

Abstract
Brassica napus (cultivar Reston) seed proteins were analyzed at 2, 3, 4, 5, and 6 weeks after flowering in biological quadruplicate using two-dimensional gel electrophoresis. Developmental expression profiles for 794 protein spot groups were established and hierarchical cluster analysis revealed 12 different expression trends. Tryptic peptides from each spot group were analyzed in duplicate using matrix-assisted laser desorption ionization time-of-flight mass spectrometry and liquid chromatography-tandem mass spectrometry. The identity of 517 spot groups was determined, representing 289 nonredundant proteins. These proteins were classified into 14 functional categories based upon the Arabidopsis (Arabidopsis thaliana) genome classification scheme. Energy and metabolism related proteins were highly represented in developing seed, accounting for 24.3% and 16.8% of the total proteins, respectively. Analysis of subclasses within the metabolism group revealed coordinated expression during seed filling. The influence of prominently expressed seed storage proteins on relative quantification data is discussed and an in silico subtraction method is presented. The preponderance of energy and metabolic proteins detected in this study provides an in-depth proteomic view on carbon assimilation in B. napus seed. These data suggest that sugar mobilization from glucose to coenzyme A and its acyl derivative is a collaboration between the cytosol and plastids and that temporal control of enzymes and pathways extends beyond transcription. This study provides a systematic analysis of metabolic processes operating in developing B. napus seed from the perspective of protein expression. Data generated from this study have been deposited into a web database (http://oilseedproteomics.missouri.edu) that is accessible to the public domain.
Brassica napus (also known as rape and oilseed rape) is the third largest oilseed crop in the world, providing approximately 13% of the world's supply of vegetable oil. B. napus seeds produce oil and protein as the main storage compounds out of the three principal storage reserves (proteins, oil [triacylglycerols], and carbohydrates [starch]) found in plant seeds (Norton and Harris, 1975; Murphy et al., 1989; King et al., 1997; Schwender and Ohlrogge, 2002). These compounds support early seedling growth, and in nature the relative proportions of these compounds vary dramatically among different plant seeds. Seeds of legume species such as soybean (Glycine max) and pea (Pisum sativum) produce seeds with 20% to as much as 40% protein and 20% oil, while B. napus produces seeds with approximately 40% oil and 15% protein (Gunstone et al., 1995).
Biochemical and molecular studies are beginning to define the biosynthetic pathways responsible for accumulation of these storage components in B. napus seed (Rawsthorne, 2002; Schwender and Ohlrogge, 2002; Hill et al., 2003; Schwender et al., 2003; Goffman et al., 2004; Kubis et al., 2004; Ruuska et al., 2004; Schwender et al., 2004a, 2004b; Chia et al., 2005; Goffman et al., 2005). Recently, a study of B. napus developing embryos demonstrated that Rubisco acts without the Calvin cycle to increase the efficiency of carbon use during triacylglycerol production (Schwender et al., 2004a). Despite these advancements, little is known about the regulation, both translational and posttranslational, of proteins during seed development. It is quite evident from the available studies that seed-filling processes are highly complex and that many genes encoding enzymes of the respective pathways are tightly coordinated for fine-tuned regulation of each storage component. Since storage components are of nutritional and economical importance, understanding the regulatory mechanisms responsible for their synthesis has become an important challenge. To this end, it may be necessary to apply systematic and multiparallel approaches to study the association of metabolic networks with seed filling on a global scale, rather than studying an isolated enzyme or pathway using conventional biochemical and molecular approaches.
Impressive achievements in genome and cDNA sequencing have yielded a wealth of information for many model organisms, including the flowering plants Arabidopsis (Arabidopsis thaliana) and rice (Oryza sativa; Arabidopsis Genome Initiative, 2000; Goff et al., 2002; Seki et al., 2002; Yu et al., 2002). As a consequence, there have been tremendous advances in high-throughput technologies, such as transcriptomics and proteomics to profile genome-wide expression of genes at the mRNA and protein levels, respectively. These technologies have provided a unique opportunity to study the biological systems on a genome-wide scale. Arabidopsis was the first plant that was used for genome-wide analysis of seed filling at the transcriptome level (Girke et al., 2000; Ruuska et al., 2002). These studies cataloged temporal changes in gene expression and provided insight into the primary transcriptional networks that coordinate the genome-wide response to seed developmental programs and lead to the distribution of carbon among oil, protein, and carbohydrate reserves. Nevertheless, it should be noted that cellular signaling and metabolic events are also driven by protein-protein interactions, posttranslational protein modifications, and enzymatic activities that cannot be predicted accurately or described by transcriptional profiling approaches alone. Therefore, to better understand gene function and to address biochemical and physiological questions, it is imperative to include proteomics in any systems biological approach (Aebersold and Mann, 2003; Huber, 2003). Improvements in high-resolution two-dimensional gel electrophoresis (2-DE), imaging and analysis, development of protein and nucleotide databases, and methods for protein identification using modern mass spectrometry (MS), such as matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) and liquid chromatography-tandem MS (LC-MS/MS), have made the large-scale profiling and identification of proteins a dynamic new area of research in plant biology (Aebersold and Mann, 2003; Gorg et al., 2003; Agrawal et al., 2005).
In recent years, proteomics has been applied to investigate seed germination and seed development using a proteomics workflow including Medicago truncatula (Gallardo et al., 2003), pea (Schiltz et al., 2004), and soybean (Hajduch et al., 2005). Soybean represents the most systematic study conducted to date; the high-density reference map established in the study includes the developmental expression profiles of 679 protein spots and identification of 422 protein spots representing 216 nonredundant proteins and 14 protein functional classes.
Here we have conducted an in-depth analysis of B. napus (cv Reston) during five sequential stages of seed filling using 2-DE coupled with MS (MALDI-TOF and LC-MS/MS), with the main objective to characterize metabolic networks. We present high-resolution reference maps of pH 3 to 10 and pH 4 to 7, along with expression profiles of 794 protein spots that reveal 12 principal expression trends during seed development. Using both MALDI-TOF and LC-MS/MS, the identity of 517 protein spots were obtained representing 289 nonredundant proteins. One of the surprising findings of this proteomic study is the preponderance of proteins related to metabolism and energy production. We present and discuss the regulation of these metabolic networks at the protein level, by mapping the protein components and their expression profiles on to the pathways of carbon assimilation. Data generated from this study have been deposited into a database (http://oilseedproteomics.missouri.edu) that is accessible to the public domain.
RESULTS
Characterization of Developing B. napus Seed
Developing B. napus (var. Reston) seeds were staged precisely at 2, 3, 4, 5, and 6 weeks after flowering (WAF), the period when major metabolic changes occur within the embryo. At each developmental stage, seed fresh weight and protein content were measured (Fig. 1). At 2 WAF the liquid endosperm dominates while the embryo accounts for a small portion of the seed. The embryo begins to take significance within the seed at 3 WAF. This can be estimated by the pale green appearance of seeds at 2 and 3 WAF with increasing darker portion representing the developing embryo. At 4, 5, and 6 WAF, the embryo accounts for most of the seed mass and the seed coat begins to senesce, as visualized by the change in color, when approaching the desiccation stage (Bewley and Black, 1994).
Figure 1.

Characterization of B. napus seeds during seed filling. Sampling was carried out at 2, 3, 4, 5, and 6 WAF. The top section shows seed stages. Seed fresh weight, FA content, and total seed protein were measured and presented graphically in the bottom sections, respectively. Fresh weight values are the average of 10 seeds, FA and protein determinations are the average of three biological replicates. sd is denoted by error bars.
Seed fresh weight increased from 2 until 5 WAF followed by a decrease in weight at 6 WAF, indicating that developing seeds enter into the desiccation phase after 5 WAF. This is in agreement with previous studies on B. napus seed development where the authors concluded that the aqueous soluble fraction increased to a maximum at 5 WAF and then declined (Norton and Harris, 1975). After this period, protein content increased dramatically, reaching approximately 10% of fresh seed weight at 6 WAF, also in agreement with a previous report (Norton and Harris, 1975). Measurements of seed fresh weight and protein content at five different stages indicate that selected time points are representative of the seed-filling phase of development.
To further characterize developing B. napus seed, seed fatty acids (FAs) were quantified by gas chromatography (GC; Table I). Accumulation of total FAs increased significantly during seed development (Fig. 1) and by 6 WAF accounted for over 20% of the seed dry mass. Compositional analysis revealed major fluctuations in the individual FA species (Table I). Twelve different FAs accounted for nearly all of the acyl chains present in developing B. napus seed. Interestingly, the accumulation of oleic acid (18:1) and eicosenoic acid (20:1) showed maximum abundance at 5 WAF, while erucic acid (22:1) peaked later at 6 WAF. This is not surprising, given that 18:1 is the precursor for elongation to 22:1, the prominent FA in B. napus Reston.
Table I.
FA composition in developing B. napus cv Reston seeds
Twelve principal B. napus seed FAs were quantified in biological triplicate from 2, 3, 4, 5, and 6 WAF seed. The seed FAs were derivatized to FA methyl esters for analysis by gas chromatography (GC). Identification of FAs was performed by on-line mass spectral analysis while quantification was performed by flame ionization. The amount of each (and total) FAs is expressed in microgram per milligram of seed dry weight. Values are the average of biological triplicates and sd is shown. ND, Not detected.
| FA | WAF
|
||||
|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | |
| 16:0 | 3.5 ± 0.4 | 9.5 ± 5.0 | 9.2 ± 3.4 | 17.3 ± 1.8 | 19.2 ± 5.3 |
| 18:0 | 1.9 ± 0.3 | 4.5 ± 4.1 | 4.4 ± 3.3 | 9.3 ± 0.8 | 10.6 ± 2.4 |
| 18:1Δ9 | 0.6 ± 0.1 | 13.0 ± 2.9 | 36.7 ± 9.7 | 49.7 ± 16.4 | 34.6 ± 0.9 |
| 18:1Δ11 | 0.6 ± 0.1 | 7.0 ± 2.0 | 6.4 ± 1.0 | 10.0 ± 2.0 | 8.1 ± 1.3 |
| 18:2 | 12.1 ± 1.7 | 19.4 ± 2.1 | 19.5 ± 2.0 | 31.9 ± 6.4 | 36.1 ± 9.0 |
| 18:3 | 4.6 ± 0.7 | 6.5 ± 0.5 | 9.2 ± 1.1 | 13.7 ± 2.3 | 16.1 ± 2.7 |
| 20:0 | 0.2 ± 0.1 | 0.5 ± 0.1 | 0.8 ± 0.4 | 1.5 ± 0.7 | 1.3 ± 0.4 |
| 20:1Δ11 | ND | 3.4 ± 1.9 | 11.9 ± 9.1 | 22.7 ± 7.4 | 15.2 ± 1.9 |
| 20:1Δ13 | ND | 0.9 ± 0.6 | 1.6 ± 0.9 | 2.5 ± 0.4 | 3.7 ± 0.6 |
| 22:0 | ND | 0.9 ± 1.3 | 1.2 ± 1.0 | 2.1 ± 0.3 | 2.5 ± 1.3 |
| 22:1Δ13 | ND | 2.3 ± 2.8 | 33.6 ± 11.8 | 36.0 ± 10.2 | 61.4 ± 13.6 |
| 22:1Δ15 | ND | 0.4 ± 0.1 | 1.5 ± 0.3 | 1.3 ± 0.2 | 4.2 ± 1.0 |
| Total FA content | 23.2 ± 3.4 | 68.0 ± 15.6 | 135.9 ± 43.0 | 197.9 ± 45.1 | 213.1 ± 38.6 |
Medium-Range Isoelectric Focusing Is Necessary for Reduction of Spot Overlap in Two-Dimensional Gels of Developing B. napus Seed
Initial 2-DE analysis of proteins from B. napus seed was performed using an isoelectric focusing (IEF) range of pH 3 to 10. As seen in Figure 2A, the region between pH 4 to 7 is protein rich and overlapping spots are prevalent. Use of medium pH gradients is one effective way to reduce spot overlap (Campostrini et al., 2005). Therefore, additional 2-DE with pH 4 to 7 immobiline pH gradient (IPG) strips was performed to ameliorate this problem (Fig. 2B). Visual inspection of 2-DE gels revealed highly dynamic changes in the seed proteome. Each 2-DE gel contained approximately 1,200 to 1,300 Coomassie-stained protein spots with a dynamic range in spot volume ranging from 1 to 4.5 × 106. However, spots below a volume of 7 × 103 were difficult to quantify, therefore, the quantitative, dynamic range varied from 7 × 103 to 4.5 × 106.
Figure 2.

Two-dimensional gel electrophoresis analysis of proteins (1 mg) isolated from B. napus seeds at 2, 3, 4, 5, and 6 WAF. A, Protein analysis using wide-range IPG strips with pH range from 3 to 10. B, Protein analysis using medium-range IPG strips with pH range from 4 to 7. Reference gel is a composite of all five investigated stages obtained by pooling of 0.2 mg of protein from each stage.
Protein Quantification Established Developmental Expression Profiles for 794 Protein Spots
For accurate determination of protein expression, the five developmental stages of B. napus seed development were analyzed in biological quadruplicate (Fig. 3A). Four sample harvesting events were followed by four independent protein extractions. This naturally resulted in both biological and technical variation in the quantification results that is noted by the sds presented in Supplemental Tables I and II. It was noticed that low abundance spots, as well as those spots present in only two biological replicates, typically exhibited the highest level of variation. To select only high-quality protein spots for expression profiling, the following threshold criteria were applied. Each protein spot was present in at least three biological replicate gels and detected in at least two developmental stages. Using this approach, a total of 794 protein spot groups were matched, manually validated, and quantified (Supplemental Table I). Moreover, coefficient of variation (CV) value was calculated to allow direct comparison of significance in acquired quantification data (Supplemental Table I).
Figure 3.

Proteomics experimental design used in this study. A, Each investigated seed stage was analyzed in quadruplicate by 2-DE. Only those spots present in at least three gels were considered for analysis. Relative quantification and expression profiles were determined using ImageMaster 2-DE Platinum software version 5.0. Spots from analyzed gels were matched to a reference gel and relative volumes were calculated for each group. Average relative volumes and sds for matched spots were calculated and data were plotted onto a line graph. Expression profiles were then deposited into a Web-based data repository (http://oilseedproteomics.missouri.edu). B, Protein spots with expression profiles were excised from reference (pooled) gels of pH ranges of 3 to 10 and 4 to 7. Each spot was trypsin digested and peptides were analyzed by MALDI-TOF and LC-MS/MS. Spectra were searched against NCBI and TIGR databases using MS Fit program of Protein Prospector for MALDI-TOF spectra and SEQUEST for data acquired by LC-MS/MS. At this point, four independent protein assignments for each spot were obtained: (1) MALDI-TOF data searched against NCBI; (2) MALDI-TOF searched against TIGR; (3) LC-MS/MS searched against NCBI; and (4) LC-MS/MS searched against TIGR. Two-step data consolidation approach was applied (see “Materials and Methods”) that resulted in 517 highly confident protein assignments.
Seed Storage Proteins Can Be Subtracted in Silico to Eliminate the Bias of Highly Expressed Proteins
Seed storage proteins (SSPs) are highly abundant at 4, 5, and 6 WAF. As these proteins accumulate, the relative volume of other spots concomitantly decrease, which may bias composite and individual expression profiles. To test this hypothesis, all spots corresponding to SSPs were subtracted in silico and expression of the remaining spots were renormalized (Supplemental Table II). To view possible influence of in silico SSP subtraction, the composite expression profiles (discussed below) were created using both original and SSP-subtracted datasets (Fig. 6). Although the shape of expression trends was not greatly affected, slight changes were observed. For instance, maximum accumulation of photosynthetic proteins occurred at 3 WAF. After SSP removal, the peak moved to 4 WAF.
Figure 6.

Chronological characterization of processes during seed filling in B. napus. The composite expression profiles (pooled relative volumes) for all functional subclasses of proteins (according to classification scheme by Bevan et al., 1998) containing 10 or more proteins are shown. Profiles were generated using relative two quantification datasets: total protein spot groups (full line) and spot groups minus in silico subtracted SSPs (dashed line). Name of functional subclass, number of unique proteins (in parentheses), and expression trend with maximum y axis values are shown.
Combination of MALDI-TOF MS and LC-MS/MS Resulted in 517 High-Confidence Protein Assignments
In theory, B. napus is a nonideal organism for proteomic analyses, due to the lack of comprehensive sequence databases. The National Center for Biotechnology Information (NCBI) nonredundant protein database contained only 2,207 entries for Brassica species, as of March, 2005. The largest database for B. napus is available at The Institute for Genomic Research (TIGR), containing 5,568 tentative consensus entries. Due to these limitations, we decided to acquire MS data in duplicate using two different mass spectrometers and to perform comprehensive data mining using both NCBI and TIGR databases.
Figure 3B gives an overview of data acquisition and data mining strategy. Altogether 794 spots were excised from 2-DE gels of pH 4 to 7 and pH 3 to 10 (pH 7–10 region only), digested with trypsin, and analyzed by MALDI-TOF MS and LC-MS/MS in parallel. Peptide mass fingerprint (PMF) analysis using MALDI-TOF MS resulted in 175 protein assignments, while LC-MS/MS resulted in 479 assignments. Protein assignments by MALDI-TOF and LC-MS/MS were integrated to reveal 137 identical and 46 nonidentical assignments. In total, 517 assignments were made (65.1% identification efficiency), of which 289 were nonredundant proteins (Supplemental Table III).
LC-MS/MS Yielded Higher Percentage of Protein Assignments Than MALDI-TOF PMF
It was previously reported that MALDI-TOF and LC-MS/MS are equally efficient at protein identification when comprehensive databases are available (Lim et al., 2003; Person et al., 2003). With plant species such as B. napus, for which database resources are limited, application of both MS methods may improve the identification rate. The protein identification workflow (Fig. 3B) used here allows for direct comparison of these two ubiquitous MS methods for protein identification. Interrogation of MALDI-TOF data against two databases (nonredundant NCBInr and TIGR) resulted in 175 protein assignments out of 794 protein spots, representing a 22.0% identification frequency. When compared to results obtained with other plants such as soybean (Herman et al., 2003; Mooney et al., 2004; Hajduch et al., 2005), Arabidopsis (Gallardo et al., 2001, 2002a, 2002b), or Medicago truncatula (Gallardo et al., 2003; Watson et al., 2003) the identification efficiency is low. In contrast, searching LC-MS/MS data against the same databases resulted in 479 assignments (60.3% identification frequency). These data support the notion that LC-MS/MS is superior to MALDI-TOF for protein identification. However, our results indicate that parallel MS identification strategies can improve the overall identification. In this study, the identification rate increased from 60.3% using LC-MS/MS alone to 65.1% using a combination of MALDI-TOF and LC-MS/MS. From the 517 identified proteins, 38 (7.3%) were identified exclusively by MALDI-TOF, 342 by LC-MS/MS only (66.1%), and 137 by both methods (26.5%). Although LC-MS/MS clearly produced a higher number of assignments, identification by MALDI-TOF may be more successful for some proteins.
Manual Validation of Protein Annotation Improves Data Analysis
In general, database annotations infrequently provide information about intracellular localization. Since enzymes may have different functional roles depending upon subcellular location, proteins that may potentially have multiple intracellular isoforms were analyzed by three different algorithms to predict subcellular targeting. For example, in this study, six protein spots were identified simply as malate dehydrogenase (MDH). In this case, MDH can be located either in the cytosol where it is a part of gluconeogenesis, in the mitochondria where it is involved in the TCA cycle, or in the plastids where it is involved in redox shuttling or the photosynthetic C4 cycle. Applying subcellular localization algorithms, it was suggested that four spots (884, 5,218, 5,222, and 5,225) are cytosolic MDH, and the remaining two spots (5,185 and 5,292) are mitochondrial MDH isoforms. However, it should be noted that subcellular assignments based upon localization algorithms are at best predictive, experimental evidence is required for confirmation.
A large percentage of proteins identified in this study were annotated as unknown. At an early stage of this investigation, unknown proteins accounted for as much as 38% of all the identified proteins (data not shown). Although some databases provide current annotation information about sequence homology/similarity in the identifier tag, many do not. Therefore, all assignments lacking this information were subjected to homology search using BLASTP against the NCBI nonredundant database. This manual validation strategy reduced the percentage of unknown proteins from 38% to 2.1%.
Proteins Associated with Energy and Metabolism Are Prevalent in Developing B. napus Seed
All identified proteins were classified into functional classes as originally established by Bevan et al. (1998) for the Arabidopsis genome project. However, classes were slightly modified to be more suitable for a seed development study. With respect to the metabolism class, a polysaccharide catabolism subclass was added and the lipid and sterols subclass of metabolism was separated into individual subclasses. An additional subclass, seed maturation, was added in the cell growth and division class. Supplemental Table III provides a list of all identified proteins sorted into plant functional classes with details of protein assignments. On a relative spot volume basis, proteins associated with protein destination and storage are the most abundant class followed by proteins associated with energy production and metabolism (Fig. 4). However, when isoform redundancy is taken into account, proteins associated with energy are the most represented followed by metabolism-related proteins.
Figure 4.

Functional classification of identified B. napus developing seed proteins according to scheme by Bevan et al. (1998). Out of a total of 517 identified proteins, 289 had nonredundant function. Black bars show distribution of all 517 proteins and gray bars show distribution of 289 nonredundant proteins into functional classes.
Hierarchical Clustering of 794 Quantified Spot Groups Resulted in 12 Cluster Groups
Hierarchical clustering reduced 794 expression profiles into 12 expression cluster groups (Fig. 5). Visual inspection of these expression groups suggests diverse and complex patterns of regulation. The two most abundant groups, 3 and 11, had negative slopes, indicating decreasing abundance with seed age. These groups were largely composed of proteins involved in energy and protein destination and storage, representing 21.2% and 20.1% of proteins, respectively (Fig. 5). The third most abundant group, cluster 10, representing 13.7% of proteins, showed a positive slope during seed filling; the majority of these proteins were involved in protein destination and storage.
Figure 5.

Hierarchical clustering of 794 protein spot expression profiles using SAS statistical software. Relative quantification data without SSP removal were used for these analyses (Supplemental Table I). Expression trend, number of clustered expression profiles, and functional composition are shown for each of the 12 established clusters. Vertical lines indicate inverse degree of relatedness of clusters.
Figure 5 also revealed that the distribution of plant functional classes in protein expression clusters is not homogenous. For instance, proteins involved in protein destination and storage were mainly grouped in clusters 10 and 11 with negative and positive expression slopes, respectively. This is due to the heterogeneous composition of this functional class that contains SSPs, which increase during seed fill, but also proteins involved in folding and stability that decreased with seed age (Supplemental Table III).
Composite Expression Profiles of Functional Subclasses Revealed System-Wide Trends
To characterize cellular activities during seed filling, composite expression profiles were established for individual subclasses. For statistical reasons, only those functional subclasses containing 10 or more proteins were considered for analysis. Based upon these criteria, 384 of 517 identified proteins (74.7%) were grouped into 13 functional subclasses (Fig. 6). The most prevalent subclass of proteins in terms of relative spot volume, storage proteins (71 proteins), gradually increased in abundance beginning at 3 WAF. The second most abundant subclass, amino acid metabolism (42 proteins), decreased in abundance from 2 WAF until midpoint of seed filling and remained at a constant level thereafter. Relative abundance of detoxification proteins, the third most abundant group with 36 proteins, rapidly increased beginning at 5 WAF. Proteins related to photosynthesis are the fourth abundant cluster and revealed a gradual increase in relative abundance until 3 WAF, followed by a slight decrease.
Collectively, these functional subclasses exhibited three different expression trends. The first group included proteins expressed mainly at early stages of seed filling. These proteins are involved in glycolysis, respiration, metabolism of sugars, signal transduction, metabolism of amino acids, proteolysis, and defense (Fig. 6). Proteins of the second group, involved in photosynthesis and lipid metabolism, exhibited highest expression at midpoint of seed filling. Finally, detoxification, seed maturation, and SSPs were highly abundant at later stages of development (Fig. 6). Although perhaps overly simplistic, the expression trends agree with previous observations that seed filling in B. napus begins with sugar mobilization, and is followed by sequential surges in amino acid, lipid, and storage protein synthesis (Fig. 6).
DISCUSSION
Within the larger goal of profiling protein expression globally in developing B. napus seeds, the aim of this study was to characterize the metabolic pathways operating during seed filling. The major economic value of B. napus lies in its oil, and any insight into the regulation of oil accumulation during seed filling would be useful. Therefore, we focus our discussion on the pathways leading to de novo FA synthesis. However, it is important to mention that this study has also found protein components for a number of other pathways (Supplemental Table III). For instance, 8% and 5% of the total identified proteins are involved in amino acid metabolism and proteolysis, respectively. Interestingly, the majority of proteins identified here were represented by multiple isoelectric forms, suggestive of posttranslational modification. Thus, as genome resources of Brassica crops improve, the high-resolution 2-DE maps reported here can be used as a predictive tool to search for unexpected isoelectric species to begin uncovering posttranslation regulation.
As demonstrated in this study, the main advantage of 2-DE is the detection of different isoelectric species of various proteins. For example, 17 isoelectric species were detected for the glycolytic enzyme Fru bisphosphate aldolase (FBA). This is an unusually high number of isoelectric species, which is strongly suggestive of posttranslational modification(s). This type of information cannot be acquired using transcriptomic or metabolomic approaches. Although adept at resolving isoelectric species, the technique of 2-DE is somewhat restricted at quantifying low abundance proteins. For instance, Glc 6-P dehydrogenase, transketolase, transaldolase, ribulose 5-P epimerase, and Rib 5-P isomerase were analyzed in a recent study of enzyme activities of oxidative pentose phosphate pathway in developing B. napus embryos (Hutchings et al., 2005). However, this proteomics study did not detect these proteins, presumably due to their low abundance. Despite the known limitations of current 2-DE methodology at detecting underrepresented proteins, the expression and identity of 517 protein species expressed during seed filling of B. napus were characterized in this investigation, representing the largest integrated dataset for any oilseed.
Rubisco Is Highly Expressed throughout Seed Filling
Eleven Rubisco large subunits were detected and can be divided into two groups, based on their peak of accumulation. The first group showed maximum abundance at early stages of seed filling (2 or 3 WAF) and includes seven protein spots (511, 520, 536, 4,885, 4,931, 4,937, and 5,009). The second group (spots 4,919, 4,922, and 4,924) with higher protein abundance at midpoint of seed filling (4 WAF), showed bell-shape expression profiles, very similar to the data acquired for pyruvate dehydrogenase (PDH; spots 5,148 and 5,811; Fig. 7). Interestingly, expression profiles of the second group (spots 4,919, 4,922, and 4,924) and PDH (spots 5,148 and 5,811) are also very similar to the composite expression profile of enzymes involved in lipid metabolism (Fig. 5). A similar increase in the Rubisco small subunit was also reported in Arabidopsis by microarray analysis (Ruuska et al., 2002).
Figure 7.

Pathways for carbon assimilation during seed filling in B. napus. Proteins involved in sugar breakdown are displayed on the corresponding metabolic pathways. Graph shows expression of protein spots during seed filling. Expression profiles were generated using the total protein spot dataset after in silico SSP subtraction. Number above each graph shows maximum value y axis (relative volume). Dashed arrows are used when no protein was detected. Abbreviations for metabolites: UDP-G, UDP-Glc; G-1-P, Glc 1 phosphate; G-6-P, Glc 6 phosphate; F-6-P, Fru 6 phosphate; F-1,6bisP, Fru 1,6 bis phosphate; GAP, glyceraldehyde 3-P; DHAP, dihydroxyacetone phosphate; 1,3-bis PGA, 1,3 bis phosphoglyceric acid; 3-PGA, 3 phosphoglyceric acid; 2-PGA, 2 phosphoglyceric acid; PEP, phosphoenolpyruvate. Abbreviations for enzymes: SuSy, Suc synthase; UGP, UDP-Glc pyrophosphorylase; PGM, phosphoglucomutase; PGI, phosphoglucose isomerase; PFK, pyrophosphate-dependent phosphofructokinase; FBA, Fru bisphosphate aldolase; GAPDH, glyceraldehyde 3-P dehydrogenase; TPI, triose-phosphate isomerase; PGK, phosphoglycerate kinase; iPGAM, 2,3-bisphosphoglycerate-independent phosphoglycerate mutase; PRK, phosphoribulokinase; Rubisco, ribulose-1,5-bisphosphate carboxylase; PK, pyruvate kinase; PDH, pyruvate dehydrogenase.
The high abundance of Rubisco subunits is in contrast with low abundance of other enzymes of the Calvin cycle, many of which were below the detection limit of this proteomics study. A possible explanation for this disparity in protein abundance may be found in a recent stable isotope labeling study of B. napus embryos (Schwender et al., 2004a). The Calvin cycle is the cyclic regeneration of ribulose-1,5-bisphosphate from 3-phosphoglyceric acid (3-PGA), which results in carbon dioxide distribution into all carbon positions of the cycle's intermediates, including 3-PGA (Bassham et al., 1954). The labeling experiment showed that 13C was incorporated mainly into the C1 carbon position of 3-PGA, while FAs derived from C2 and C3 of 3-PGA were labeled at extremely low levels (Schwender et al., 2004a). Based upon these data the authors concluded a possible role of Rubisco in carbon dioxide recycling, apart from the Calvin cycle.
Glycolytic Reactions during Seed Filling Are Principally Cytosolic
An important component of carbon assimilation in developing seeds is glycolysis. Although this ubiquitous pathway was first elucidated in the 1940's (Meyerhof and Junowicz-Kocholaty, 1943; Meyerhof, 1945), relatively little is known about the regulation and control of this pathway. This is particularly true in plants due to the added complexity of parallel pathways in both the cytosol and plastids (Plaxton, 1996; Fernie et al., 2004).
This study localized several protein spots corresponding to numerous different glycolytic enzymes both in the cytosol and plastids. Based upon quantification data acquired during seed development, it is possible to examine the apparent redundancy of glycolytic pathways between the cytosol and plastids. Suc synthase (SuSy) catalyzes the initial release of sugar for glycolysis by converting Suc into UDP-Glc and Fru. A total of three SuSy spots (202, 204, and 4,628) were identified (Fig. 7). The overall abundance of all three detected protein spots is similar although their expression profiles differ. While two SuSy spots (spots 202 and 204) shared almost identical expression profile with maximum abundance at 3 WAF, the abundance of a third form (spot 4,628) reached a maximum at 5 WAF followed by a dramatic decrease thereafter. This suggests the presence of two types of SuSy that are perhaps active during early (type I) and late (type II) phases of seed filling.
UDP-Glc pyrophoshorylase (UGP) catalyzes the reversible production of Glc-1-P from UDP-Glc. In Arabidopsis there are two homologous UGP genes located on two different chromosomes (Kleczkowski et al., 2004). We identified their homologs in B. napus, spot 557 (homolog to At5g17310) and spot 4,932 (homolog to At3g03250). Despite 92% amino acid homology sequence, their expression profiles during seed filling are slightly different (Fig. 7). Spot 4,932 was found to be highly expressed at 2 WAF after which its relative abundance declined in a gradual manner reaching a minimum at 6 WAF. Spot 557, which is about 4.4 times less abundant than spot 4,932, peaked in abundance at 3 WAF and decreased thereafter. These expression profiles suggest that the abundant UGP (spot 4,932) may play a typical role in glycolysis, because its profile is the same as the composite expression profiles of glycolytic enzymes (Fig. 7). On the other hand, protein spot 557 may also have a role outside of glycolysis during seed filling. In addition to glycolysis, UGP can be involved in cell wall biogenesis because a product of UGP, UDP-Glc, is used in the biosynthesis of cell wall polysaccharides and serves as a precursor for cell wall biogenesis (Gibeaut, 2000).
Phosphoglucomutase (PGM) catalyzes the interconversion of Glc-1-P and Glc-6-P. The Arabidopsis genome contains two cytosolic and one plastidial form of PGM (Caspar et al., 1985). The plastidial form of PGM has been characterized in B. napus (Harrison et al., 2000). We have identified six protein spots as cytosolic PGM matching to three different sequences in the database (Supplemental Table III). Spot 337 matched to cytosolic PGM from Populus tomentosa, three others (4,724, 4,731, and 4,740) to B. napus homologs of cytoplasmic PGM from Arabidopsis, and two (4,705 and 4,741) matching to another cytoplasmic PGM from Arabidopsis. The overall expression trend of each PGM was found to be similar, higher expression at early stages of seed filling and lower at 6 WAF (Fig. 7). However, differences between protein abundances at the early stages were observed. For instance, spots 4,740 and 4,741 were most abundant at 2 WAF and spots 337, 4,705, 4,724, and 4,731 at 3 WAF.
In Arabidopsis two isozymes of phosphoglucose isomerase (PGI) exist, one in the plastids and the other in the cytosol (Caspar et al., 1985). We identified two protein spots as cytosolic PGI, that had similar expression trends, but one (4,883) with maximum abundance at 3 WAF and the second (4,875) at 2 and 4 WAF. Pyrophosphate-dependent phosphofructokinase (PFK) catalyzes conversion of F-6-P and F-1,6-bisP, and in plants is regulated by Fru-2,6 bisphosphate (Huber, 1986; Stitt, 1990; Nielsen et al., 2004). We identified two cytosolic isoelectric species of PFK (spots 374 and 4,801; Fig. 7) and a putative organellar form (spot 4,844). Both cytosolic and organellar PFKs are abundant, but the cytosolic spot 374 is about 3 times more abundant than organellar PFK. Cytosolic and organellar PFKs also differ in their developmental expression trends. The two detected cytosolic spots of PFK share similar expression profiles: high abundance at 2 WAF followed by dramatic decrease, and undetectable at 5 and 6 WAF. On the other hand, the organellar PFK is detectable throughout seed filling showing a maximum abundance at 4 WAF.
FBA catalyzes the aldol cleavage of Fru 1,6-bisP to glyceraldehyde 3-P (GAP) and dihydroxyacetone phosphate (DHAP). Surprisingly, a relatively large number of cytosolic and plastidial FBA spots were identified (Fig. 7). Most of the nine cytosolic spots and eight plastidial spots shared similar expression profiles, high abundance at early stages followed by rapid decrease between 3 and 4 WAF. However, two differences between cytosolic and plastidial FBA can be noted; the cytosolic forms were generally more abundant and only two cytosolic FBA (spots 5,157 and 5,189) peaked in expression at 4 WAF. The high number of protein spots suggests these activities may be posttranslationally modified.
Triose-P isomerase (TPI) catalyzes the interconversion of GAP and DHAP. This reaction is reversible although the equilibrium favors DHAP. Seven cytosolic spots of TPI (spots 1,156, 5,515, 5,520, 5,524, 5,525, 5,528, and 5,530) were identified, but no plastidial TPI spot could be detected (Fig. 7). Interestingly, one protein spot (spot 5,515) showed maximum abundance at 6 WAF (Fig. 7).
Glyceraldehyde 3-P dehydrogenase (GAPDH) reversibly catalyzes the conversion of GAP into 1,3-bis PGA. Five cytosolic spots (spots 821, 1,427, 5,184, 5,186, and 5,206) and one plastidial (spot 820) GAPDH were identified. The cytosolic and plastidial GAPDH were almost equally abundant during seed filling and shared very similar expression profiles (Fig. 7). Two spots of cytosolic phosphoglycerate kinase (PGK; spots 5,144 and 5,145) and two plastidial PGK (5,054 and 5,055) were identified. Like TPI, only cytosolic forms of 2,3-bisphosphoglycerate-independent phosphoglycerate mutase (iPGAM) and enolase were identified (Fig. 7). Expression profile of iPGAM (spot 4,747) was present in abundance at 2 WAF. Expression profiles of enolases (spots 4,898 and 4,903) were also high at 2 WAF, but they accumulated strongly until 4 WAF followed by a rapid decrease in abundance.
The detection of multiple isoelectric species for cytosolic and plastidial glycolytic enzymes and strong similarities in their expression profiles suggest possible posttranslational modifications as well as coordination between cytosolic and plastidial glycolysis during seed filling.
Proteomics Data Suggest That Phosphoenolpyruvate Is a Direct Precursor for de Novo FA Synthesis in Plastids
The current model of metabolite flux between cytosol and plastids has established that either phosphoenolpyruvate (PEP) or pyruvate is transported into plastids for further processing into acetyl-CoA (Weber, 2004; Weber et al., 2005). Moreover, a previous microarray analysis of Arabidopsis developing seeds has indicated that plastid uptake of cytosolic PEP is a more likely possibility than the uptake of cytosolic pyruvate during seed development (Ruuska et al., 2002), particularly since a plastid pyruvate translocator has yet to be identified. Furthermore, a flux model for central carbon metabolism of developing B. napus embryos constructed based on stable isotope labeling of sugars has also suggested that the main carbon flux into FAs is through plastid uptake of cytosolic PEP (Schwender et al., 2003; Kubis et al., 2004).
In this study, we identified almost all enzymes involved in cytosolic and many for plastid glycolysis. The notable exceptions are cytosolic pyruvate kinase, plastid iPGAM, plastid enolase, and plastid TPI (Fig. 7). One possible explanation is low expression levels, which is supported by the observation that plastidial iPGAM and enolase were previously determined to have low specific activities (Eastmond and Rawsthorne, 2000). Since we did not detect plastid iPGAM and enolase, 3-PGA produced by plastidial glycolysis, and more importantly by Rubisco bypass, would need to be transported into the cytosol by triose phosphate/phosphate translocator, converted to PEP, then transported back into plastids by PEP translocator before conversion to acetyl-CoA for FA synthesis. However, the absence of these plastid glycolytic enzymes in this proteomic study do not preclude the potential for low level expression, due to the limited dynamic range of this analysis.
FA Biosynthesis Machinery Expressed Prominently at Midpoint of Seed Filling
Conversion of acetyl-CoA into malonyl-CoA is catalyzed by acetyl-CoA carboxylase. This plastid complex is comprised of four subunits, the biotin carboxylase, biotin carboxyl carrier protein, and carboxyltransferase subunits (α and β; Shorrosh et al., 1996). Detection of two spots corresponding to biotin carboxylase (spots 4,940 and 4,952) indicates high expression from 3 to 5 WAF. Previous investigations showed that acetyl-CoA carboxylase is highly expressed during embryo development in B. napus (Elborough et al., 1996; Thelen et al., 2001). The peak of protein accumulation for malonyl-CoA transacylase suggests that the reversible conversion of malonyl-CoA into malonyl-acyl-carrier protein (ACP) is highly active at 3 WAF. Expression profiles of enzymes involved in remaining reactions toward FA synthesis are highly expressed at 4 or 5 WAF. Ketoacyl-ACP synthetase I, the enzyme that catalyzes the condensation of malonyl-ACP into 3-ketoacyl-ACP, reached high abundance at 3 WAF and remained high through 4 WAF. Four detected spots of enoyl-ACP reductase shared similar expression trends. Surprisingly, we detected seven spots corresponding to stearoyl-ACP desaturase. Four proteins (spots 5,166, 5,193, 5,194, and 5,198) are highly abundant and share almost identical expression profiles, with peak of protein abundance at 4 WAF. Two low abundant protein spots (spots 5,169 and 5,180) are expressed differently during seed filling. Spot 5,169 was found to be highly abundant only at 4 WAF, whereas spot 5,180 was constitutively and highly expressed from 2 until 4 WAF. The abundance of stearoyl-ACP desaturase during seed filling suggests low catalytic turnover of this enzyme as well as the importance of 18:1 export from the plastids.
In summary, this investigation represents a systematic proteomics study of whole-seed proteins expressed during seed filling in B. napus. Multiple categories of proteins were observed, although protein storage, energy, and metabolism associated proteins were most abundant. The preponderance of metabolic proteins presented a unique opportunity to map activities (and isoelectric species therein) for carbon assimilation. Surprisingly, carbon flow from Suc to acetyl-CoA could be entirely predicted based upon the representation of proteins for each enzymatic step. The expression levels of cytosolic pyruvate kinase, plastid enolase, and most of the enzymes of the Calvin cycle were below the detection limit of this proteomics study, except Rubisco and phosphoribulokinase that were both highly expressed. Thus, carbon flow from Suc appears to primarily follow a cytosolic glycolytic track until PEP, at which point carbon is likely imported into plastids and converted into pyruvate and acetyl-CoA for de novo FA synthesis.
MATERIALS AND METHODS
Plant Material and Growth Conditions
B. napus (cv Reston) was grown in a growth chamber (16-h light/8-h dark cycle, 23°C day/20°C night, 50% humidity and light intensity of 8,000 LUX). Flowers were tagged upon opening and the developing seeds were collected at precisely 2, 3, 4, 5, and 6 WAF, in the middle of a light cycle. The dry weight and total protein content were measured at each developmental stage. Total protein was quantified using the dye-binding Coomassie protein assay using chicken γ-globulin as the standard (Bio-Rad).
FA Analysis
Developing seeds of B. napus at 2, 3, 4, 5, and 6 WAF were divided to three test glass tubes per stage (5–10 seeds per tube) and dried at 80°C overnight. After dry weight determination, 1 mL of 14% boron trifluoride was added to each tube along with 17:0 FA standard in toluene (0.5% of dry mass exactly). Total volume of toluene was brought to 150 μL and samples were incubated at 95°C for 90 min, with vortexing every 10 min. After incubation, samples were cooled to room temperature. To each tube, 1 mL of water and 3 mL of hexane were added. Tubes were vortexed and centrifuged at 3,000 rpm for 5 min. Top phase was removed and transferred to a new conical glass tube. Samples were reextracted with additional 3 mL of hexane, dried under nitrogen stream, and resuspended in 400 μL of hexane before analysis by GC. Analysis of FA was carried on Agilent Technologies model 689N Network GC system gas chromatograph with a DB-23 column (30 m × 0.25 mm; film thickness 0.25 μm; Agilent 122–2,332). The GC conditions were: injector temperature and flame ionization detector temperature, 250°C; running temperature program, 150°C for 1 min, then increasing at 2°C/min to 200°C followed by a 5 min hold at 200°C.
Protein Isolation and 2-DE
Total protein was isolated from developing seed and subjected to 2-DE as described previously (Hajduch et al., 2005). Briefly, the protein pellet was resuspended in IEF sample extraction buffer (8 m urea, 2 m thiourea, 2% (w/v) CHAPS, 2% (v/v) Triton X-100, and 50 mm dithiothreitol) with vortex mixing for 30 min at room temperature followed by centrifugation for 15 min at 14,000g to remove insoluble material. Protein quantification was performed in triplicate using the Coomassie dye binding assay (Bio-Rad) against standard curve of chicken γ-globulin. One milligram of total protein was mixed with 2.25 μL of appropriate IPG buffer (Amersham Biosciences) in a total volume of 450 μL and subjected to IEF followed by SDS-PAGE, as described previously (Hajduch et al., 2005).
Image Acquisition, Analysis, and in Silico SSP Subtraction
Coomassie G-250 (colloidal) stained gels were imaged by scanning densitometry. Digitized 2-DE images (300 dpi, 16-bit grayscale pixel depth) of five developmental stages in biological quadruplicate were analyzed using ImageMaster 2-D Platinum software (version 5.0, GE Healthcare) as described previously (Hajduch et al., 2005). A two-step data normalization method was employed to obtain an integrated dataset for the entire investigation. First, protein abundance was expressed as relative volume according to the normalization method provided by ImageMaster software that compensates for slight variations in sample loading, gel staining, and destaining. Second, relative spot volumes were adjusted using correction constants, as described previously (Hajduch et al., 2005), to allow direct data comparison between the two gel datasets: pH 4 to 7 and pH 3 to 10 sets. To enable direct spot-to-spot comparison of significance levels of acquired protein spot quantifications, coefficient of variation (CV) for each protein spot was calculated using following formula:
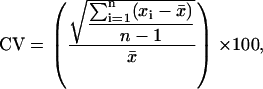 |
where 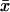 is the average of relative volumes (x) of spots in biological quadruplicate analysis and n is the sample size (four in case of biological quadruplicate).
is the average of relative volumes (x) of spots in biological quadruplicate analysis and n is the sample size (four in case of biological quadruplicate).
To subtract SSP in silico, spot volumes were calculated for each of 794 protein spots using ImageMaster 2-D Platinum software. In total, 71 identified SSP were removed from the dataset and two-step normalization approach was applied as described above.
Hierarchical Cluster Analysis of Expression Profiles
For cluster analysis of expression profiles, hierarchical clustering was performed using SAS statistical software (SAS Institute). The procedure contained two steps. First, the program established the number of classes that is best for a present dataset. The CLUSTER keyword was used with options STANDARD METHOD=AVERAGE CCC PSEUDO as the command for step 1, in which STANDARD means to normalize the variables; AVERAGE means a certain clustering method in contrast to the other 10 methods that are included in SAS IDE; CCC and PSEUDO are both options for calculating some statistical variables that are used to determine the class number. Second, the program clustered expression profiles into each of the established classes. Expression profile data were normalized in two steps. In the first step, any zero between two nonzero points was replaced with the average of two neighbor values. In the second step, a linear transformation was used to normalize expression profiles of different spots to uniform scale. The SAS program used the procedure FASTCLUS for the real clustering and the maximum number of clusters established earlier. For each spot, the variable distance parameter was generated by SAS.
Protein Identification by MS
Arraying of 2-DE gel spot, in-gel digestion, and C18 microbed chromatograph were each performed as described previously (Hajduch et al., 2005). Mass spectral analysis of trypsin-digested protein samples were carried out on a Voyager-DE Pro MALDI-TOF mass spectrometer (Applied Biosystems) and on a linear ion trap tandem mass spectrometer (ProteomeX LTQ, Thermo-Finnigan) using LC and nano-spray ionization. The MALDI-TOF instrument was operated as described previously (Hajduch et al., 2005). The LC-MS/MS was operated according to manufacturer's instructions for high-throughput protein identification. Briefly, on-line capillary LC included two polymeric sample traps (2 μg capacity each) and a fast-equilibrating C18 capillary column (Micro-Tech Scientific; 150 μm i.d. × 10 cm). The method alternated between loading/equilibration and elution using the two peptide traps (one trap is being equilibrated while the other is being eluted) to reduce time required for each on-line LC-MS/MS. For analysis, 10 μL of sample in 0.1% (v/v) formic acid was loaded. For sample elution, a 15 min gradient with 40% of solution A (0.1% formic acid in water) and 60% of solution B (0.1% formic acid in acetonitrile) was followed by a 5 min gradient with 20% solution A and 80% solution B. The column was reset for 2 min and reequilibrated for 10 min with 100% of solution A before sample previously absorbed onto the second trap was eluted. Eluted tryptic peptides were directly analyzed by LC-MS/MS using 75 μm i.d., 360 μm, o.d. 15 μm tip needles (New Objective) with a 1.7 kV nano-spray voltage. Manufacturer's recommended scan method for high-throughput protein identification consisted of double-play analysis mode, a full MS scan (400–1,600 mass-to-charge ratio), followed by data-dependent triggered MS/MS scan for the most intense ion.
Database Searching with Spectral Data and Uploading to the Oilseed Proteome Database
Searches against the NCBI (ftp://ftp.ncbi.nih.gov/blast/) nonredundant database (as of March, 2005) and TIGR tentative consensus database for B. napus (http://www.tigr.org/tigr-scripts/tgi/T_index.cgi?species=oilseed_rape) were independently performed using a two-step approach to mine maximum information from MALDI-TOF MS and LC-MS/MS. PMF-based protein identification was performed on local copy of version 3.2.1 of the MS-Fit program of Protein Prospector (http://prospector.ucsf.edu; Clauser et al., 1999) as previously described (Hajduch et al., 2005). Analysis of LC-MS/MS data was performed on a local license copy of SEQUEST software (Eng et al., 1994; Yates et al., 1995) as part of the BioWorks 3.1SR1 software suite. Search parameters were set as follows: enzyme, trypsin; number of internal cleavage sites, 2; mass range, 400 to 1,600; threshold, 500; minimum ion count, 35; peptide mass tolerance, 1.50; variable modifications, oxidation (M); static modification, carboxyamidomethylation (C). Matching peptides were filtered according to correlation scores (XCorr at least 1.5, 2.0, and 2.5 for +1, +2, and +3 charged peptides, respectively). For all protein assignments, a minimum of two unique peptides was required. Proteins with three or more unique peptides matching to protein sequence were automatically considered as a positive identification. In a situation where assignments with two unique peptides matched to the sequence, the difference between theoretical and experimental MW/pI should not exceed ±25% variance to be considered a match.
This approach resulted in four independent sets of protein identification data: (1) MALDI-TOF, TIGR search; (2) MALDI-TOF, NCBI search; (3) LC-MS/MS, TIGR search; and (4) LC-MS/MS, NCBI search. To reach consensus in protein assignments, a two-step data reduction strategy was employed. The first step combined the search results from TIGR and NCBI databases for each MS method. If two different protein assignments for one protein spot were noted, the one with the highest number of matching peptides was taken. If number of matching peptides was the same, the assignment with the highest coverage was taken. The second step combined the integrated protein assignments assigned by MALDI-TOF and MS/MS (from step 1). If two different protein identifications were assigned, preference was given to MS/MS-based protein assignment.
Assignments annotated as unknown and without specific homology/similarity descriptions in identifier tag were BLASTP searched against the NCBI nonredundant database (as of March, 2005) to further query their homology. This study uses terminology as follows: homology for results, where E-value of BLAST search was 0.0; in all other cases similarity is used.
Subcellular localizations of assigned proteins were predicted using three independent programs: TargetP (http://www.cbs.dtu.dk/services/TargetP/; Emanuelsson et al., 2000), iPSORT (http://hc.ims.u-tokyo.ac.jp/iPSORT/; Bannai et al., 2002), and Predotar v. 1.03 (http://genoplante-info.infobiogen.fr/predotar/predotar.html; Small et al., 2004). Information about subcellular localization was incorporated into protein description if at least two programs predicted the same subcellular destination.
All data from this investigation are available from the oilseed proteomics server (http://oilseedproteomics.missouri.edu). Programming for the web database was performed, as described previously (Hajduch et al., 2005). Data are viewable through 2-DE gels and a protein identification table. The spots on 2-DE gel and protein numbers in the protein table are hyperlinked to display expression profile and protein identification data. Expression profiles of all proteins except SSP were generated using relative abundance data with in silico SSP subtraction (Supplemental Table II). However, expression profiles of individual SSPs may represent valuable data due to the number of different isoelectric species identified in this study. For this purpose, expression profiles of 71 identified SSPs were generated using the original dataset (Supplemental Table I). Thus, online database represents compromise, where two independent datasets of expression profiles can be viewed together.
Supplementary Material
This work was supported by the National Science Foundation-Plant Genome Research Program Young Investigator Award (grant no. DBI–0332418).
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Jay J. Thelen (thelenj@missouri.edu).
The online version of this article contains Web-only data.
Article, publication date, and citation information can be found at www.plantphysiol.org/cgi/doi/10.1104/pp.105.075390.
References
- Aebersold R, Mann M (2003) Mass spectrometry-based proteomics. Nature 422: 198–207 [DOI] [PubMed] [Google Scholar]
- Agrawal GK, Yonekura M, Iwahashi Y, Iwahashi H, Rakwal R (2005) System, trends and perspectives of proteomics in dicot plants Part I: technologies in proteome establishment. J Chromatogr B 815: 109–123 [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815 [DOI] [PubMed] [Google Scholar]
- Bassham JA, Benson AA, Kay LD, Harris AZ, Wilson AT, Calvin M (1954) The path of carbon in photosynthesis XXI: the cyclic regeneration of carbon dioxide acceptor. J Am Chem Soc 76: 1760–1770 [Google Scholar]
- Bevan M, Bancroft I, Bent E, Love K, Goodman H, Dean C, Bergkamp R, Dirkse W, Van Staveren M, Stiekema W, et al (1998) Analysis of 19 Mb of contiguous sequence from chromosome 4 of Arabidopsis thaliana. Nature 391: 485–488 [DOI] [PubMed] [Google Scholar]
- Bewley JD, Black M (1994) Seeds: Physiology of Development and Germination. Plenum Press, New York
- Campostrini N, Areces LB, Rappsilber J, Pietrogrande MC, Dondi F, Patorino F, Ponzoni M, Righetti PG (2005) Spot overlapping in two-dimensional maps: a serious problem ignored for much too long. Proteomics 5: 2385–2395 [DOI] [PubMed] [Google Scholar]
- Caspar T, Huber SC, Sommerville C (1985) Alterations in growth photosynthesis and respiration in a starchless mutant of Arabidopsis thaliana (L) deficient in chloroplast phosphoglucomutase activity. Plant Physiol 79: 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia TY, Pike MJ, Rawsthorne S (2005) Storage oil breakdown during embryo development of Brassica napus (L). J Exp Bot 56: 1285–1296 [DOI] [PubMed] [Google Scholar]
- Clauser KR, Baker PR, Burlingame AL (1999) Role of accurate mass measurement (+/− 10 ppm) in protein identification strategies employing MS or MS/MS and database searching. Anal Chem 71: 2871–2882 [DOI] [PubMed] [Google Scholar]
- Eastmond PJ, Rawsthorne S (2000) Coordinate changes in carbon partitioning and plastidial metabolism during the development of oilseed rape embryo. Plant Physiol 122: 767–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elborough KM, Winz R, Deka RK, Markham JE, White AJ, Rawsthorne S, Slabas AR (1996) Biotin carboxyl carrier protein and carboxyltransferase subunits of the multi-subunit form of acetyl-CoA carboxylase from Brassica napus: cloning and analysis of expression during oilseed rape embryogenesis. Biochem J 315: 103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300: 1005–1016 [DOI] [PubMed] [Google Scholar]
- Eng J, McCormack AL, Yates JR III (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5: 976–989 [DOI] [PubMed] [Google Scholar]
- Fernie AR, Carrari F, Sweetlove LJ (2004) Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol 7: 254–261 [DOI] [PubMed] [Google Scholar]
- Gallardo K, Job C, Groot SP, Puype M, Demol H, Vandekerckhove J, Job D (2001) Proteomic analysis of Arabidopsis seed germination and priming. Plant Physiol 126: 835–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo K, Job C, Groot SP, Puype M, Demol H, Vandekerckhove J, Job D (2002. a) Importance of methionine biosynthesis for Arabidopsis seed germination and seedling growth. Physiol Plant 116: 238–247 [DOI] [PubMed] [Google Scholar]
- Gallardo K, Job C, Groot SP, Puype M, Demol H, Vandekerckhove J, Job D (2002. b) Proteomics of Arabidopsis seed germination: a comparative study of wild-type and gibberellin-deficient seeds. Plant Physiol 129: 823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo K, Le Signor C, Vandekerckhove J, Thompson RD, Burstin J (2003) Proteomics of Medicago truncatula seed development establishes the time frame of diverse metabolic processes related to reserve accumulation. Plant Physiol 133: 664–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibeaut DM (2000) Nucleotide sugars and glucosyltransferases for synthesis of cell wall matrix polysaccharides. Plant Physiol Biochem 38: 69–80 [Google Scholar]
- Girke T, Todd J, Ruuska S, White J, Benning C, Ohlrogge J (2000) Microarray analysis of developing Arabidopsis seeds. Plant Physiol 124: 1570–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff SA, Ricke D, Lan T-H, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, et al (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296: 92–100 [DOI] [PubMed] [Google Scholar]
- Goffman FD, Alonso AP, Schwender J, Shachar-Hill Y, Ohlrogge JB (2005) Light enables a very high efficiency of carbon storage in developing embryos of rapeseed. Plant Physiol 138: 2269–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffman FD, Ruckle M, Ohlrogge J, Shachar-Hill Y (2004) Carbon dioxide concentrations are very high in developing oilseeds. Plant Physiol Biochem 42: 703–708 [DOI] [PubMed] [Google Scholar]
- Gorg A, Weiss W, Dunn MJ (2003) Current two-dimensional electrophoresis technology for proteomics. Proteomics 4: 3665–3685 [DOI] [PubMed] [Google Scholar]
- Gunstone DF, Harwood JL, Padley FB (1995). The Lipid Handbook. Chapman & Hall, London
- Hajduch M, Ganapathy A, Stein JW, Thelen JJ (2005) A systematic proteomic study of seed filling in soybean: establishment of high-resolution two-dimensional reference maps, expression profiles, and an interactive proteome database. Plant Physiol 137: 1397–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison CJ, Mould R, Leech MJ, Johnson S, Turner L, Schreck SL, Baird K, Jack P, Rawsthorne S, Hedley CL, et al (2000) The rug3 locus of pea encodes plastidial phosphoglucomutase. Plant Physiol 122: 1187–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman EM, Helm RM, Jung R, Kinney AJ (2003) Genetic modification removes an immunodominant allergen from soybean. Plant Physiol 132: 36–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill LM, Morley-Smith ER, Rawsthorne S (2003) Metabolism of sugars in the endosperm of developing seeds of oilseed rape. Plant Physiol 131: 228–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber LA (2003) Is proteomics heading in the wrong direction? Nat Rev Mol Cell Biol 4: 74–80 [DOI] [PubMed] [Google Scholar]
- Huber SC (1986) Fructose-26-bisphosphate as a regulatory metabolite in plants. Annu Rev Plant Physiol Plant Mol Biol 37: 233–246 [Google Scholar]
- Hutchings D, Rawsthorne S, Emes MJ (2005) Fatty acid synthesis and the oxidative pentose phosphate pathway in developing embryos of oilseed rape (Brassica napus L.). J Exp Bot 56: 577–585 [DOI] [PubMed] [Google Scholar]
- King SP, Lunn JE, Furbank RT (1997) Carbohydrate content and enzyme metabolism in developing canola siliques. Plant Physiol 114: 153–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleczkowski LA, Geisler M, Ciereszko I, Johansson H (2004) UDP-glucose pyrophosphorylase: an old protein with new tricks. Plant Physiol 134: 912–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubis SE, Pike MJ, Everett CJ, Hill LM, Rawsthorne S (2004) The import of phosphoenolpyruvate by plastids from developing embryos of oilseed rape Brassica napus (L) and its potential as a substrate for fatty acid synthesis. J Exp Bot 55: 1455–1462 [DOI] [PubMed] [Google Scholar]
- Lim H, Eng J, Yates JR III, Tollaksen SL, Giometti CS, Holden JF, Adams MW, Reich CI, Olsen GJ, Hays LG (2003) Identification of 2D-gel proteins: a comparison of MALDI/TOF peptide mass mapping to mu LC-ESI tandem mass spectrometry. J Am Soc Mass Spectrom 14: 957–970 [DOI] [PubMed] [Google Scholar]
- Meyerhof O (1945) The origin of the reaction of Harden and Young in cell-free alcoholic fermentation. J Biol Chem 157: 105–120 [Google Scholar]
- Meyerhof O, Junowicz-Kocholaty R (1943) The equilibria of isomerase and aldolase, and the problem of the phosphorylation of glyceraldehydes phosphate. J Biol Chem 149: 71–92 [Google Scholar]
- Mooney BP, Krishnan HB, Thelen JJ (2004) High-throughput peptide mass fingerprinting of soybean seed proteins: automated workflow and utility of UniGene expressed sequence tag databases for protein identification. Phytochemistry 65: 1733–1744 [DOI] [PubMed] [Google Scholar]
- Murphy DJ, Cummins I, Kang AS (1989) Synthesis of the major oil-body membrane protein in developing rapeseed (Brassica napus) embryos: integration with storage-lipid and storage-protein synthesis and implications for the mechanism of oil-body formation. Biochem J 258: 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen TH, Rung JH, Villadsen D (2004) Fructose-2,6-bisphosphate: a traffic signal in plant metabolism. Trends Plant Sci 9: 556–563 [DOI] [PubMed] [Google Scholar]
- Norton G, Harris JF (1975) Composition changes in developing rape seed (Brassica napus L). Planta 123: 163–174 [DOI] [PubMed] [Google Scholar]
- Person MD, Lo HH, Towndrow KM, Jia Z, Monks TJ, Lau SS (2003) Comparative identification of prostanoid inducible proteins by LC-ESI-MS/MS and MALDI-TOF mass spectrometry. Chem Res Toxicol 16: 757–767 [DOI] [PubMed] [Google Scholar]
- Plaxton WC (1996) The organization and regulation of plant glycolysis. Annu Rev Plant Physiol Plant Mol Biol 47: 185–214 [DOI] [PubMed] [Google Scholar]
- Rawsthorne S (2002) Carbon flux and fatty acid synthesis in plants. Prog Lipid Res 41: 182–196 [DOI] [PubMed] [Google Scholar]
- Ruuska SA, Girke T, Benning C, Ohlrogge JB (2002) Contrapuntal networks of gene expression during Arabidopsis seed filling. Plant Cell 14: 1191–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruuska SA, Schwender J, Ohlrogge JB (2004) The capacity of green oilseeds to utilize photosynthesis to drive biosynthetic processes. Plant Physiol 136: 2700–2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiltz S, Gallardo K, Huart M, Negroni L, Sommerer N, Burstin J (2004) Proteome reference maps of vegetative tissues in pea: an investigation of nitrogen mobilization from leaves during seed filling. Plant Physiol 135: 2241–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwender J, Goffman F, Ohlrogge JB, Shachar-Hill Y (2004. a) Rubisco without the Calvin cycle improves the carbon efficiency of developing green seeds. Nature 432: 779–782 [DOI] [PubMed] [Google Scholar]
- Schwender J, Ohlrogge JB (2002) Probing in vivo metabolism by stable isotope labeling of storage lipids and proteins in developing Brassica napus embryos. Plant Physiol 130: 347–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwender J, Ohlrogge JB, Shachar-Hill Y (2003) A flux model of glycolysis and the oxidative pentosephosphate pathway in developing Brassica napus embryos. J Biol Chem 278: 29442–29453 [DOI] [PubMed] [Google Scholar]
- Schwender J, Ohlrogge J, Shachar-Hill Y (2004. b) Understanding flux in plant metabolic networks. Curr Opin Plant Biol 7: 309–317 [DOI] [PubMed] [Google Scholar]
- Seki M, Narusaka M, Kamiya A, Ishida J, Satou M, Sakurai T, Nakajima M, Enju A, Akiyama K, Oono Y, et al (2002) Functional annotation of a full-length Arabidopsis cDNA collection. Science 296: 141–145 [DOI] [PubMed] [Google Scholar]
- Shorrosh BS, Savage LJ, Soll J, Ohlrogge JB (1996) The pea chloroplast membrane-associated protein, IEP96, is a subunit of acetyl-CoA carboxylase. Plant J 10: 261–268 [DOI] [PubMed] [Google Scholar]
- Small I, Peeters N, Legeai F, Lurin C (2004) Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4: 1581–1590 [DOI] [PubMed] [Google Scholar]
- Stitt M (1990) Fructose-2-6-bisphosphate as a regulatory molecule in plants. Annu Rev Plant Physiol Plant Mol Biol 41: 153–185 [Google Scholar]
- Thelen JJ, Mekhedov S, Ohlrogge JB (2001) Brassicaceae express multiple isoforms of biotin carboxyl carrier protein in a tissue-specific manner. Plant Physiol 125: 2016–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson BS, Asirvatham VS, Wang L, Sumner LW (2003) Mapping the proteome of barrel medic (Medicago truncatula). Plant Physiol 131: 1104–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber AP (2004) Solute transporters as connecting elements between cytosol and plastid stroma. Curr Opin Plant Biol 7: 247–253 [DOI] [PubMed] [Google Scholar]
- Weber AP, Schwacke R, Flugge UI (2005) Solute transporters of the plastid envelope membrane. Annu Rev Plant Biol 56: 133–164 [DOI] [PubMed] [Google Scholar]
- Yates JR III, Eng JK, McCormack AL, Schieltz D (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem 67: 1426–1436 [DOI] [PubMed] [Google Scholar]
- Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, et al (2002) A draft sequence of the rice genome (Oryza sativa L ssp indica). Science 296: 79–92 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.