–15664. doi: 10.1073/pnas.95.26.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Batterham R. L., Cowley M. A., Small C. J., Herzog H., Cohen M. A., Dakin C. L., Wren A. M., Brynes A. E., Low M. J., Ghatei M. A., et al. Nature. 2002;418:650–654. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- 30.Sainsbury A., Schwarzer C., Couzens M., Fetissov S., Furtinger S., Jenkins A., Cox H. M., Sperk G., Hökfelt T., Herzog H. Proc. Natl. Acad. Sci. USA. 2002;99:8938–8943. doi: 10.1073/pnas.132043299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naveilhan P., Hassani H., Canals J. M., Ekstrand A. J., Larefalk A., Chhajlani V., Arenas E., Gedda K., Svensson L., Thoren P., et al. Nat. Med. 1999;5:1188–1193. doi: 10.1038/13514. [DOI] [PubMed] [Google Scholar]
- 32.Hill J. O., Peters J. C. Science. 1998;280:1371–1374. doi: 10.1126/science.280.5368.1371. [DOI] [PubMed] [Google Scholar]
- 33.Arner P. Br. J. Nutr. 2000;83(Suppl. 1):S9–S16. doi: 10.1017/s0007114500000891. [DOI] [PubMed] [Google Scholar]
- 34.Astrup A., Buemann B., Western P., Toubro S., Raben A., Christensen N. J. Am. J. Clin. Nutr. 1994;59:350–355. doi: 10.1093/ajcn/59.2.350. [DOI] [PubMed] [Google Scholar]
- 35.Sims E. A. H. Med. Clin. North Am. 1989;73:97–110. doi: 10.1016/s0025-7125(16)30694-0. [DOI] [PubMed] [Google Scholar]
- 36.Kanatani A., Ishihara A., Iwaasa H., Nakamura K., Okamoto O., Hidaka M., Ito J., Fukuroda T., MacNeil D. J., Van der Ploeg L. H. T., et al. Biochem. Biophys. Res. Commun. 2000;272:169–173. doi: 10.1006/bbrc.2000.2696. [DOI] [PubMed] [Google Scholar]