

Abstract
Purpose
To investigate the effects of insulin and leptin on in vitro wound healing of transformed human corneal epithelial cell monolayers and to identify cellular (migration versus proliferation) and intracellular signaling mechanisms.
Methods
Scratch wounds were created in monolayers of an immortalized human corneal epithelial cell (HCEC) line. The wounded monolayers were exposed to insulin and leptin. Wound areas were measured every hour after wounding for up to 8 hours. Phosphoinositide 3-kinase (PI3-kinase) and mitogen-activated protein (MAP)-kinase signaling was analyzed with Western blot. The actions of insulin were also examined after incubation with inhibitors to extracellular signal regulated kinase (ERK 1/2) and PI3-kinase.
Results
The presence of insulin, but not leptin facilitated closure of wounds created in corneal epithelial cell monolayers. Phosphorylation of ERK 1/2 and Akt was stimulated after exposure of the monolayers to insulin. Inhibitors of PI3-kinase and ERK 1/2 prevented or reduced insulin-induced corneal wound healing, respectively.
Conclusions
Exposure of corneal epithelium to insulin facilitated closure of in vitro small wounds through enhanced cell migration instead of proliferation, which depended on ERK 1/2 and PI3-kinase signaling. These data suggest a mechanism by which insulin may influence corneal wound healing in vitro. In vivo, disruptions to the insulin signaling pathway observed in diseases such as diabetes might account for the delayed wound healing and corneal defects.
Continuous renewal, a process relying on proliferation, migration, and shedding of corneal epithelial cells (CECs) from the surface1 is essential for the integrity of the cornea and maintenance of corneal transparency. The events underlying corneal re-epithelialization are complex and are regulated by factors such as chemical gradients (chemotaxis) and extracellular matrix molecules that guide the epithelium to fill sites of injury. Several growth factors play a role in directional cell migration leading to wound repair. Growth factors implicated in cell migration and corneal re-epithelialization include epidermal growth factor (EGF), insulin-like growth factor (IGF), hepatocyte growth factor (HGF), and keratinocyte growth factor (KGF).2–7 Each of these growth factors has been shown to enhance corneal wound healing after injury, both in vivo and in vitro, but the underlying mechanisms of their actions have yet to be fully elucidated.
Insulin, a peptide closely related to IGF, is another growth factor implicated in wound repair, but its role is less well documented. Insulin stimulates haptotactic migration of human epidermal keratinocytes8 and topical insulin therapy has previously been shown to be beneficial to the healing of ulcerations9,10 and of burns.11 In addition, diabetes mellitus a commonly occurring pathologic disorder resulting from insulin deficiency or resistance to insulin is associated with impaired wound healing.12 Insulin therapy may be beneficial for the treatment of wounds in diabetic patients; and, in diabetic mice, treatment of dermal wounds with insulin restores the wound-healing response to a level not different from normal, non-insulin-treated mice.13
Most recently, it has been shown that insulin is present in human tear film, and receptors to insulin have been detected in the human ocular surface,14 the cornea,15 and neuronal and vascular tissues of the retina.16–18 The functions of insulin receptors within structures of the eye are not known, but diabetes is the major cause of blindness in people of working age and is often associated with disorders of the corneal epithelium.19,20 Epithelial defects include recurrent epithelial erosions, delayed re-epithelialization, abnormal wound healing, and increased susceptibility to injury. Numerous theories have been postulated to account for the complications associated with diabetes, but the mechanisms remain unclear. We examined the role of insulin and the underlying signaling mechanisms in corneal re-epithelialization. In mice, mutations in either the obese (ob) gene encoding the protein leptin or the diabetic (db) gene encoding the leptin receptor leads to a phenotype similar to diabetes.21 The actions of insulin are often mirrored by leptin and leptin has been postulated to play a role in wound healing.22–25 Thus, the actions of this cytokine on corneal wound healing were also examined. In an in vitro monolayer model of wound healing, the ability of insulin but not leptin to enhance re-epithelialization of transformed human corneal epithelial cells (HCECs)26 was demonstrated. Exposure of corneal epithelial cells to insulin increased phospho-Akt levels and phosphorylation of extracellular signal-regulated kinase (ERK)-1/2. In addition, the actions of insulin were prevented by the presence of inhibitors to PI3-kinase and reduced in the presence of PD 98059, an inhibitor of ERK 1/2 activation. Together with previous reports that insulin receptors are present in structures of the eye,14 –18 including HCECs,14 these data suggest a role for insulin in the stimulation of HCEC wound healing and that PI3-kinase and ERK 1/2 signaling may underlie this action. Pharmacological agents targeted at the downstream signaling pathways of the insulin receptor may be of therapeutic value in the management of corneal injury and the prevention of blindness occurring as its result.
Materials and Methods
Materials and Cell Culture
Tissue culture plastic dishes (35 × 10 mm) and flasks (75 cm2; Falcon, Wiesbaden, Germany) were used. Insulin, LY 293002 and bromodeoxyuridine (BrdU) were obtained from Sigma-Aldrich (Poole, UK). PD 98059 and wortmannin were obtained from Calbiochem, Nottingham, UK). Anti-BrdU (bromodeoxyuridine) was obtained from Roche Diagnostics (Lewes, UK), and FITC rabbit anti mouse was obtained from Sigma-Aldrich. Simian virus (SV)40 immortalized HCECs were cultured in supplemental hormonal epithelial medium (SHEM; DMEM/F12), supplemented with 15% fetal bovine serum (FBS), 5 μg/mL insulin, 0.1 μg/mL cholera toxin, 10 ng/mL human epidermal growth factor, and 40 μg/mL gentamicin (all from Invitrogen-Gibco, Paisley, UK).26 The cells were kindly provided by Kaoru Araki-Sasaki (Osaka University, School of Medicine, Osaka, Japan). Once 70% to 80% confluent, cells were subcultured onto 24-well plates. Cells were further incubated for 4 to 7 days (37°C, 5% CO2) to form confluent monolayers. SHEM was changed every 2 to 3 days.
Immunocytochemistry and Confocal Fluorescence Microscopy
After fixation and permeabilization, HCECs were blocked with 10% skimmed milk for 15 minutes, washed, and incubated with a rabbit anti-insulin receptor antibody directed at the β-subunit portion of the insulin receptor (0.5 μg/mL; Santa Cruz Biotechnologies, Santa Cruz, CA) overnight at 4°C. Immunolabeling was visualized with a donkey anti rabbit Cy3 secondary antibody (Molecular Probes, Eugene, OR; 8 μg/mL) at room temperature for 40 minutes. Images were obtained with a laser scanning confocal imaging system (MicroRadiance; Bio-Rad, Herts, UK). For excitation of Cy3, a dedicated 568-nm line was used.
Wound-Healing Protocol
Wound-healing experiments were performed on monolayers of HCECs plated onto 24-well plates. Cells were serum and growth factor starved for 24 hours before experimentation. Immediately before wounding, cells were incubated in fresh medium that either contained or lacked serum and growth factors and enzyme inhibitors, as indicated in the Results section. Two basic control media were used: SHEM and serum and growth factor-free medium (S/GF-free medium, which is SHEM without addition of serum, insulin, and EGF). Confluent monolayers of HCECs were wounded using a handheld, fire-polished glass pipette. The initial wound size and shape were similar and therefore shared the same mechanistic features in healing.27 Statistics showed that the initial wound area was not significantly different between experimental conditions. Images were captured hourly and wound size analyzed (Quantiment 500MC imaging system; Leica, Cambridge, UK). Cells were kept in a CO2 incubator at 37°C, 5% CO2, and 95% humidity, except when images were being captured for a short period at room temperature. Data were expressed as area resurfaced by the epithelial cells, as opposed to total wound area remaining. Statistical analysis of re-epithelialization was performed on computer (Excel and Origin, ver 6.0; Microsoft, Redmond, WA). Unpaired Student’s t-tests were performed to determine statistical significance (n = individual wounds).
BrdU Labeling
Starved HCEC were wounded with a fire-polished glass pipette and incubated in BrdU (10 μM) for 30 minutes and 1, 2, 4, and 8 hours at 34°C. Immediately after exposure to BrdU, cells were fixed with 4% paraformaldehyde for 15 minutes and denatured with 0.07 N NaOH for 2 minutes at room temperature. Cells were incubated overnight with a monoclonal anti-BrdU antibody (1:200) and treated with an FITC rabbit anti mouse secondary antibody for 40 minutes at room temperature. As a control, preconfluent HCECs were also labeled with the BrdU antibody using the same labeling protocol outlined for confluent monolayers.
Western Blot Analysis
A monolayer of HCECs that had been starved for 24 hours was stimulated with insulin (5 μg/mL) for 10, 60, and 90 minutes. The cells were washed twice with TBS (20 mM Tris-HCl, 150 mM NaCl [pH 7.4] and 0.05% Tween 20) and lysed with lysis buffer. Identical amounts of protein lysates were resolved by 4% to 12% SDS-PAGE followed by electroblotting onto nitrocellulose membrane (Invitrogen, Carlsbad, CA). The membrane was rinsed twice in TBS, blocked with 5% nonfat milk for 1 hour at room temperature, and probed with the antibodies specified in each experiment.
Results
Insulin Receptor Expression
HCEC expressed insulin receptor immunoreactivity as determined with a specific antibody directed against the β-subunit of the insulin receptor. All HCECs showed positive staining (Fig. 1), while negative control with normal rabbit serum instead of specific first antibody and/or secondary antibody alone did not show any staining. Insulin receptors were present mainly in the cytoplasm and nucleus.
Figure 1.
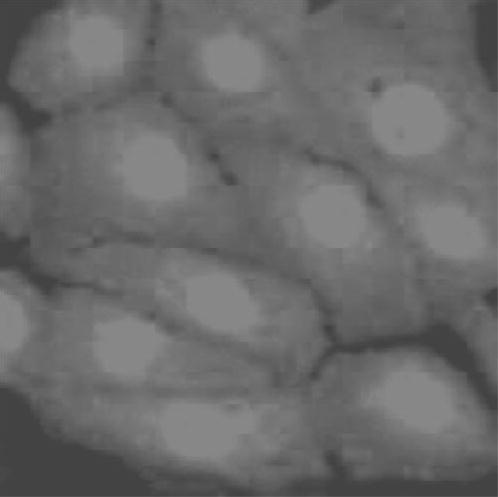
HCECs expressed insulin receptors. HCECs were labeled with a rabbit anti-insulin receptor antibody directed at the β-subunit portion of the receptor. Cytoplasmic and nuclear staining were evident. Control staining with normal rabbit serum and/or secondary antibody alone did not show positive staining.
Effect of Insulin in Monolayer Wound Healing
When exposed to SHEM, most wounds were completely resurfaced, with epithelial cells migrating into and resurfacing the denuded area 8 to 10 hours after wounding (Fig. 2a–c). The average area covered over the 8-hour period examined was 57,441 ± 2,332 μm2 (Figs. 2a–c, 3; n = 130).
Figure 2.
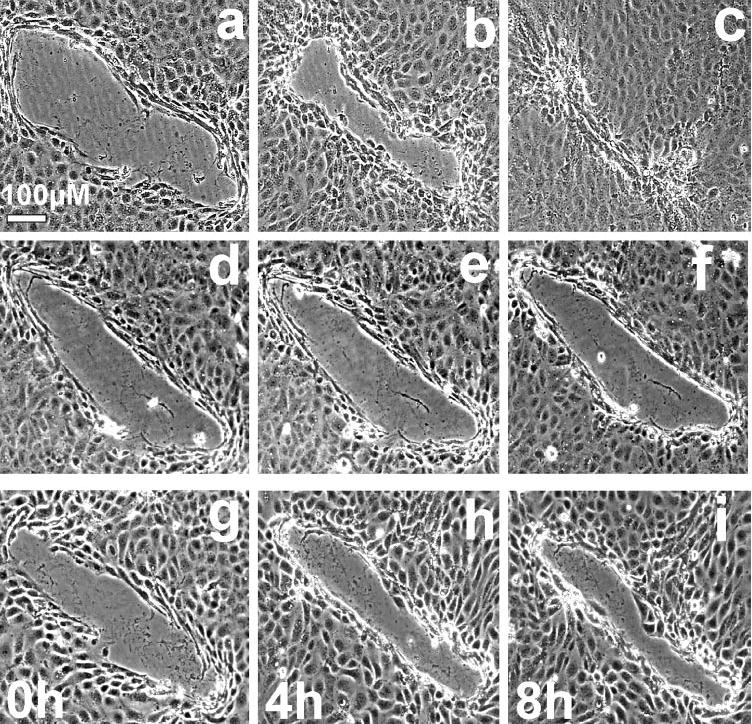
HCECs migrated to cover a denuded area in monolayer culture. Time-lapse images show the movement of the sheet of cells to heal a monolayer wound. Cells were incubated in (a–c) SHEM, (d–f) S/GF-free medium, and (g–i) S/GF-free medium with insulin added.
Figure 3.
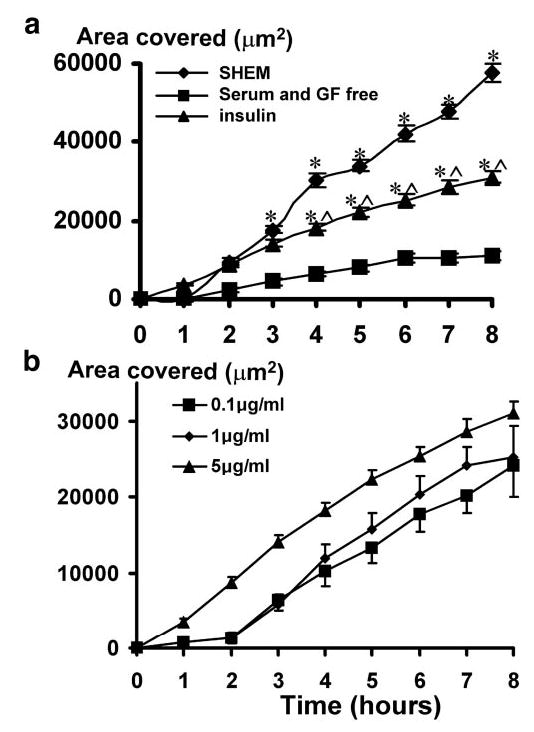
(a) Insulin significantly restored migration of HCECs into the wound-denuded area in S/GF-free medium. (b) Dose–response curve of the healing rate at different insulin concentrations. A monolayer culture of HCECs was wounded as in Figure 2. Time-lapse images were taken, and the area covered by migrating cells was quantified. Withdrawal of serum and growth factors from the SHEM (S/GF-free) significantly reduced healing (the area covered). Addition of insulin significantly restored healing: *P < 0.01 when compared with that in S/GF-free medium; ∧P < 0.01 when compared with that in SHEM.
Growth factors are known to be involved in cell migration and proliferation and to play an important role in wound healing.29 Omission of both serum and growth factors from the SHEM significantly impaired HCEC wound repair, and after 8 hours, the resurfaced wound area was reduced to 11,218 ± 1,269 μm2, approximately five times less than in control wounds (Figs. 2d–f, 3. n = 89; P < 0.00001 compared with that in SHEM).
To determine whether insulin plays a role in the healing process, cells were exposed to insulin (0.1–5 μg/mL) in S/GF-free medium. At all concentrations tested, insulin partially and significantly enhanced the rate of wound closure. No significant difference in the degree of facilitation was observed within the concentration range examined (P > 0.05; Student’s t-test). After 8 hours, the total wound area that was resurfaced with the addition of insulin at 0.1, 1, and 5 μg/mL to the S/GF-free control was 24,176 ± 2,660 μm2 (n = 20; P = 2.06−4), 25,217 ± 4,108 μm2 (n = 20, P = 2.1−5) and 31,067 ± 1,490 μm2 (n = 104; P = 2.4−9), respectively (Figs. 2g–i, 3; Student’s t-test insulin versus S/GF-free control).
During the 8-hour healing period, there was no significant BrdU labeling of cells in SHEM, S/GF-free medium, or S/GF-free medium with insulin (n = 104; data not shown). The absence of BrdU uptake is in line with previous findings that re-epithelialization of small wounds occurs apparently independent of proliferation.27,28
Effect of Leptin on Monolayer Wound Healing of HCECs
In most cell types, the actions of insulin are closely related to or opposed by the actions of the cytokine leptin. Accumulating evidence suggests a possible role for leptin in wound healing,22–25 and the leptin protein has been detected in wound fluid.24 The concentration of leptin used in this study was within the range reported in wound fluid (50–250 ng/mL)24 and is also within the range previously reported to stimulate wound closure in ob/ob and normal mice.22,25 However, leptin (100 ng/mL) did not alter the healing rate of wounded monolayers of HCECs. After 8 hours, and in the presence of leptin, the area resurfaced was not statistically different from that covered under control S/GF-free conditions. The total area resurfaced in the presence of leptin 1, 2, 3, 4, 5, 6, 7, and 8 hours after wounding were: 342 ± 657; 729 ± 713; 2,935 ± 1,033; 7,392 ± 551; 8,926 ± 972; 11,460 ± 738; 13,521 ± 1,291; and 14,021 ± 1,622 μm2, respectively (n = 27). This completely overlaps with the healing curve in S/GF-free control conditions (Fig. 3). Thus, unlike insulin, leptin, at a concentration (100 ng/mL) within the range detected in wound fluid22 and similar to that which induces closure of the wound epithelium during skin repair,24,25 did not facilitate re-epithelialization in this wound-healing model.
Insulin-Induced Wound Closure through Activation of PI3-Kinase
Activation of PI3-kinase was assessed with Western blot techniques, using its downstream target Akt. Very little phospho-Akt was detected in starved HCECs (cultured in S/GF-free SHEM). After 10 minutes of incubation with insulin, phosphorylation of Akt increased and persisted even 90 minutes after insulin exposure (from two independent experiments; Fig. 4).
Figure 4.
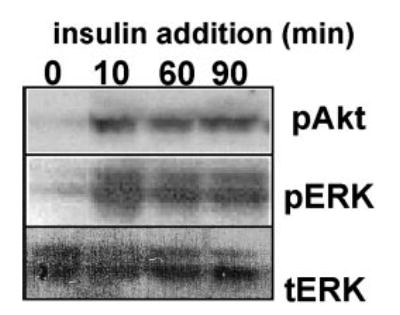
Insulin activated PI3-kinase and ERK signaling pathways. Confluent monolayers of HCECs starved of serum and growth factors for 24 hours were incubated with insulin (5 μg/mL) for 0, 10, 60, and 90 minutes. Cells were then lysed. Phosphorylated Akt (pAkt) and ERK (pERK) were determined with Western blot. Total ERK 1/2 probed with the same membrane after stripping showed that total levels of ERK remained the same in all samples (tERK).
To examine the role of PI3-kinase in insulin-induced promotion of wound healing, the effect of the PI3-kinase inhibitors LY 29400230 and wortmannin31 on the actions of insulin were investigated. To further confirm the results, these experiments were performed in parallel with exposure to both insulin and the PI3-kinase inhibitors alone. Exposure of corneal cells to LY 294002 (10 μM) per se did not significantly affect the rate of wound closure in S/GF-free medium (P = 0.12; n = 58; data not shown). However, in the presence of LY 294002 the ability of insulin to facilitate wound healing was prevented, and the rate of wound healing was reduced to levels little different from S/GF-free conditions (n = 57; Fig. 5). Thus, in the presence of insulin and LY 294002, 16,189 ± 1,651 μm2 of the wound area was resurfaced after 8 hours (compared with 40,881 ± 1,657 μm2 in the presence of insulin but absence of LY 294002). Similarly, the ability of insulin to stimulate wound closure was reduced by the presence of the structurally unrelated PI3-kinase inhibitor wortmannin. Like LY 294002, wortmannin (10 nM) itself had no effect on the wound healing that occurred in S/GF-free medium (n = 45; P = 0.77; data not shown) but its presence prevented the wound closure stimulated by insulin (Fig. 5). After 8 hours 14,204.0 ± 1,463.0 μm2 of the denuded area was resurfaced compared with 11,217.7 ± 1,269.2 μm2 under S/GF-free conditions. Thus, the resurfaced area was not significantly different when compared with the control S/GF-free condition (n = 94; P = 0.13). These results suggest that PI3-kinase is a likely signaling intermediate involved in the HCEC wound healing stimulated by insulin.
Figure 5.
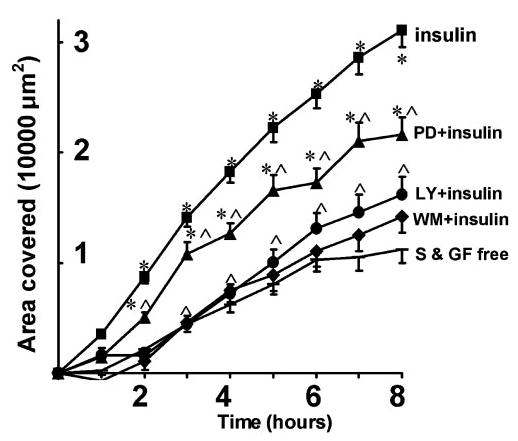
Insulin-restored migration of HCECs in S/GF-free medium was dependent on PI3-kinase and MAP-kinase signaling. The PI3-kinase inhibitors wortmannin (WM) and LY294002 (LY) completely abolished the restoration of the migration of cells into the denuded area in S/GF-free medium. MAP kinase inhibition with PD98059 (PD) partially, but significantly inhibited the restoration of the migration of cells into the denuded area in S/GF-free medium. *P < 0.01 when compared with that in S/GF-free medium; ∧P < 0.01 when compared with that in S/GF-free medium with insulin.
Insulin-Induced Wound Closure through ERK Signaling
MAP kinases are known to control cell motility7,32,33 and two members of the MAP kinase family, ERK-1 and ERK-2, are rapidly activated after mechanical injury.34 In the absence of insulin, phospho-ERK 1/2 was not detected. Exposure to insulin for 10, 60, or 90 minutes resulted in significant phosphorylation of ERK 1 and ERK 2 (Fig 4).
In the absence of serum or growth factors, incubation with PD 98059 (10 μg/mL), an inhibitor of ERK activation, did not significantly affect re-epithelialization of the corneal epithelium (n = 43; P = 0.64). The curve of the area covered in this condition virtually overlaps that in the S/GF-free medium (data not shown). The presence of PD 98059 did not prevent insulin-induced wound closure, but it did significantly reduce the ability of insulin to do so (n = 74; P = 0.0002). In the presence of insulin alone, 31,067 ± 1,490 μm2 of the denuded area was resurfaced 8 hours after wounding, compared with 21,630 ± 1,541 μm2 after incubation with PD 98059. This suggests the involvement of the MAP kinase pathway in the actions of insulin on monolayer wound healing in human HCECs.
Comparison between Insulin- and EGF-Stimulated Wound Closure
EGF has been characterized as a potent stimulator of wound closure. The actions of insulin were compared with those of EGF. EGF (10 ng/mL) significantly enhanced, but did not fully restore wound closure compared with closure stimulated in SHEM. After 8 hours 40,881 ± 1,657 μm2 of the denuded area was resurfaced (n = 76; Fig. 6). Thus, like insulin, EGF stimulated wound closure. However, compared with insulin, EGF, at the concentrations examined in this study, was a more potent stimulator of epithelial wound closure (P = 0.002). Although insulin and EGF both stimulated closure of small wounds created in monolayers of HCECs, when used alone, neither fully supported the degree of closure promoted in SHEM containing EGF and insulin as well as fetal calf serum. To examine the effects of coapplication of insulin and EGF, both growth factors were added to the medium in the absence of serum. The presence of 10 ng/mL EGF and 5 μg/mL insulin fully restored the rate of wound healing. After 8 hours, 53,520 ± 2,421 μm2 of the denuded area was re-epithelialized, an area not significantly different from that covered in SHEM (P = 0.24; n = 79; Fig. 6).
Figure 6.
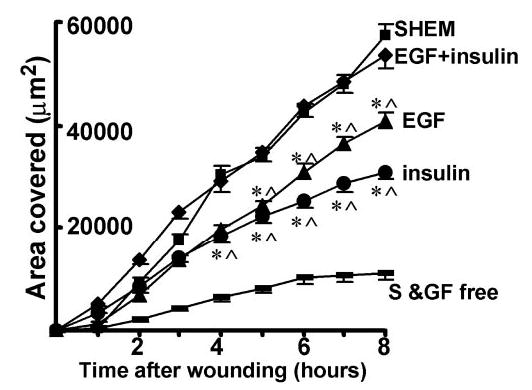
Additive restoration of migration of HCECs by insulin and EGF in S/GF-free medium. EGF or insulin alone did not fully restored migration, but in combination, they fully restored the migration of cells into the denuded area in S/GF-free medium. *P < 0.01 when compared with that in S/GF-free medium; ∧P < 0.01 when compared with that in S/GF-free medium with insulin.
Discussion
Insulin therapy is known to be beneficial for the treatments of burns11 and epidermal wounds in humans.35 Diabetes is often associated with delayed wound healing12 and impaired corneal re-epithelialization.36,37 Although the beneficial properties of insulin in wound repair have been recognized, the underlying cellular and signaling mechanisms have yet to be determined. The present investigation showed that, in transformed HCECs, (1) addition of insulin to S/GF-free medium significantly restored cell migration into the denuded area, a process independent of cell proliferation; (2) insulin stimulated Akt and ERK1/2 activation; and (3) the restoration of migration of monolayer cells into the denuded area by insulin depended on PI3-kinase and partially involved ERK signaling. In addition, combination of EGF with insulin fully restored the migration of epithelium in this in vitro monolayer wound-healing model of HCECs. When added at a concentration within the range detected in wound fluid24 and within the range previously shown to simulate wound closure during skin repair in ob/ob and normal mice,22,25 leptin did not have any effect.
The insulin receptor, which is a membrane-spanning glycoprotein belonging to a family of ligand-gated receptor tyrosine kinases, is present in human corneal epithelial tissue.14 Insulin receptor immunofluorescence was found in the cytoplasm and nucleus (Fig. 1). The intranuclear staining may represent nuclear translocation of insulin receptors under our culture conditions.37 This is consistent with immunohistochemical results in the human cornea.14 Although Rocha et al.14 also detected insulin receptor immunofluorescence in the plasma membrane, staining was most prominent in the cytoplasm. Thus, the data presented in this study suggest that HCEC lines may be a valuable tool to study the effects and the mechanism of insulin action on HCECs.
In the presence of SHEM, small wounds in corneal sheets healed rapidly (over a period of 8–10 hours). Consistent with the involvement of growth factors in cell migration, wound healing was significantly impaired in the absence of growth factors and serum; however, re-epithelialization was not completely abolished. The low rate of wound repair may be attributable to the release of endogenous growth factors from the epithelial sheets in response to injury. To determine the effects of insulin on wound healing with minimal interference from other growth factors, we examined the actions of insulin in the presence of medium lacking growth factors and serum. Addition of insulin (0.1–5 μg/mL) to the S/GF-free medium restored re-epithelialization to more than 50% of that observed in the presence of SHEM. Given the lack of BrdU uptake in insulin-containing SHEM, this action of insulin is attributable to an effect on cell migration, as opposed to the proliferation and differentiation in lens cells.39,40 These data are consistent with previous reports that insulin facilitates migration of human keratinocytes.41 Proliferation of corneal epithelial cells has been shown to occur at least 12 to 24 hours after wounding.42,43,44 Thus, although insulin did not induce migration by proliferation in this study, the possibility that proliferation plays a role in the healing of larger wounds or at later stages in the healing process cannot be ruled out.
One of the best-characterized growth factors involved in CEC migration is EGF,45,46 and endogenous agonists to the EGF receptor are abundant at a site of injury. During migration, EGF receptors are activated at the leading edge of wounded corneal epithelium, and inhibition of EGF receptor tyrosine kinase activity slows migration.6 In an electric field, EGF receptors accumulate cathodally and appear to direct migration by reorganization of F-actin.47 Fang et al.41 have shown that EGF, at the same concentration used in the present study, fully restores the migration rate of single human keratinocytes in an electric field. Our findings suggest that although EGF significantly enhances wound closure, it is insufficient to restore the wound-healing response of CECs fully. The actions of insulin and EGF, however, were additive, and coapplication of insulin and EGF fully restored the wound-healing response.
Leptin, an ob gene product, is a hypoxia-inducible pleiotropic cytokine known to participate in multiple cellular and physiological processes, including wound healing of skin.22,23,48 In this in vitro wound-healing model of small wounds of the corneal epithelial monolayer, leptin did not stimulate closure despite being examined at concentrations detected in wound fluid24 and reports that it facilitate healing of skin.22,23,48 Enhancement of wound healing by leptin observed in vivo may be attributable to stimulation of proliferation that played no part in wound healing in this study or by angiogenesis. Indeed, leptin induces proliferation of the keratinocyte cell line HaCaT, as well as human primary keratinocytes22 and induces neovascularization in rat corneas,22 cultured human endothelial venous cells, and porcine aortic endothelial cells.49
PI3-kinase is involved in wound healing, and EGF stimulates PI3-kinase activation and expression during CEC wound closure.50,51 In normally insulin-sensitive tissues, insulin signals through the insulin receptor substrate (IRS)/PI3-kinase pathway.52 In the present investigation, insulin stimulated the phosphorylation of Akt, the downstream target of PI3-kinase. Furthermore, insulin-mediated wound closure was prevented by the presence of LY 294002 and wortmannin. PI3-kinase regulates actin cytoskeleton reorganization possibly involving activation of the small Rho guanosine triphosphatases (GTPases) that produce protrusions of the plasma membrane and formation of lamellipodia.53 Increasing evidence suggests that growth factor-induced modulation of the actin cytoskeleton is essential for wound healing. One possible mechanism by which insulin may facilitate corneal would healing is through activation of PI3-kinase and subsequent modulation of the actin cytoskeleton.
The ERK family of MAP kinases also plays an important role in wound healing. One of the downstream effectors of activated ERK is myosin light chain kinase (MLCK). It is through activation of MLCK that ERK is thought to stimulate cell migration.33 ERK is a common downstream signaling protein activated after insulin receptor stimulation, and ERK can also be activated downstream of PI3-kinase activation. Exposure of HCECs to insulin led to phosphorylation of ERK 1/2, and exposure to PD 98059 significantly reduced the ability of insulin to augment wound healing, suggesting the involvement of ERK activation for the actions of insulin.
Insulin has been shown to bind, albeit weakly, to the insulin-like growth factor (IFG)-1 receptor,54–56 a receptor tyrosine kinase with extensive similarity to the insulin receptor.57,58 To exclude the possibility that at higher concentrations insulin exerts some of its effects through the IGF-1 receptor, lower concentrations of insulin were tested (Fig. 3B). It is most likely that insulin facilitates wound closure through action on the insulin receptors.
In this in vitro wound-healing model, insulin has an evident beneficial effect on small wounds in the human cornea epithelial monolayer, apparently through enhanced migration of cell sheets. Nakamura et al.,59 using a rabbit corneal section where the cells were grown on denuded cornea, found no effect of insulin, but IGF-1, added in combination with substance P (SP) had a stimulatory effect on migration of corneal epithelium. This discrepancy may be accounted for by differences in extracellular matrix molecules in the different models used. One clinical case report showed nonhealing, severe, neurotrophic and anhidrotic keratopathy did not respond to 2 weeks’ treatment with insulin, but healed gradually in the following 2 weeks treatment with SP in combination with IGF-1.60 The interaction of extracellular matrix and insulin on migration of cornea epithelial cells needs further investigation.
In summary, at a concentration ranging from 0.1 to 5 μg/mL, insulin augments re-epithelialization of small wounds created in a monolayer of the HCEC line, apparently through cell migration. This effect is probably mediated by the PI3-kinase and MAP kinase signaling pathways.
Footnotes
Supported by the Wellcome Trust Grant 068012. MZ is a Wellcome Trust University Award Senior Lecturer (Grant 058551).
Disclosure: L.J. Shanley, None; C.D. McCaig, None; J.V. Forrester, None; M. Zhao, None
References
- 1.Throft R, Friend J. The X, Y, Z hypothesis of corneal epithelial maintenance. Invest Ophthalmol Vis Sci. 1983;24:1442–1443. [PubMed] [Google Scholar]
- 2.Nakagawa S, Nishida T, Manabe R. Actin organization in migrating corneal epithelium of rabbits in situ. Exp Eye Res. 1985;41:335–343. doi: 10.1016/s0014-4835(85)80024-5. [DOI] [PubMed] [Google Scholar]
- 3.Watanabe K, Nakagawa S, Nishida T. Stimulatory effects of fibronectin and EGF on migration of corneal epithelial cells. Invest Ophthalmol Vis Sci. 1987;28:205–211. [PubMed] [Google Scholar]
- 4.Wilson SE, He YG, Weng J, Zieske JD, Jester JV, Schultz GS. Effect of epidermal growth factor, hepatocyte growth factor, and keratinocyte growth factor, on proliferation, motility and differentiation of human corneal epithelial cells. Exp Eye Res. 1984;59:665–678. doi: 10.1006/exer.1994.1152. [DOI] [PubMed] [Google Scholar]
- 5.Nishida T, Nakamura M, Ofuji K, Reid TW, Mannis MJ, Murphy CJ. Synergistic effects of substance P with insulin-like growth factor-1 on epithelial migration of the cornea. J Cell Physiol. 1996;169:159–166. doi: 10.1002/(SICI)1097-4652(199610)169:1<159::AID-JCP16>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 6.Zieske JD, Takahashi H, Hutcheon AE, Dalbone AC. Activation of epidermal growth factor receptor during corneal epithelial migration. Invest Ophthalmol Vis Sci. 2000;41:1346–1355. [PubMed] [Google Scholar]
- 7.McBain VA, Forrester JV, McCaig CD. HGF, MAPK, and a small physiological electric field interact during corneal epithelial cell migration. Invest Ophthalmol Vis Sci. 2003;44:540–547. doi: 10.1167/iovs.02-0570. [DOI] [PubMed] [Google Scholar]
- 8.Benoliel AM, Kahn-Perles B, Imbert J, Verrando P. Insulin stimulates haptotactic migration of human epidermal keratinocytes through activation of NF-kappa B transcription factor. J Cell Sci. 1987;110:2089–2097. doi: 10.1242/jcs.110.17.2089. [DOI] [PubMed] [Google Scholar]
- 9.Rosenthal SP. Acceleration of primary wound healing by insulin. Arch Surg. 1968;96:53–55. doi: 10.1001/archsurg.1968.01330190055012. [DOI] [PubMed] [Google Scholar]
- 10.Van Ort SR, Gerber RM. Topical application of insulin in the treatment of decubitus ulcers: a pilot study. Nurs Res. 1976;25:9–12. [PubMed] [Google Scholar]
- 11.Pierre EJ, Barrow RE, Hawkins HK, et al. Effects of insulin on wound healing. J Trauma. 1998;44:342–345. doi: 10.1097/00005373-199802000-00019. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg CS. Wound healing in the patient with diabetes mellitus. Nurs Clin North Am. 1990;25:247–261. [PubMed] [Google Scholar]
- 13.Weringer EJ, Kelso JM, Tamai IY, Arguilla ER. Effects of insulin on wound healing in diabetic mice. Acta Endocrinol (Copenh) 1982;99:101–108. doi: 10.1530/acta.0.0990101. [DOI] [PubMed] [Google Scholar]
- 14.Rocha EM, Cunha DA, Carneiro EM, Boschero AC, Saad MJ, Velloso LA. Identification of insulin in the tear film and insulin receptor and IGF-1 receptor on the human ocular surface. Invest Ophthalmol Vis Sci. 2002;43:963–967. [PubMed] [Google Scholar]
- 15.Naeser P. Insulin receptors in human ocular tissues: immunohistochemical demonstration in normal and diabetic eyes. Ups J Med Sci. 1997;102:35–40. doi: 10.3109/03009739709178930. [DOI] [PubMed] [Google Scholar]
- 16.Thomopoulos P, Pessac B. Insulin receptors in cultured mouse retinal cells. Diabetologia. 1979;16:275–279. doi: 10.1007/BF01221955. [DOI] [PubMed] [Google Scholar]
- 17.Im JH, Pillion DJ, Meezan E. Comparison of insulin receptors from bovine retinal blood vessels and nonvascular retinal tissue. Invest Ophthalmol Vis Sci. 1986;27:1681–1690. [PubMed] [Google Scholar]
- 18.Rodrigues M, Waldbillig RJ, Rajagopalan S, Hackett J, LeRoith D, Chader GJ. Retinal insulin receptors: localization using a polyclonal anti-insulin receptor antibody. Brain Res. 1988;443:389–394. doi: 10.1016/0006-8993(88)91639-3. [DOI] [PubMed] [Google Scholar]
- 19.Zagon IS, Jenkins JB, Sassani JW, et al. Naltrexone, an opioid antagonist, facilitates reepithelialization of the cornea in diabetic rat. Diabetes. 2002;51:3055–3062. doi: 10.2337/diabetes.51.10.3055. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg ME, Tervo TM, Immonen IJ, Muller LJ, Gronhagen-Riska C, Vesaluoma MH. Corneal structure and sensitivity in type 1 diabetes mellitus. Invest Ophthalmol Vis Sci. 2000;41:2915–2921. [PubMed] [Google Scholar]
- 21.Zhang Y, Proence R, Maffei M, et al. Positional cloning of mouse obese gene and its human homologue. Nature. 1994;372:452–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 22.Frank S, Stallmeyer B, Kampfer H, Kolb N, Pfeilschifter J. Leptin enhances wound re-epithelialization and constitutes a direct function of leptin in skin repair. J Clin Invest. 2000;106:501–509. doi: 10.1172/JCI9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ring BD, Scully S, Davis CR, et al. Systemically and topically administered leptin both accelerate wound healing in diabetic ob/ob mice. Endocrinology. 2000;141:446–449. doi: 10.1210/endo.141.1.7373. [DOI] [PubMed] [Google Scholar]
- 24.Marikovsky M, Rosenblum CI, Faltin Z, Friedman-Einat M. Appearance of leptin in wound fluid as a response to injury. Wound Repair Regen. 2002;10:302–307. doi: 10.1046/j.1524-475x.2002.10505.x. [DOI] [PubMed] [Google Scholar]
- 25.Stallmeyer B, Pfeilschifter J, Frank S. Systemically and topically supplemented leptin fails to reconstitute a normal angiogenic response during skin repair in diabetic ob/ob mice. Diabetologia. 2001;44:471–479. doi: 10.1007/s001250051645. [DOI] [PubMed] [Google Scholar]
- 26.Araki-Sasaki K, Ohashi Y, Sasabe T, et al. An SV40-immortalized human corneal epithelial cell line and its characterization. Invest Ophthalmol Vis Sci. 1995;36:614–621. [PubMed] [Google Scholar]
- 27.Fenteany G, Janmey PA, Stossel TP. Signaling pathways and cell mechanics involved in wound closure by epithelial cell sheets. Curr Biol. 2000;10:831–838. doi: 10.1016/s0960-9822(00)00579-0. [DOI] [PubMed] [Google Scholar]
- 28.Sponsel HT, Guzelian PS, Brown SE, et al. Mechanisms of recovery from mechanical injury of cultured rat hepatocytes. Am J Physiol. 1996;271:C721–C727. doi: 10.1152/ajpcell.1996.271.3.C721. [DOI] [PubMed] [Google Scholar]
- 29.Schultz G, Chegini N, Grant M, Khaw P, MacKay S. Effects of growth factors on corneal wound healing. Acta Ophthalmol Suppl. 1992;202:60–66. doi: 10.1111/j.1755-3768.1992.tb02170.x. [DOI] [PubMed] [Google Scholar]
- 30.Vlahos CJ, Matter WF, Huin KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY 294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 31.Ui M, Okada T, Haszeki K, Hazeki O. Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem Sci. 1995;20:303–307. doi: 10.1016/s0968-0004(00)89056-8. [DOI] [PubMed] [Google Scholar]
- 32.Glise B, Bourbon H, Noselli S. Hemipterous encodes a novel Drosophila MAP kinase kinase, required for epithelial cell sheet movement. Cell. 1995;83:451–461. doi: 10.1016/0092-8674(95)90123-x. [DOI] [PubMed] [Google Scholar]
- 33.Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dieckgraefe BK, Weems DM, Santoro SA, Alpers DH. ERK and p38 MAP kinase pathways are mediators of intestinal epithelial wound-induced signal transduction. Biochem Biophys Res Commun. 1997;233:389–394. doi: 10.1006/bbrc.1997.6469. [DOI] [PubMed] [Google Scholar]
- 35.Greenway SE, Filler LE, Greenway FL. Topical insulin in wound healing: a randomised, double-blind, placebo-controlled trial. J Wound Care. 1999;8:526–528. doi: 10.12968/jowc.1999.8.10.26217. [DOI] [PubMed] [Google Scholar]
- 36.Lee CH, Whiteman AL, Murphy CJ, Barney NP, Taylor PB, Reid TW. Substance P, insulin like growth factor 1, and surface healing. Arch Ophthalmol. 2002;120:215–217. [PubMed] [Google Scholar]
- 37.Nakamura M, Kawahara M, Nakata K, Nishida T. Restoration of corneal epithelial barrier function and wound healing by substance P and IGF-1 in rats with capsaicin-induced neurotrophic keratopathy. Invest Ophthalmol Vis Sci. 2003;44:2937–2940. doi: 10.1167/iovs.02-0868. [DOI] [PubMed] [Google Scholar]
- 38.Gletsu N, Dixon W, Clandinin MT. Insulin receptor at the mouse hepatocyte nucleus after a glucose meal induces dephosphorylation of a 30-kDa transcription factor and a concomitant increase in malic enzyme gene expression. J Nutr. 1999;129:2154–2161. doi: 10.1093/jn/129.12.2154. [DOI] [PubMed] [Google Scholar]
- 39.Piatigoorsky J. Insulin initiation of lens fiber differentiation in culture: elongation of embryonic lens epithelial cells. Dev Biol. 1973;30:214–216. doi: 10.1016/0012-1606(73)90060-2. [DOI] [PubMed] [Google Scholar]
- 40.Wride MA. Cellular and molecular features of lens differentiation: a review of recent advances. Differentiation. 1996;61:77–93. doi: 10.1046/j.1432-0436.1996.6120077.x. [DOI] [PubMed] [Google Scholar]
- 41.Fang KS, Farboud B, Nuccitelli R, Isseroff RR. Migration of human keratinocytes in electric fields requires growth factors and extracellular calcium. J Invest Dermatol. 1998;111:751–756. doi: 10.1046/j.1523-1747.1998.00366.x. [DOI] [PubMed] [Google Scholar]
- 42.Kitazawa T, Kinoshita S, Fujita K, et al. The mechanism of accelerated corneal epithelial healing by human epidermal growth factor. Invest Ophthalmol Vis Sci. 1990;31:1773–1778. [PubMed] [Google Scholar]
- 43.Beuerman RW, Thompson HW. Molecular and cellular responses if the corneal epithelium to wound healing. Acta Ophthalmol Suppl. 1992;202:7–12. doi: 10.1111/j.1755-3768.1992.tb02161.x. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida K, Nakaymam K, Nagahama H, et al. Involvement of p27(KIP1) degradation by Skp2 in the regulation of proliferation in response to wounding of corneal epithelium. Invest Ophthalmol Vis Sci. 2002;43:364–370. [PubMed] [Google Scholar]
- 45.Frati L, Daniel S, Delogu A, Covelli I. Selective binding of the epidermal growth factor and its specific effects on the epithelial cells of the cornea. Exp Eye Res. 1972;14:135–141. doi: 10.1016/0014-4835(72)90059-0. [DOI] [PubMed] [Google Scholar]
- 46.Savage CR, Jr, Cohen S. Proliferation of corneal epithelium induced by epidermal growth factor. Exp Eye Res. 1973;15:361–366. doi: 10.1016/0014-4835(73)90151-6. [DOI] [PubMed] [Google Scholar]
- 47.Zhao M, Pu J, Forrester JV, McCaig CD. Membrane lipids, EGF receptors, and intracellular signals colocalize and are polarized in epithelial cells moving directionally in a physiological electric field. FASEB J. 2002;16:857–859. doi: 10.1096/fj.01-0811fje. [DOI] [PubMed] [Google Scholar]
- 48.Murad A, Nath AK, Cha ST, Demir E, Flores-Riveros J, Sierra-Honigmann MR. Leptin is an autocrine/paracrine regulator of wound healing. FASEB J. 2003;17:1895–1897. doi: 10.1096/fj.03-0068fje. [DOI] [PubMed] [Google Scholar]
- 49.Bouloumie A, Drexler HC, Lafontan M, Busse R. Leptin, the product of Ob gene, promotes angiogenesis. Circ Res. 1998;83:1059–1066. doi: 10.1161/01.res.83.10.1059. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, Akhter RA. Epidermal growth factor stimulation of phosphatidylinositol 3-kinase during wound closure in rabbit corneal epithelial cells. Invest Ophthalmol Vis Sci. 1997;38:1139–1148. [PubMed] [Google Scholar]
- 51.Zhang Y, Liou GI, Gulati AK, Akhtar RA. Expression of phosphatidylinositol 3-kinase during EGF-stimulated wound repair in rabbit corneal epithelium. Invest Ophthalmol Vis Sci. 1999;40:2819–2826. [PubMed] [Google Scholar]
- 52.Shepherd PR, Withers DJ, Siddle K. Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochem J. 1998;1(333):471–490. doi: 10.1042/bj3330471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nobes CD, Hall A. Rho, Rac and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 54.Czech MP. Signal transmission by the insulin-like growth factors. Cell. 1989;59:235–238. doi: 10.1016/0092-8674(89)90281-x. [DOI] [PubMed] [Google Scholar]
- 55.Weiland M, Bahr F, Hohne M, Schunnann A, Ziehm D, Joost HG. The signaling potential of the receptors for insulin and insulin-like growth factor-1 (IGF-1) in 3T3–L1 adipocytes: comparison of glucose transport activity, induction of oncogene c-fos glucose transporter mRNA, and DNA synthesis. Cell Physiol. 1991;149:428–435. doi: 10.1002/jcp.1041490311. [DOI] [PubMed] [Google Scholar]
- 56.Lund S, Flyvbjerg A, Holman GD, Larsen FS, Pedersen O, Schmitz O. Comparative effects of IGF-1 and insulin on glucose transporter system in rat muscle. Am J Physiol. 1994;267:E461–E466. doi: 10.1152/ajpendo.1994.267.3.E461. [DOI] [PubMed] [Google Scholar]
- 57.Ullrich A, Grey A, Tam AW, et al. Insulin-like growth factor receptor primary structure: comparison with insulin receptor structural determinants that define functional specificity. EMBO J. 1986;5:2503–2512. doi: 10.1002/j.1460-2075.1986.tb04528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yarden Y, Ullrich A. Molecular analysis of signal transduction by growth factors. Biochem. 1988;27:3113–3119. doi: 10.1021/bi00409a001. [DOI] [PubMed] [Google Scholar]
- 59.Nakamura M, Chikama T, Nishada T. Characterisation of insulin-like growth factor-1 receptors in rabbit corneal epithelial cells. Exp Eye Res. 2000;70:199–204. doi: 10.1006/exer.1999.0775. [DOI] [PubMed] [Google Scholar]
- 60.Brown SM, Lamberts DW, Reid TW, Nishida T, Murphy CJ. Neurotrophic and anhidrotic keratopathy treated with substance P and insulin-like growth factor-1. Arch Ophthalmol. 1997;115:926–927. doi: 10.1001/archopht.1997.01100160096021. [DOI] [PubMed] [Google Scholar]