

Abstract
Complete atrioventricular canal (CAVC), also referred to as complete atrioventricular septal defect, is characterised by an ostium primum atrial septal defect, a common atrioventricular valve and a variable deficiency of the ventricular septum inflow. CAVC is an uncommon congenital heart disease, accounting for about 3% of cardiac malformations. Atrioventricular canal occurs in two out of every 10,000 live births. Both sexes are equally affected and a striking association with Down syndrome was found. Depending on the morphology of the superior leaflet of the common atrioventricular valve, 3 types of CAVC have been delineated (type A, B and C, according to Rastelli's classification). CAVC results in a significant interatrial and interventricular systemic-to-pulmonary shunt, thus inducing right ventricular pressure and volume overload and pulmonary hypertension. It becomes symptomatic in infancy due to congestive heart failure and failure to thrive. Diagnosis of CAVC might be suspected from electrocardiographic and chest X-ray findings. Echocardiography confirms it and gives anatomical details. Over time, pulmonary hypertension becomes irreversible, thus precluding the surgical therapy. This is the reason why cardiac catheterisation is not mandatory in infants (less than 6 months) but is indicated in older patients if irreversible pulmonary hypertension is suspected. Medical treatment (digitalis, diuretics, vasodilators) plays a role only as a bridge toward surgery, usually performed between the 3rd and 6th month of life.
Disease name and synonyms
Complete atrioventricular canal (CAVC); Common atrioventricular canal; Complete atrioventricular septal defect.
European paediatric cardiac code
Reference of Complete atrioventricular canal is 06.06.09.
Definition
CAVC is a complex cardiac malformation characterised by a variable deficiency of the atrioventricular area (crux cordis) in the developing heart. The malformation involves the atrial, ventricular and atrioventricular septa and both atrioventricular valves.
Diagnosis criteria
Diagnosis of CAVC might be clinically suspected in patients presenting in the first few months of life with congestive heart failure, cardiomegaly on chest X-ray and left axis deviation, bi-atrial enlargement and bi-ventricular pressure and volume overload on electrocardiogram (ECG). Echocardiography is the key tool for the diagnosis and anatomic classification of this malformation. It shows the ostium primum atrial septal defect, with the underlying common atrioventricular valve, and the defect of the ventricular septal inflow (Figure 1).
Figure 1.
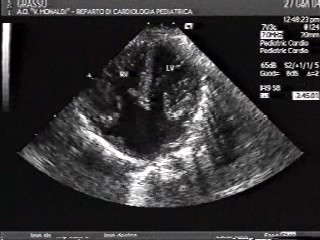
Echocardiography of Complete atrioventricular canal.
The anatomic subgroups (Rastelli's type A, B and C) can be classified on the basis of the chordal insertions and morphology of the superior bridging leaflet of the common atrioventricular valve (Table 1). Similarly, a thorough echocardiographic examination shows the degree of dysfunction of the common atrioventricular valve, as well as the presence of associated cardiac malformations. To date, cardiac catheterisation is not considered as mandatory for the diagnosis, but can be indicated in patients older than 6 months with suspected irreversible pulmonary hypertension. Cardiac catheterisation allows accurate quantification of the left-to-right shunt as well as assessment of the degree of pulmonary hypertension and the reversibility of the pulmonary artery resistances by hyperoxia and/or pharmacological tests. On left ventricular angiography, the appearance of the "gooseneck deformity" of the left ventricular outflow tract is peculiar of atrioventricular canal malformations.
Table 1.
Anatomic classification of CAVC [4]
|
Type A the superior bridging leaflet is almost completely adherent to the left ventricle and is firmly attached on the ventricular septum by multiple chordal insertions |
|
Type B the superior bridging leaflet is attached over the ventricular septum by an anomalous papillary muscle of the right ventricle |
|
Type C the superior bridging leaflet is not attached to the ventricular septum (free-floating leaflet) |
Differential diagnosis
Differential diagnosis of CAVC involves mainly the unrestrictive ventricular septal defect, associated or not to mitral valve insufficiency. The clinical picture of congestive heart failure, the bi-atrial and bi-ventricular overload on ECG, and cardiomegaly and pulmonary congestion on chest X-ray are common to the ventricular septal defect. However, on ECG the left axis deviation of QRS at -30°, usually combined with a various degree of right bundle branch block, appears to be suggestive of CAVC. Eventually, the echocardiographic examination is the cornerstone for diagnosis and is of help for that of any further associated cardiac malformation.
Epidemiology
CAVC accounts for about 3% of all cardiac malformations. Atrioventricular canal occurs in two out of every 10,000 live births. Both sexes are equally affected, with a slightly higher frequency in female (female/male ratio 1.3/1) and a striking association with Down syndrome was found [1,2].
Pathology
The complete form of AVC shows an ostium primum atrial septal defect, a common atrioventricular valve and a variable deficiency of the interventricular septum inlet [2,3]. This anatomic arrangement gives a scooped out appearance to the ventricular inlet and a long and narrow morphology to the left ventricular outlet. The key finding for the anatomic classification in type A, B or C of this malformation is the morphology of the common atrioventricular valve [4]. It is basically built-up of five leaflets (superior, inferior, mural in the right and left ventricle and antero-superior), embryologically derived from the original endocardial cushions. The size of the antero-superior leaflet is reciprocal with the extent of bridging of the superior leaflet. In type A, the superior bridging leaflet is almost completely adherent to the left ventricle and is firmly attached on the ventricular septum by multiple chordal insertions. In type B, the superior bridging leaflet is larger and overhangs the ventricular septum more than in type A, attached over it by an anomalous papillary muscle of the right ventricle. In type C, the superior bridging leaflet is larger and is not attached to the ventricular septum (free-floating leaflet), thus provoking an unrestricted interventricular communication. Type A CAVC is most frequently associated with left-sided obstructions. Type B is the least common form of atrioventricular canal. Type C is often associated with other complex cardiac malformations such as tetralogy of Fallot. Other cardiac malformations are the left ventricular inflow and outflow obstructions, mainly due to anomaly of the left component of the common atrioventricular valve, and to ventricular imbalance, with right ventricular dominance. These additional left-sided anomalies are more frequent in children without Down syndrome [5-7].
A "partial variant" of AVC exists (also known as ostium primum atrial septal defect). Nevertheless, some authors observed how "complete" and "partial" are inappropriate adjectives to describe these variants [8-11]. Indeed, despite the septal deficiency, the essence of the atrioventricular canal malformations (or atrioventricular septal defects) is the common atrioventricular junction [8-11]. Within this common junction, there may be a common atrioventricular valvar orifice (so called "complete" defects), or separate right and left valvar orifices for the right and left ventricles (so called "partial" defects).
The space between the left ventricular components of the superior and inferior bridging leaflets is traditionally called "cleft". Morphological studies suggested that this gap functions as a commissure, even though it is not supported by a papillary muscle [12]. In Rastelli's type A malformation, the space in the common anterior leaflet is also called "cleft". Again, it potentially functions as a commissure, being supported by the medial papillary muscle of the right ventricle [12].
Clinical description
Symptoms occur in infancy as a result of high pulmonary blood flow associated with pulmonary hypertension, and often complicated by insufficiency of the common atrioventricular valve. Failure to thrive, as well as congestive heart failure and frequent pulmonary infections, are invariably seen. Thus, patients with CAVC often have feeding problems and are virtually symptomatic in the first few months of life. Signs of congestive heart failure consist in feeding difficulties, excessive sweating, tachycardia, tachypnea, subcostal and intercostal retractions, mild wheezing, hepatic enlargement and poor peripheral blood perfusion [13]. If a significant regurgitation of the common atrioventricular valve is present, a systolic cardiac murmur and gallop rhythm are frequently heard. Over time, irreversible pulmonary hypertension develops, improving the signs of congestive heart failure but worsening tolerance to effort. When pulmonary artery resistances becomes higher than systemic artery resistances, the intracardiac shunt reverses and cyanosis develops, further decreasing the exercise capacity.
Natural history
Half of children with untreated CAVC die in the first year of life [1,13,14]. The main cause of death in infancy is either heart failure or pneumonia. In surviving patients with unrepaired complete atrioventricular canal, irreversible pulmonary vascular disease becomes increasingly common, and affects virtually all patients older than 2 years of age [15]. Long-term prognosis in patients with irreversible pulmonary hypertension is poor.
Treatment
Medical treatment
Medical therapy aims to improve the signs and symptoms of congestive heart failure. Thus, it should be just considered as a bridge toward surgery. Pharmacological therapy is based on digitalis, diuretics and vasodilators. Oral therapy with digoxin starts with a loading dose of 20–40 μg/kg (depending on the patient's age, from premature to child) in 3 doses over 24 h, continuing with a maintenance dose of 8–10 μg/kg/day in 2 doses. Diuretic therapy is mainly based on furosemide, at the dose of 1–6 mg/kg/day, and spironolactone, at the dose of 2–3.5 mg/kg/day. Vasodilator therapy consists chiefly in the angiotensin converting enzyme inhibitors, captopril (0.5–3 mg/kg/day, tid) or enalapril (0.1–0.4 mg/kg/day, bid). Increasing interest is raising regarding the utilisation and potential benefits of beta-blockers (mainly, propanolol, metoprolol and carvedilol) in infants and children heart failure due to congenital heart defects with left to right shunt, although long-term results are needed. Finally, the new generation of pulmonary vasodilators dramatically improved the post-operative course and the overall prognosis of the patients [16].
Surgical treatment
Surgical treatment is preferably scheduled before 6–12 months of life. Generally, the great majority of surgeons perform the repair between the 3rd (to reduce the incidence of pulmonary hypertension crisis) and the 6th month of life. Surgical palliation with pulmonary artery banding is now seldom indicated in high-risk infants (very low weight and/or in critical conditions). It reduces the pulmonary artery flow and pressure, so controlling the congestive heart failure, promoting the patient's growth and preventing the development of pulmonary vascular disease, but is contra-indicated in patients with severe atrioventricular valve regurgitation. However, more frequently complete intracardiac repair is indicated. It consists in closure of the intracardiac communications with a single or separate atrial and ventricular patches, in construction of two separate and competent atrioventricular valves using the available tissue from the common atrioventricular valve leaflet, and in repair of associated cardiac anomalies [13,17,18]. An alternative technique, using a direct suture closure of the ventricular component, accompanied by pericardial patch closure of the atrial component, was first suggested by Wilcox et al. [19]. Depending on the specific anatomic findings (i.e., in absence of severe "scooping" of the ventricular septum), the lesion can be adequately repaired in most instances by sewing down the bridging leaflets to the crest of the ventricular septum.
Risk factors for surgical repair include the patient's age, the severity of pre-operative common valve incompetence, the presence of associated cardiac malformations and the degree of the functional class [20][21][22][23]. The prognosis is directly related to the repair of the left A-V valve [20]. To date, the overall mortality for primary repair of CAVC is below 5–10%. Long-term survival is good and in 80%–95% of cases there is no need for reoperation [18,21]. Of note, the closure of the cleft results in longer times before a reoperation is necessary [24].
Aetiology
Formation of atrioventricular canal results from complex interactions of components of the extracellular matrix. Septation of the atrioventricular junction is brought about by downgrowth of the primary atrial septum, fusion of the endocardial cushions and forward expansion of the vestibular spine between atrial septum and cushions [3]. Thus, atrioventricular canal can result from arrest or interruption of the normal endocardial cushion development [25,26]. Experimental studies showed that environmental teratogens [27] or endogenous metabolic abnormalities [28] might result in abnormal development of the atrioventricular area, which may be due to altered apoptosis of these forming cells [29]. Trancription factors (TBX2, Foxp1 among the others) and signal pathways (ErbB receptor activation) involved during embryogenesis in the heart development process have been strongly suggested to have a role in atrioventricular septation [30-32].
Epidemiological studies [2,33] showed that complete atrioventricular canal tends to be associated with chromosomal abnormalities, mainly Down syndrome [10], del [8p] syndrome [34], trisomy 9, trisomy 18 [6,35]. Furthermore, CAVC with Down syndrome has been less frequently associated with left cardiac anomalies than the isolated form [5,7]. In this latter subset of patients, the analysis of potential risk factors revealed an association with maternal diabetes and antitussive drugs [36]. However, in patients with Down syndrome and complete AVC, no strong association other than maternal age has been found. In the asplenia syndrome, the CAVC is virtually always present, while it occurs in about 25% of patients with polisplenia [37]. An association between nonchromosomal defects and atrioventricular malformations have also been reported [5,6].
Genetic counselling and antenatal diagnosis
In the presence of a single affected family member, the risk of siblings of inheriting the defect is about 2%, with a higher percentage for the offspring of an affected parent. Concordance for atrioventricular malformations among siblings is higher than for other types of congenital heart defects [38].
Due to the strict association with Down syndrome and other chromosomal anomalies, genetic antenatal counselling after the foetal echocardiographic diagnosis of CAVC is mandatory. At present, prenatal diagnosis of CAVC has been associated with a 58% risk of aneuploidy, mainly trisomy 21 [39]. Again, due to the strong association between chromosomal abnormalities and CAVC, when this malformation seems isolated at antenatal echocardiography, the risk of trisomy 21 is significantly higher than when other associated cardiac lesions are diagnosed.
Contributor Information
Raffaele Calabrò, Email: raffaele.calabro@unina2.it.
Giuseppe Limongelli, Email: limongelligiuseppe@libero.it.
References
- Flyer DC. Endocardial cushion defects. In: Fyler DC, editor. Nadas' Pediatric Cardiology. Hanley & Belfus, Inc., Philadelphia; 1992. pp. 577–589. [Google Scholar]
- Ferencz C, Loffredo CA, Correa-Villasenor A, Wilson PD. In: Genetic and Environmental Risk Factors of Major Cardiovascular Malformations: The Baltimore-Washington Infant Study 1981–1989. Anderson RH Armonk N, editor. Futura Publishing Co Inc; 1997. [Google Scholar]
- Kim JS, Viragh S, Moorman AF, Anderson RH, Lamers WH. Development of the myocardium of the atrioventricular canal and the vestibular spine in the human heart. Circ Res. 2001;88:395–402. doi: 10.1161/01.res.88.4.395. [DOI] [PubMed] [Google Scholar]
- Rastelli GC, Kirklin JW, Titus JL. Anatomic observations on complete form of common atrioventricular canal with special reference to atrioventricular valves. Mayo Clinic Proc. 1966;41:296. [PubMed] [Google Scholar]
- Marino B, Vairo U, Corno A, Nava S, Guccione P, Calbro R, Marcelletti C. Atrioventricular canal in Down syndrome. Prevalence of associated cardiac malformations compared with patients without Down syndrome. Am J Dis Child. 1990;144:1120–1122. doi: 10.1001/archpedi.1990.02150340066025. [DOI] [PubMed] [Google Scholar]
- Digilio MC, Marino B, Toscano A, Giannotti A, Dallapiccola B. Atrioventricular canal defect without Down syndrome: a heterogeneous malformation. Am J Med Genet. 1999;85:140–146. doi: 10.1002/(SICI)1096-8628(19990716)85:2<140::AID-AJMG8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- De Biase L, Di Ciommo V, Ballerini L, Bevilacqua M, Marcelletti C, Marino B. Prevalence of left-sided obstructive lesions in patients with atrioventricular canal without Down's syndrome. J Thorac Cardiovasc Surg. 1986;91:467–479. [PubMed] [Google Scholar]
- Penkoske PA, Neches WH, Anderson RH, Zuberbuhler JR. Further observations on the morphology of atrioventricular septal defects. J Thorac Cardiovasc Surg. 1985;90:611–622. [PubMed] [Google Scholar]
- Falcão S, Daliento L, Ho SY, Rigby ML, Anderson RH. Cross sectional echocardiographic assessment of the extent of the atrial septum relative to the atrioventricular junction in atrioventricular septal defect. Heart. 1999;81:199–205. doi: 10.1136/hrt.81.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebels T, Ho SY, Anderson RH, Meijboom EJ, Eijgelaar A. The surgical anatomy of the left ventricular outflow tract in atrioventricular septal defect. Ann Thorac Surg. 1986;41:483–488. doi: 10.1016/s0003-4975(10)63023-8. [DOI] [PubMed] [Google Scholar]
- Anderson RH, Ho SY, Falcao S, Daliento L, Rigby ML. The diagnostic features of atrioventricular septal defect with common atrioventricular junction. Cardiol Young. 1998;8:33–49. doi: 10.1017/s1047951100004613. [DOI] [PubMed] [Google Scholar]
- Anderson RH, Zuerbuhler JR, Penkoske PA, Neches WH. Of clefts, commisssures, and things. J Thorac Cardiovasc Surg. 1985;90:605–610. [PubMed] [Google Scholar]
- Marsico F, Violini R, Calabrò R, et al. Atrioventricular septal defects. Natural history and clinical picture. In: Quero Jimenez M, Arteaga Martinez M, editor. Pediatric Cardiology – Atrioventricular Septal Defects. Ediciones Norma, Madrid; 1988. pp. 194–203. [Google Scholar]
- Santoro G, Marino B, Di Carlo D, Formigari R, Santoro G, Marcelletti C, Pasquini L. Patient selection for repair of complete atrioventricular canal guided by echocardiography. Eur J Cardio-Thorac Surg. 1996;10:439–442. doi: 10.1016/S1010-7940(96)80112-6. [DOI] [PubMed] [Google Scholar]
- Berger TJ, Blackstone EH, Kirklin JW. Survival and probability of cure without and with surgery in complete atrioventricular canal. Ann Thorac Surg. 1979;27:104–111. doi: 10.1016/s0003-4975(10)63249-3. [DOI] [PubMed] [Google Scholar]
- Trachte AL, Lobato EB, Urdaneta F, Hess PJ, Klodell CT, Martin TD, Staples ED, Beaver TM. Oral sildenafil reduces pulmonary hypertension after cardiac surgery. Ann Thorac Surg. 2005;79:194–197. doi: 10.1016/j.athoracsur.2004.06.086. [DOI] [PubMed] [Google Scholar]
- Crawford FA, Jr, Stroud MR. Surgical repair of complete atrioventricular septal defect. Ann Thorac Surg. 2001;72:1621–1628. doi: 10.1016/S0003-4975(01)03170-8. [DOI] [PubMed] [Google Scholar]
- Kirklin JW, Barratt-Boyes BG, editor. Cardiac Surgery. Churchill Livingstone, UK; 1993. Atrioventricular canal defect; pp. 749–825. [Google Scholar]
- Wilcox BR, Jones DR, Frantz EG, Brink LW, Henry GW, Mill MR, Anderson RH. Anatomically sound, simplified approach to repair of "complete" atrioventricular septal defect. Ann Thorac Surg. 1997;64:487–493. doi: 10.1016/S0003-4975(97)00566-3. [DOI] [PubMed] [Google Scholar]
- Boening A, Scheewe J, Heine K, Hedderich J, Regensburger D, Kramer HH, Cremer J. Long term results after surgical correction of atrioventricular septal defects. Eur J Cardiothorac Surg. 2002;22:167–173. doi: 10.1016/S1010-7940(02)00272-5. [DOI] [PubMed] [Google Scholar]
- Merrill WH, Hoff SJ, Bender HW., Jr . The surgical treatment of atrioventricular septal defects. In: Mavroudis C, Backer CL, editor. Pediatric Cardiac Surgery. 2. Mosby Year Book, Inc. St Louis, Missouri, USA; 1994. pp. 225–237. [Google Scholar]
- Gunther T, Mazzitelli D, Haehnel CJ, Holper K, Sebening F, Meisner H. Long-term results after repair of complete atrio-ventricular septal defects: analysis of risk factors. Ann Thorac Surg. 1998;65:754–759. doi: 10.1016/S0003-4975(98)00028-9. [DOI] [PubMed] [Google Scholar]
- Najm HK, Coles JG, Endo M, Stephens D, Rebeyka IM, Williams WG, Freedom RM. Complete atrioventricular septal defects: results of repair, risk factors and freedom from reoperation. Circulation. 1997;96:311–315. [PubMed] [Google Scholar]
- Bando K, Turrentine MW, Sun K, Sharp TG, Ensing GJ, Miller AP, Kesler KA, Binford RS, Carlos GN, Hurwitz RA, et al. Surgical management of complete atrioventricular septal defects. A twenty-year experience. J Thorac Cardiovac Surg. 1995;110:1543–1552. doi: 10.1016/S0022-5223(95)70078-1. [DOI] [PubMed] [Google Scholar]
- de la Cruz MV, Markwald RR. In: Living Morphogenesis of the Heart. Markwald R, de La Cruz M, Markwald R, editor. Springer-Verlag; New York; 1998. p. 223. [Google Scholar]
- Pierpont ME, Markwald RR, Lin AE. Genetic aspects of atrioventricular septal defects. Am J Med Genet. 2000;974:289–296. doi: 10.1002/1096-8628(200024)97:4<289::AID-AJMG1279>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Bouman HG, Broekhuizen ML, Baasten AM, Gittenberger-de Groot AC, Wenink AC. Diminished growth of atrioventricular cushion tissue in stage 24 retinoic-treated chicken embryos. Dev Dyn. 1998;213:50–58. doi: 10.1002/(SICI)1097-0177(199809)213:1<50::AID-AJA5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Santoro G, Ambrosio G, Formigari R, Marcelletti C, Chiariello M, Marino B. Low level of myocardial Superoxide Dismutase in patients with atrioventricularcanal. J Am Coll Cardiol. 1994;23 [Google Scholar]
- Saphier CJ, Yeh J. Altered apoptosis levels in hearts of human fetuses with Down syndrome. Am J Obstet Gynecol. 1998;179:962–965. doi: 10.1016/S0002-9378(98)70197-8. [DOI] [PubMed] [Google Scholar]
- Harrelson Z, Kelly RG, Goldin SN, Gibson-Brown JJ, Bollag RJ, Silver LM, Papaioannou VE. Tbx2 is essential for patterning the atrioventricular canal and for morphogenesis of the outflow tract during heart development. Development. 2004;131:5041–5052. doi: 10.1242/dev.01378. [DOI] [PubMed] [Google Scholar]
- Wang B, Weidenfeld J, Lu MM, Maika S, Kuziel WA, Morrisey EE, Tucker PW. Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development. 2004;131:4477–4487. doi: 10.1242/dev.01287. [DOI] [PubMed] [Google Scholar]
- Camenisch TD, Schroeder JA, Bradley J, Klewer SE, McDonald JA. Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat Med. 2002;8:850–855. doi: 10.1038/nm742. [DOI] [PubMed] [Google Scholar]
- Carmi R, Boughman JA, Ferencz C. Endocardial cushion defect: further studies of "isolated" versus "syndromic" occurrence. Am J Med Genet. 1992;43:569–575. doi: 10.1002/ajmg.1320430313. [DOI] [PubMed] [Google Scholar]
- Marino B, Reale A, Giannotti A, Digilio MC, Dallapiccola B. Nonrandom association of atrioventricular canal and del(8p) syndrome. Am J Med Genet. 1992;42:424–427. doi: 10.1002/ajmg.1320420404. [DOI] [PubMed] [Google Scholar]
- Francalanci P, Marino B, Boldrini R, Abella R, Iorio F, Bosman C. Morphology of the atrioventricular valve in asplenia syndrome: a peculiar type of atrioventicular canal defect. Cardiovasc Pathol. 1996;5:145–151. doi: 10.1016/1054-8807(95)00117-4. [DOI] [PubMed] [Google Scholar]
- Loffredo CA, Hirata J, Wilson PD, Ferencz C, Lurie IW. Atrioventricular septal defects: possible etiologic differences between complete and partial defects. Teratology. 2001;63:87–93. doi: 10.1002/1096-9926(200102)63:2<87::AID-TERA1014>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Rose V, Izukawa T, Moes CA. Syndromes of asplenia and polysplenia. A review of cardiac and non-cardiac malformations in 60 cases with special reference to diagnosis and prognosis. Br Heart J. 1975;37:840–852. doi: 10.1136/hrt.37.8.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora JJ, Nora AH. Maternal transmission of congenital heart diseases: new recurrence risk figures and the questions of cytoplasmic inheritance and vulnerability to teratogens. Am J Cardiol. 1987;59:459–463. doi: 10.1016/0002-9149(87)90956-8. [DOI] [PubMed] [Google Scholar]
- Delisle MF, Sandor GG, Tessier F, Farquharson DF. Outcome of fetuses diagnosed with atrioventricular septal defect. Obstet Gynecol. 1999;94:763–767. doi: 10.1016/S0029-7844(99)00389-0. [DOI] [PubMed] [Google Scholar]