

Abstract
A serine (Ser-700) amino acid rather than an asparagine (Asn-700) at residue 700 of thrombospondin-1 has been linked to an increased risk for development of premature, familial heart attacks. We now have identified both functional and structural differences between the Ser-700 and Asn-700 thrombospondin-1 variants. The Ser-700 variant increased the rate and extent of platelet aggregation and showed increased surface expression on platelets compared with the Asn-700 variant. These differences could be ascribed to an enhanced interaction of the Ser-700 variant with fibrinogen on the platelet surface and are consistent with a prothrombotic phenotype in Ser-700 individuals. The Ser-700 variant thrombospondin-1 was conformationally more labile than the Asn-700 variant as demonstrated by increased susceptibility to proteolytic digestion and enhanced susceptibility to unfolding by denaturants. These data suggest a potential molecular and cellular basis for a genetic risk factor associated with early onset myocar-dial infarction.
Genome-wide scan and high throughput single nucleotide polymorphism (SNP)1 association studies have begun to identify specific genes and polymorphisms that underlie complex diseases such as acute myocardial infarction (MI) (1), asthma (2-4), and inflammatory bowel diseases (5). Through one such high throughput analysis, which entailed the evaluation of 72 SNPs in 62 vascular biology genes in 398 sib-pairs, we identified three members of the thrombospondin gene (THBS) family encoding for thrombospondin protein (TSP)-1, -2, and -4, which are associated with familial, premature MI (6). This very aggressive form of heart attack occurs before age 40 in men and 45 in women. The TSP-1 SNP is a rare N700S recessive trait (1% frequency) but has the highest association with disease (odds ratio for MI = 8.66). This rarity leads to a low likelihood of successful replication in small studies (7, 8), but in one subsequent preliminary report (9) this disease association of the Ser-700 variant was corroborated.
TSP-1 is synthesized by a variety of cells and is a component of the extracellular matrix (10). It is stored within secretory granules of platelets (11, 12) and is the major protein released from these cells when they are stimulated with agonists (11-13). Although a variety of functions have been ascribed to TSP-1 and its domains (14, 15), the region harboring the N700S SNP is devoid of such known functional assignments with respect to platelet and other vascular cell responses (14, 16). Moreover, TSP-1 has not been previously regarded as a significant contributor to thrombotic diseases. Although TSP-1 has been identified as a constituent of human atherosclerotic plaques (17), mice in which the TSP-1 gene has been inactivated do not exhibit an overt hemostatic phenotype (18). The goal of the current study was to determine how the structure and function of N700S SNP in TSP-1 are altered and, thereby, to identify potential mechanisms by which this substitution predisposes individuals to familial, premature MI.
EXPERIMENTAL PROCEDURES
Protein Purification and Radioiodination
Asn-700 and Ser-700 variants of TSP-1 were purified from the platelets of individual donors homozygous for each variant, and samples from at least three different donors of each genotype were used. The TSP-1 from these donors was isolated according to methods described previously (13) but using 0.5–1 unit of blood as starting material. Platelets were washed by centrifugation in the presence of prostaglandin E1 and activated with thrombin to induce secretion. TSP-1 in the releasate was bound onto a heparinSepharose column (Amersham Biosciences) and eluted with an NaCl gradient (see Fig. 3). The TSP-1 variants were radioiodinated with 125I using a modified chloramine-T procedure (19). The purity of the TSP-1 preparations was assessed by SDS-PAGE (see under “Results”). In addition, the presence of active transforming growth factor-β1, which is bound and activated by TSP-1 (20, 21), was assessed as a likely contaminant with biological activity using a commercially available enzyme-linked immunosorbent assay (Promega). The levels of the growth factor were: Asn-700 rTSP-1, 1 pg/μg protein; Ser-700 rTSP-1, <0.5 pg/μg; Asn-700 TSP-1, <0.5 pg/μg; and Ser-700 TSP-1, 3 pg/μg.
Fig. 3.
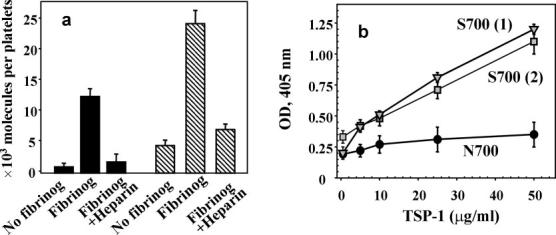
Effect of fibrinogen on TSP-1 binding to platelets. a, binding of isolated Asn-700 (black bars) and Ser-700 (striped bars) TSP-1 to platelets. Isolated platelets were stimulated with 0.25 unit/ml thrombin, with or without 300 nm fibrinogen, and fixed in 1% paraformaldehyde. After a 30-min incubation in the presence of 20 μg/ml 125I-Ser-700 TSP-1 or 125I-Asn-700 TSP-1 at room temperature, unbound ligand was separated by centrifugation through sucrose. The platelet-bound radioactivity was measured using a γ-counter. Data are means ± S.D. of four separate experiments using platelets from two Ser-700 patients and three Asn-700 donors. b, binding of Ser-700 (two donors) or Asn-700 TSP-1 to fibrinogen. Fibrinogen (20 μg/ml) was immobilized onto microtiter plate wells (2HB Immulon) overnight at 4 °C. 0.5% polyvinyl alcohol was used as a post-coat (1 h at 22 °C). The TSP-1 preparations were allowed to bind at the indicated concentrations for 1 h at 22 °C, and TSP-1 mAb 6G6 and secondary antibodies labeled with alkaline phosphatase were used for quantitation. Results were quantified at 405 nm. Data are means ± S.D. of four separate experiments.
Baculovirus encoding Asn-700 TSP-1, kindly provided by Dr. Mosher (University of Wisconsin, Madison, WI), was used to produce recombinant (r)TSP-1 in Sf9 insect cells. The rSer-700 variant was made in our laboratory in collaboration with Virus Core of Cleveland Clinic Foundation. The Sf9 cells expressing rTSP-1 were grown in the serum-free medium to minimize contamination with serum growth factors. Protein concentration was measured at 280 nm using an extinction coefficient of 13 for a 1% solution. Fibrinogen was purified as described previously (22). TSPI-1 and 6G6, monoclonal antibodies to TSP-1, were produced as described previously (23).
Platelet Studies
Platelet aggregation in platelet-rich plasma (PRP), in the presence or absence of either recombinant or naturally occurring TSP-1 variants, was induced by 20 mm ADP. TSP-1 (10 μg/ml) was added to the PRP and preincubated for 10 min prior to addition of the agonist. Using an aggregometer (Bio/Data Corp., Horsham, PA), aggregation was monitored as the value of the decrease of light transmission, setting the minimum and maximum responses with PRP and platelet-poor plasma, respectively. The approximate diameters of aggregates were estimated with a Leica microscope using a scale bar. Washed platelets were obtained by differential centrifugation followed by gel filtration. Fixed platelets were prepared (24) by stimulating washed platelets with a solution of 10 μm ADP plus 20 μm epinephrine and then fixing them with 1% paraformaldehyde. This protocol fixes integrin αIIbβ3 in an activated state, thereby limiting the effects of TSP-1 added to the aggregation rather than the activation step of the platelet-platelet interaction (25, 26). Aggregation of these cells was induced by the addition of 20 μg/ml fibrinogen in the presence or absence of the TSP-1 variants.
To measure platelet surface expression of endogenous TSP-1, washed platelets without stimulation or after 5 min of thrombin stimulation were reacted with FITC-labeled TSPI-1 mAb or with a labeled mAb to P-selectin as a control. Surface expression was analyzed by flow cytometry using the FacsCalibur instrument (BD Biosciences) with CellQuest software to quantify mean fluorescence intensities.
For binding of radioiodinated TSP-1 to platelets, isolated platelets were stimulated with 0.25 unit/ml thrombin and fixed in 1% paraformaldehyde. After washing, 20 μg/ml 125I-TSP-1 variants were added, with or without fibrinogen (300 μm) or heparin (1 mm, sodium salt, from porcine intestinal mucosa). After 30 min at room temperature, unbound ligand was separated by centrifugation through 20% sucrose, and platelet-bound radioactivity was measured in a γ-counter.
Solid Phase Binding Assays
Fibrinogen (2 μg/well in 50 mm NaHCO3, 150 mm NaCl, pH 8.4) was immobilized onto 96-well microtiter plates (Immulon 2HB, ThermoLabsystems, Franklin, MA) overnight at 4 °C. The plates were washed, post-coated with 0.5% polyvinyl alcohol for 1 h at 22 °C, and then Asn-700 or Ser-700 TSP-1 were incubated in a solution of 20 mm Tris·HCl, 150 mm NaCl, 2 mm MgCl2, and 1 mm CaCl2, to which 0.05% Tween 20 was added. After incubation for 2 h at 22 °C, wells were washed and incubated with 6G6 (0.5 μg/ml) mAb to TSP-1 for 1 h at 22 °C. A secondary goat anti-mouse antibody labeled with alkaline phosphatase (Pierce) and p-nitrophenyl phosphate substrate (Sigma) was used to assess the amount of bound TSP-1.
Trypsin Digestion of TSP-1
Digestion was performed at an enzyme: substrate molar ratio of 1:20 at 37 °C. At selected time points, samples were removed, treated with protease inhibitors, and subjected to SDS-PAGE followed by transfer and Western blotting with mAb 6G6. Intensities of bands were quantified using the ScanImage program.
Biophysical Measurements
Circular dichroism measurements were carried out in a JASCO-700 Spectropolarimeter (Tokyo, Japan), using a protein concentration of 0.25 mg/ml in a 1-mm path-length cell. Fluorescent spectra were obtained using a protein concentration of 10 μg/ml and an excitation wavelength of 280 nm in a PerkinElmer LS-50B luminescence spectrometer.
RESULTS
In view of the prominence of TSP-1 in platelets, the dominant role of these cells in thrombus formation leading to MI (27), and recent studies implicating platelets in the development of atherosclerotic lesions (28, 29), we focused on the influence of TSP-1 on platelet aggregation. Low dose (5 μm) ADP was used as both a model and a physiologically relevant agonist to stimulate a submaximal aggregation response (∼35% of maximum) of platelets in plasma. A low concentration (10 μg/ml) of Asn-700 rTSP-1 (>95% pure, based on SDS-PAGE, as shown in Fig. 4b) enhanced the aggregation response to 80% of maximum (Fig. 1a). Addition of the same amount (in moles) of fibrinogen or bovine serum albumin failed to enhance the response. Observed microscopically, TSP-1 increased the size of the platelet aggregates by 3–4-fold. This effect was evident when washed platelets were stimulated (20 μm ADP), treated with paraformaldehyde to fix the fibrinogen receptor, αIIbβ3, in an activated state (25), and naturally occurring TSP-1 was added. Aggregation was more extensive when induced by fibrinogen with TSP-1 than when TSP-1 was omitted (Fig. 1b). The use of fixed platelets excludes an effect of TSP-1 or possible contaminants on platelet activation and indicates that TSP-1 exerts an influence on the post-stimulation stage of platelet-platelet cohesion. With conditions established under which TSP-1 exerted a notable influence on platelet aggregation, we analyzed the effect of isolated Asn-700 and Ser-700 TSP-1 from the platelets of individual donors. Under the conditions used, maximum aggregation of stimulated, fixed platelets responding to addition of fibrinogen was 23.4 ± 1.3% with Asn-700 TSP-1 (Fig. 1c) compared with 34.6 ± 3.9% and 32.8 ± 2.8% with Ser-700 TSP-1 from the two different donors (p = 0.018 and 0.03, respectively, compared with the Asn-700 variant). Of note, platelet aggregation in the presence of Ser-700 TSP-1 began immediately on stirring, whereas a 40–60-s lag was observed with the Asn-700 TSP-1 (Fig. 1d). These data indicate that Ser-700 TSP-1 enhances platelet aggregation, consistent with the prothrombotic phenotype of these individuals.
Fig. 4.
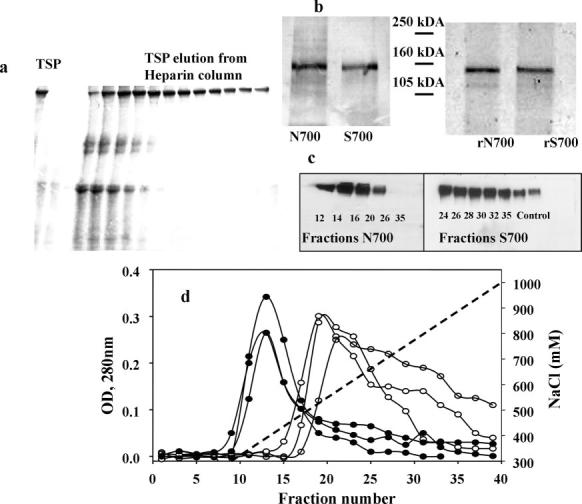
TSP-1 purification. a, 4–15% gradient SDS-PAGE of Asn-700 TSP-1 fractions were eluted using a heparin-Sepharose CL-6B column. b, SDS-PAGE (7%) analysis of TSP-1 variants under reducing conditions (left panel) represents Asn-700 and Ser-700 TSP-1 variants isolated from platelets and recombinant Asn-700 and Ser-700 TSPs (right panel). The gel was developed by silver staining. c, Western blot analysis of Asn-700 and Ser-700 TSP-1 fractions eluted from heparin-Sepharose CL-6B columns using TSPI-1 monoclonal antibodies. d, elution profiles of three Asn-700 (filled circles) and three Ser-700 (open circles) TSP-1 isolated from heparin-Sepharose CL-6B columns equilibrated with TBS (20 mm Tris·HCl; 150 mm NaCl), 2 mm MgCl2, 1 mm CaCl2, using an NaCl gradient (dotted line).
Fig. 1.
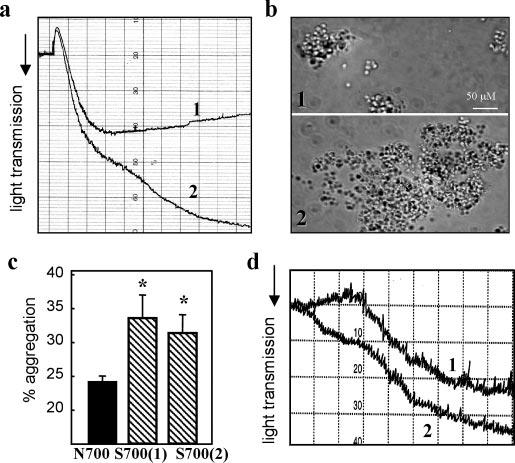
TSP-1 variants and platelet aggregation. a, platelet aggregation in PRP in the absence (curve 1) or presence (curve 2) of rTSP-1 (Asn-700) was induced by 20 μm ADP. PRP was preincubated with 10 μg/ml rTSP-1 for 10 min. Aggregation was monitored by the decrease of light transmission, with the minimum and maximum set with platelet-poor and platelet-rich plasma, respectively. b, micrographs show aggregates of ADP-stimulated platelets fixed with 0.5% paraformaldehyde, washed, and then aggregated by addition of fibrinogen (300 nm) in the absence (panel 1) or presence (panel 2) of purified rTSP-1 at 100 μm. c, aggregation of fixed platelets stimulated by 10 μm ADP/20 μm epinephrine plus 20 μg/ml fibrinogen in the absence or presence of 10 μg/ml TSP-1 isolated from one Asn-700 donor (black bar) or from two different Ser-700 donors (striped bars). The maximum extent of aggregation, recorded at 5 min after addition of ADP/epinephrine, is presented as the means ± S.D. of five separate experiments using platelets from five Asn-700 donors. d, representative aggregation curves of platelets prepared as in c in the presence of Ser-700 (curve 2) and Asn-700 (curve 1) TSP-1 (10 μg/ml).
When it is secreted from platelet α granules, a significant portion of the TSP-1 becomes associated with the surface of stimulated platelets (30, 31). We assessed basal and thrombin-stimulated TSP-1 surface expression by FACS, using a TSPI-1 mAb that was verified to react equally with Asn-700 and Ser-700 TSP-1 and platelets from three Ser-700 and three Asn-700 donors. Little Asn-700 TSP-1 was detected on the surface of resting platelets, but stimulation of the cells with thrombin, a strong agonist, resulted in a substantial increase in surface-bound TSP-1. Under identical conditions, the signal with Ser-700 TSP-1 platelets was 2.4-fold higher (Fig. 2a), and even basal Ser-700 TSP-1 expression was higher. These differences were observed despite the Ser-700 donor patients' daily dosage of aspirin, administered to blunt platelet reactivity. The observed differences in TSP-1 surface expression were not the results of different amounts of the TSP-1 variants released from or within the platelets as determined by enzyme-linked immunosorbent assay (data not shown). Furthermore, exposure of P-selectin, another α granule release marker (32), was not elevated in the Ser-700 individuals, with or without platelet stimulation (Fig. 2b). In fact, P-selectin expression was slightly lower with the Ser-700 versus the Asn-700 platelets and was similar to that obtained when an Asn-700 individual ingested aspirin for 3 consecutive days prior to the analysis (Fig. 2b).
Fig. 2.
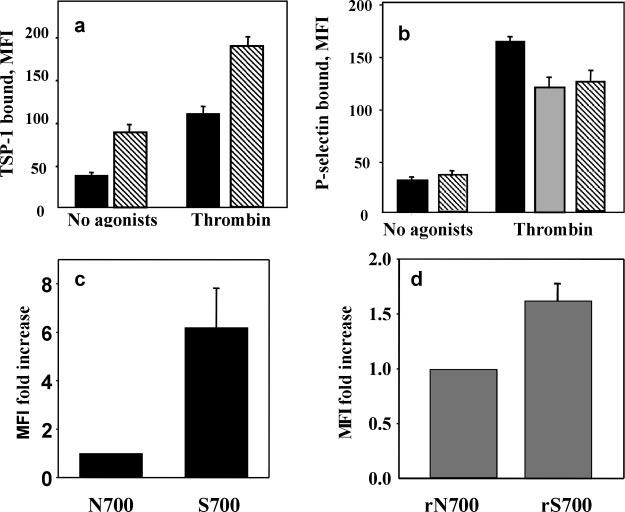
Expression of TSP-1 variants on the platelet surface. Endogenous TSP-1 (a) and P-selectin (b) expression on the surface of platelets from donors with Asn-700 (black bars) or Ser-700 (striped bars) TSP-1. Isolated platelets were stimulated with thrombin or remained unstimulated, and the amount of bound TSP-1 was detected by FACS using FITC-labeled anti-TSP mAb TSPI-1. In parallel, an expression of P-selectin was detected with mAb to P-selectin. Data are means ± S.D. of five separate experiments using platelets from three N700S and three Ser-700 donors. Because patients with the Ser-700 substitution were being treated with aspirin, platelets of the Asn-700 donor, who had taken aspirin for 3 days prior to the study, were also analyzed (b, gray bar). c and d, platelets from healthy donors were stimulated with 10 mm ADP/20 mm epinephrine in the presence of 300 nm fibrinogen and fixed with 1% paraformaldehyde. Binding of TSP-1 variants isolated from platelets (c) and recombinant TSP-1 (d) was assessed by flow cytometry using FITC-labeled rabbit polyclonal antibodies to TSP-1. Data are means ± S.D. of four separate experiments. MFI, mean fluorescent intensity.
To directly assess whether the differences in surface expression of the variants arise from differential interaction between the TSP-1s and platelets, we measured the binding of naturally occurring and recombinant TSP-1 variants to platelets isolated from healthy donors by FACS analysis. TSP-1 variants were added to platelets stimulated with 10 μm ADP and fixed with paraformaldehyde to limit the measured reaction to the direct binding of the TSP-1 variants and to avoid potential platelet stimulation by the TSP-1 preparations (19). After a 40-min incubation of the ADP-fixed platelets with the TSP-1 variants, the amount of bound TSP-1 was assessed with anti-TSP-1 labeled with FITC. Under identical conditions, the Ser-700 TSP-1 variant exhibited significantly increased binding to platelets compared with the Asn-700 variant (Fig. 2c). Significant differences (p = 0.05) were observed also in the interaction of the recombinant TSP-1 variants with platelets, although the extent of the difference was less dramatic than with the naturally occurring proteins (Fig. 2d).
As an independent approach to examining the interaction of the TSP-1 variants with platelets, we iodinated the purified, naturally occurring variants of TSP-1 and measured their direct binding to platelets. Under the conditions used, minimal interaction was observed unless fibrinogen was present (Fig. 3a). This finding supports previous data showing that fibrinogen bound to integrin αIIbβ3 provides a primary pathway for TSP-1 binding to platelets (33), a reflection of the high affinity of TSP-1 for fibrinogen. In the presence of fibrinogen (300 nm, ∼Kd for αIIbβ3), the amount of Asn-700 TSP-1 bound to platelets increased by 8-fold, whereas the amount of bound Ser-700 TSP-1 increased by 16-fold. The interaction between TSP-1 and fibrinogen-occupied platelets was almost completely inhibited by heparin (Fig. 3a), which binds with high affinity to the N-terminal region of TSP-1 (34).
Our data indicate that Asn-700 and Ser-700 TSP-1 interact differently with platelets, which, in turn, may arise from a difference in the binding of the TSP-1 variants to fibrinogen. Fibrinogen was immobilized onto microtiter plates, varying amounts of Asn-700 or Ser-700 TSP-1 were added, and their binding was quantified with a 6G6 mAb against TSP-1. The binding of the two TSP-1 variants to the immobilized fibrinogen differed greatly (Fig. 3b); at the highest concentration tested (50 μg/ml), ∼5-fold more Ser-700 than Asn-700 TSP-1 was bound (Fig. 3b). It has been proposed that TSP-1 forms additional bridges between aggregating platelets and, thereby, increases the extent of platelet aggregation (33). Thus, our data showing that Ser-700 TSP-1 has increased binding to fibrinogen provides a potential explanation as to why this variant supports enhanced platelet aggregation and increases thrombus burden.
In our experiments we used TSP-1 purified from platelets as well as the recombinant TSP-1 variants. The quality of TSP-1 preparations was assessed by SDS-PAGE immediately after purification, under nonreducing (Fig. 4a) and reducing (Fig. 4b) conditions with subsequent Western blotting (Fig. 4c). Only fractions with minimum contamination (18-36) were used in experiments. Because it still was possible that TSP-1 preparations contained platelet activators that are effective at the relatively low concentrations, we performed the majority of experiments using platelets fixed with paraformaldehyde.
The isolation procedure we used to purify TSP-1 takes advantage of a binding affinity to heparin (35, 36), but Asn-700 and Ser-700 TSP-1 behaved differently when eluted from heparin-Sepharose columns (Fig. 4d). The Ser-700 TSP-1 required substantially higher NaCl concentrations to elute from the heparin columns than Asn-700 TSP-1. For the Ser-700 TSP-1 from three different individuals, the concentration of NaCl that eluted 50% of the bound TSP-1 was 650 mm compared with 470 mm for Asn-700 TSP-1. Nevertheless, the products that were isolated were similar as shown by Western blotting (Fig. 4c). Differences in heparin binding may reflect overall changes in the organization of the variants, which, in turn, might alter protease susceptibility, and we explored this possibility. As evident from Fig. 5a, cleavage fragments of Asn-700 TSP-1, detected by blotting with a 6G6 mAb against TSP-1, were evident only after 60 min of digestion with trypsin. In contrast, substantial degradation of the Ser-700 TSP-1 was observed at 15–30 min. The intensities of the immunoblotting bands below 180 kDa were quantified by densitometry. The amounts of product at 60 and 120 min of trypsin digestion were 10- and 4-fold higher with Ser-700 than Asn-700 TSP-1, respectively (Fig. 5b). The intensities of these bands with the Ser-700 variants isolated from two different donors were similar.
Fig. 5.
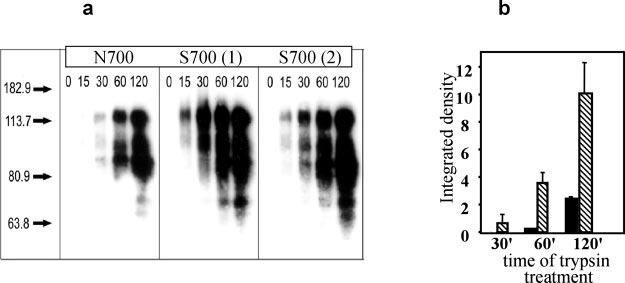
Trypsin digestion of TSP-1. Trypsin digestion of TSP-1 was performed at an enzyme:substrate molar ratio of 1:20 at 37 °C. At selected time points, samples were removed, treated with protease inhibitors, and subjected to SDS-PAGE followed by transfer and Western blot. a, Western blots of sequential trypsin digests of one Asn-700 and two Ser-700 TSP-1 preparations probed with 6G6 mAb. b, intensity of the low molecular mass (70-kDa) band was quantified for Asn-700 TSP-1 (black bars) and Ser-700 TSP-1 (striped bars) using the ScanImage program. Data are means ± S.D. of three separate experiments.
Based on the differences in heparin affinity and trypsin sensitivity, we implemented approaches to compare the conformations of the two TSP-1 molecules. The intrinsic tryptophan fluorescence spectra of the TSP-1 proteins (Fig. 6a) were identical with the broad maximum at 345 nm, indicative of a large number of tryptophan residues (22 are present in TSP-1) in environments differing slightly in exposure to solvent (37); however, as the two purified TSP-1s were unfolded with denaturants guanidine HCl (GdnHCl) or urea, differences in their intrinsic fluorescence at the 345 -nm maximum became evident (Fig. 6, b and c). Asn-700 TSP-1 retained its fluorescence signal at low concentrations of the denaturants (up to 1 m GdnHCl and 3 m urea), whereas the Ser-700 TSP-1 began to lose fluorescent intensity at very low concentrations of the denaturants. These differences suggest that the conformation of Ser-700 TSP-1 is more labile. This interpretation is supported by CD measurements (Fig. 6d). In the absence of GdnHCl, the far UV CD spectra of the two TSP-1 molecules were complex, indicating multiple secondary structural elements (38), but were essentially the same (Fig. 6d). At a high concentration (3 m) of the denaturant, the spectra of the two proteins were altered, reflecting their unfolding, but also remained superimposable. The CD spectra of the Asn-700 form in 0.5 m GdnHCl or no denaturant were the same, but the low concentration of denaturant markedly altered the spectrum of Ser-700 TSP-1 with the changes indicating a loss of secondary structure (Fig. 6d).
Fig. 6.
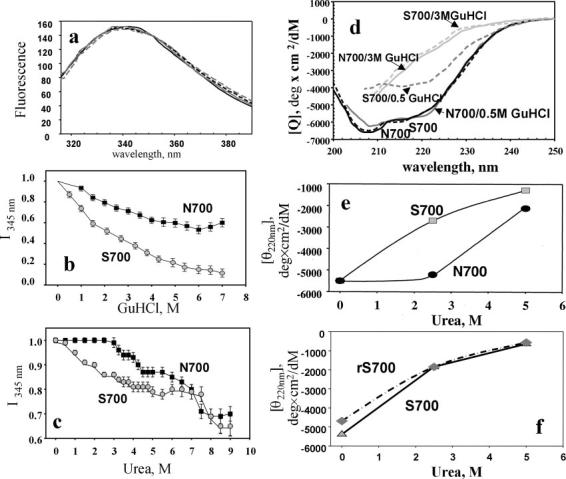
Structural characteristics of the TSP-1 variants. a, intrinsic tryptophan fluorescence spectra (excitation λ = 280 nm) of Asn-700 (solid line), Ser-700 (solid gray line), rAsn-700 (dashed line), and rSer-700 (gray dashed line) TSP-1 variants at 10 μg/ml in TBS, 2 mm MgCl2, 1 mm CaCl2. b and c, effect of GdnHCl (b) and urea (c) on the fluorescence intensity at 345 nm. Proteins were incubated with the denaturants for 5 min before recording the spectra. Data are normalized on the level of fluorescence intensity in the absence of denaturants. d, far UV CD spectra (at 23 °C) of wild-type (solid lines) and mutant (dashed lines) TSP-1 (0.25 mg/ml in TBS containing 2 mm MgCl2, 1 mm CaCl2, pH 7.2) at different concentrations of GdnHCl. e, changes of ellipticity (θ) at 220 nm in TSP variants purified from platelets. Asn-700 (black symbols); Ser-700 (gray symbols). f, changes in ellipticity at 220 nm of purified Ser-700 variants of TSP-1 at different concentrations of urea for Ser-700 from platelets (solid line) and rSer-700 (dashed line).
In the experiment shown in Fig. 6e, we quantified the changes in ellipticity (θ) at the 220-nm minimum, a reflection of secondary structure. These values for the two TSP-1 variants are similar in the absence or presence of 5 m urea. In the presence of 3 m urea, the value was unchanged for Asn-700 TSP-1 but was substantially altered for Ser-700 TSP-1. In a similar experiment, we were able to demonstrate that the behavior of Ser-700 TSP-1 purified from one of the donors behaved in a fashion that was indistinguishable from the rSer-700 TSP-1 (Fig. 6f).
DISCUSSION
One of the major challenges evolving from successful high throughput genome and SNP association studies exists at the proteomic level. How do specific gene products and their SNPs contribute to disease? In this study, we have identified both structural and functional differences between the Asn-700 and Ser-700 TSP-1 variants that may account for the increased risk of premature MI in individuals carrying the Ser-700 form of the molecule. The Ser-700 variant supports enhanced platelet aggregation compared with the Asn-700 form, and this difference could be attributed to an increase in apparent affinity of the Ser-700 variant for fibrinogen. Previous studies (33) suggest a role for TSP-1 in platelet aggregation (39, 40), which may depend on interaction with fibrinogen. The interaction of TSP-1 with fibrinogen is one of high affinity (41) and is mediated by the N-terminal region of the molecule (14), the domain that also contains the major heparin-binding site within TSP-1 (14). Our data show that the Ser-700 variant exhibits higher apparent affinities for platelets and fibrinogen. Our additional observations demonstrate that the interaction of TSP-1 with platelets is heparin-sensitive (Fig. 2c) and that this variant exhibits higher apparent affinity for heparin, i.e. the differential elution of the variants from heparin-Sepharose (Fig. 3a) are consistent with the enhanced binding of the Ser-700 variant to platelets and fibrinogen. Relative to the Asn-700 variant, the Ser-700 variant also showed increased platelet surface expression on thrombin stimulation. The increase in surface expression was selective for TSP-1 compared with P-selectin, which is consistent with the increase observed in the apparent affinity of the Ser-700 variant for platelets. Increased surface expression and enhanced platelet aggregation are both consistent with the prothrombotic phenotype predicted by the original genetic association study (6). This previous study also found that plasma TSP-1 levels are lower in individuals with the Ser-700 variant (6). Either enhanced binding to circulating platelets or increased susceptibility to proteolysis could lead to lower plasma levels of the Ser-700 variant.
Structurally, the Ser-700 variant was conformationally more labile than the Asn-700 variant as demonstrated by differences in trypsin susceptibility, tryptophan fluorescence, and CD spectral analysis. Recently, Hannah et al. (42) used the latter two techniques to demonstrate conformational differences in fragments containing the two SNPs in the presence or absence of Ca2+. We did not see Ca2+-dependent differences in the fluorescence or CD spectra of the intact variants (data not shown), but the observation that the same two reporting parameters that were altered in the fragments extended to or were accentuated in the intact molecules adds credence to the conformational differences between the TSP-1 molecules. These conformational differences affected the heparin binding functions of the TSP-1 variants, resident in residues 1–258, which is distant both in terms of primary sequence (43) as well as the conformation of the molecule based on electron microscopy (35). Thus, it appears the conformational differences induced by the substitutions at position 700 reverberate over long distances in the molecule.
The functional and structural differences between the TSP-1 forms were observed with both naturally occurring and recombinant variants. The differences between the SNP variants, however, tended to be more pronounced with the naturally occurring forms. For example, the differences in platelet surface expression of the naturally occurring SNP variants were almost 6-fold (range 3–9-fold with multiple preparations from the two patient samples used) but were only 1.6-fold with the recombinant molecules (1.3–1.9-fold with separate preparations). One explanation is that naturally occurring variants may have additional polymorphisms that affect conformation. Sequencing the THBS1 genes from these patients will be undertaken to address this possibility. We believe it more likely, however, that, even for the Asn-700 forms, the naturally occurring and recombinant molecules may not be precisely identical. This possibility is suggested based on the differential reactivity of certain mAbs with the TSP-1s isolated from the two sources in Western blots. Whereas two mAbs reacted equally (intensity of bands based on densitometric scanning) with naturally occurring and recombinant forms of TSP-1, one well characterized TSPI-1 mAb (23) reacted well with naturally occurring TSP-1 but poorly with the recombinant molecule. One possible explanation for this result is attributable to differences in glycosylation in mammalian versus insect cells, which might also affect secondary folding (44 - 46). Nevertheless, the differences in structure and function were observed with variant molecules produced from both sources, and it was the magnitude of the differences that varied.
Coronary artery disease and particularly acute MI remain the most significant causes of death and disability in Western society. Defying the general concept that common diseases are based on a common genetic polymorphism, the TSP-1 SNP was rare and recessive; nevertheless, the presence of this variant led to an increased risk of more than 8-fold for premature, familial MI in the initial GenQuest population (6). Replication of gene association studies is regarded as an important validation but has been rarely observed because of many variables, including heterogeneity of the disease under study and the population being analyzed (8, 47); however, a preliminary report (9) does appear to document an association of the TSP-1 SNP with premature MI. The data shown in the present study, i.e. an increase in the propensity of platelets to aggregate, a central step in arterial thrombosis, suggest a potential mechanism by which the Ser-700 substitution engenders prothrombotic activity to TSP-1. Our data, together with previous data of Frojmovic and co-workers (33), suggest that TSP-1 in general and the Ser-700 variant, in particular, enhance the aggregation and stability of platelet aggregates by virtue of the interaction with fibrinogen. As a next step in validating the proposed pathogenic mechanisms of the TSP-1 SNPs, the development of transgenic mice expressing the different forms could be considered. The progression of atherosclerotic lesions to thrombosis, however, is a rarity in the usual apoE or low density lipoprotein receptor-deficient mice (48) and may require very complex genetic crosses (49), of uncertain relevance to human pathology, to test the influence of the TSP-1 SNPs in MI development. Thus, multiple approaches, including transgenic mice, gene array, and proteomic analyses, as well as large scale genetic association studies, will be required to establish whether observed functional and structural differences between the TSP-1 variants are causally related to premature cardiovascular disease. Moreover, it must be kept in mind that the TSP-1 variant accounts for only a small fraction of this phenotypically aggressive form of heart attack. Nevertheless, insights that are relevant to even a fraction of this critically important disease should be a helpful step forward.
Footnotes
This work was supported in part by National Institutes of Health Grant HL071625 (to T. V. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: SNP, single nucleotide polymorphism; MI, myocardial infarction; r, recombinant; TSP, thrombospondin protein; PRP, platelet-rich plasma; FITC, fluorescein isothiocyanate; mAb, monoclonal antibody; FACS, fluorescence-activated cell sorter; GdnHCl, guanidine HCl.
REFERENCES
- 1.Weiss EJ, Bray PF, Tayback M, Schulman SP, Kickler TS, Becker LC, Weiss JL, Gerstenblith G, Goldschmidt-Clermont PJ. N. Engl. J. Med. 1996;334:1090–1094. doi: 10.1056/NEJM199604253341703. [DOI] [PubMed] [Google Scholar]
- 2.Takeoka S, Unoki M, Onouchi Y, Doi S, Fujiwara H, Miyatake A, Fujita K, Inoue I, Nakamura Y, Tamari M. J. Hum. Genet. 2001;46:57–63. doi: 10.1007/s100380170109. [DOI] [PubMed] [Google Scholar]
- 3.Hang LW, Hsia TC, Chen WC, Chen HY, Tsai FJ. J. Clin. Lab Anal. 2003;17:57–60. doi: 10.1002/jcla.10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Eerdewegh P, Little RD, Dupuis J, Del Mastro RG, Falls K, Simon J, Torrey D, Pandit S, McKenny J, Braunschweiger K, Walsh A, Liu Z, Hayward B, Folz C, Manning SP, Bawa A, Saracino L, Thackston M, Benchekroun Y, Capparell N, Wang M, Adair R, Feng Y, Dubois J, FitzGerald MG, Huang H, Gibson R, Allen KM, Pedan A, Danzig MR, Umland SP, Egan RW, Cuss FM, Rorke S, Clough JB, Holloway JW, Holgate ST, Keith TP. Nature. 2002;418:426–430. doi: 10.1038/nature00878. [DOI] [PubMed] [Google Scholar]
- 5.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 6.Topol EJ, McCarthy J, Gabriel S, Moliterno DJ, Rogers WJ, Newby LK, Freedman M, Metivier J, Cannata R, O'Donnell CJ, Kottke-Marchant K, Murugesan G, Plow EF, Stenina O, Daley GQ. Circulation. 2001;104:2641–2644. doi: 10.1161/hc4701.100910. [DOI] [PubMed] [Google Scholar]
- 7.Yamada Y, Izawa H, Ichihara S, Takatsu F, Ishihara H, Hirayama H, Sone T, Tanaka M, Yokota M. N. Engl. J. Med. 2004;347:1916–1923. doi: 10.1056/NEJMoa021445. [DOI] [PubMed] [Google Scholar]
- 8.Boekholdt SM, Trip MD, Peters RJ, Engelen M, Boer JM, Feskens EJ, Zwinderman AH, Kastelein JJ, Reitsma PH. Arterioscler. Thromb. Vasc. Biol. 2002;22:e24–e27. doi: 10.1161/01.atv.0000046235.22451.66. [DOI] [PubMed] [Google Scholar]
- 9.Peyvandi F, Palla R, Ardissino D, Foco L, Bernardinelli L, Bauer K, Mannucci PM. Blood. 2003;102:11. Abstr. 1129. [Google Scholar]
- 10.Adams JC. Int. J. Biochem. Cell Biol. 1997;29:861–865. doi: 10.1016/s1357-2725(96)00171-9. [DOI] [PubMed] [Google Scholar]
- 11.Hagen I. Biochim. Biophys. Acta. 1975;392:242–254. doi: 10.1016/0304-4165(75)90006-9. [DOI] [PubMed] [Google Scholar]
- 12.Lawler JW, Chao FC, Fang PH. Thromb. Haemostasis. 1977;37:355–357. [PubMed] [Google Scholar]
- 13.Lawler JW, Slayter HS, Coligan JE. J. Biol. Chem. 1978;253:8609–8616. [PubMed] [Google Scholar]
- 14.Adams JC. Annu. Rev. Cell Dev. Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- 15.Mosher DF. Annu. Rev. Med. 1990;41:85–97. doi: 10.1146/annurev.me.41.020190.000505. [DOI] [PubMed] [Google Scholar]
- 16.Lawler J. Curr. Opin. Cell Biol. 2000;12:634–640. doi: 10.1016/s0955-0674(00)00143-5. [DOI] [PubMed] [Google Scholar]
- 17.Riessen R, Kearney M, Lawler J, Isner JM. Am. Heart J. 1998;135:357–364. doi: 10.1016/s0002-8703(98)70105-x. [DOI] [PubMed] [Google Scholar]
- 18.Lawler J, Sunday M, Thibert V, Duquette M, George EL, Rayburn H, Hynes RO. J. Clin. Investig. 1998;101:982–992. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolff R, Plow EF, Ginsberg MH. J. Biol. Chem. 1986;261:6840–6846. [PubMed] [Google Scholar]
- 20.Schultz-Cherry S, Murphy-Ullrich JE. J. Cell Biol. 1993;122:923–932. doi: 10.1083/jcb.122.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schultz-Cherry S, Lawler J, Murphy-Ullrich JE. J. Biol. Chem. 1994;269:26783–26788. [PubMed] [Google Scholar]
- 22.Marguerie GA, Plow EF, Edgington TS. J. Biol. Chem. 1979;254:5357–5363. [PubMed] [Google Scholar]
- 23.Aiken ML, Ginsberg MH, Plow EF. Blood. 1987;69:58–64. [PubMed] [Google Scholar]
- 24.Plow EF, Marguerie G. Proc. Natl. Acad. Sci. U. S. A. 1982;79:3711–3715. doi: 10.1073/pnas.79.12.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du XP, Plow EF, Frelinger AL, III, O'Toole TE, Loftus JC, Ginsberg MH. Cell. 1991;65:409–416. doi: 10.1016/0092-8674(91)90458-b. [DOI] [PubMed] [Google Scholar]
- 26.Marguerie GA, Edgington TS, Plow EF. J. Biol. Chem. 1980;255:154–161. [PubMed] [Google Scholar]
- 27.Bhatt DL, Topol EJ. Nat. Rev. Drug Discov. 2003;2:15–28. doi: 10.1038/nrd985. [DOI] [PubMed] [Google Scholar]
- 28.Massberg S, Brand K, Gruner S, Page S, Muller E, Muller I, Bergmeier W, Richter T, Lorenz M, Konrad I, Nieswandt B, Gawaz M. J. Exp. Med. 2002;196:887–896. doi: 10.1084/jem.20012044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Nat. Med. 2003;9:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 30.Aiken ML, Ginsberg MH, Plow EF. Semin. Thromb. Hemostasis. 1987;13:307–316. doi: 10.1055/s-2007-1003506. [DOI] [PubMed] [Google Scholar]
- 31.Legrand C, Thibert V, Dubernard V, Begault B, Lawler J. Blood. 1992;79:1995–2003. [PubMed] [Google Scholar]
- 32.Stenberg PE, McEver RP, Shuman MA, Jacques YV, Bainton DF. J. Cell Biol. 1985;101:880–886. doi: 10.1083/jcb.101.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonnefoy A, Hantgan R, Legrand C, Frojmovic MM. J. Biol. Chem. 2001;276:5605–5612. doi: 10.1074/jbc.M010091200. [DOI] [PubMed] [Google Scholar]
- 34.Legrand C, Morandi V, Mendelovitz S, Shaked H, Hartman JR, Panet A. Arterioscler. Thromb. 1994;14:1784–1791. doi: 10.1161/01.atv.14.11.1784. [DOI] [PubMed] [Google Scholar]
- 35.Lawler J, Derick LH, Connolly JE, Chen JH, Chao FC. J. Biol. Chem. 1985;260:3762–3772. [PubMed] [Google Scholar]
- 36.Chen H, Herndon ME, Lawler J. Matrix Biol. 2000;19:597–614. doi: 10.1016/s0945-053x(00)00107-4. [DOI] [PubMed] [Google Scholar]
- 37.Stryer L. Science. 1968;162:526–533. doi: 10.1126/science.162.3853.526. [DOI] [PubMed] [Google Scholar]
- 38.Adler AJ, Greenfield NJ, Fasman GD. Methods Enzymol. 1973;27:675–735. doi: 10.1016/s0076-6879(73)27030-1. [DOI] [PubMed] [Google Scholar]
- 39.Bornstein P. J. Clin. Investig. 2001;107:929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dixit VM, Haverstick DM, O'Rourke KM, Hennessy SW, Grant GA, Santoro SA, Frazier WA. Proc. Natl. Acad. Sci. U. S. A. 1985;82:3472–3476. doi: 10.1073/pnas.82.10.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lahav J, Lawler J, Gimbrone MA. Eur. J. Biochem. 1984;145:151–156. doi: 10.1111/j.1432-1033.1984.tb08534.x. [DOI] [PubMed] [Google Scholar]
- 42.Hannah BL, Misenheimer TM, Annis DS, Mosher DF. J. Biol. Chem. 2003;278:8929–8934. doi: 10.1074/jbc.m211185200. [DOI] [PubMed] [Google Scholar]
- 43.Chen H, Sottile J, Strickland DK, Mosher DF. Biochem. J. 1996;318:959–963. doi: 10.1042/bj3180959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hofsteenge J, Huwiler KG, Macek B, Hess D, Lawler J, Mosher DF, Peter-Katalinic J. J. Biol. Chem. 2001;276:6485–6498. doi: 10.1074/jbc.M008073200. [DOI] [PubMed] [Google Scholar]
- 45.Gonzalez d. P., Klein D, Macek B, Hess D, Peter-Katalinic J, Hofsteenge J. Mol. Cell Proteomics. 2002;1:11–18. doi: 10.1074/mcp.m100011-mcp200. [DOI] [PubMed] [Google Scholar]
- 46.Castellino FJ, Davidson DJ, Rosen E, McLinden J. Methods Enzymol. 1993;223:168–185. doi: 10.1016/0076-6879(93)23044-n. [DOI] [PubMed] [Google Scholar]
- 47.Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. Genet. Med. 2002;4:45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Calara F, Silvestre M, Casanada F, Yuan N, Napoli C, Palinski W. J. Pathol. 2001;195:257–263. doi: 10.1002/path.915. [DOI] [PubMed] [Google Scholar]
- 49.Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, Rosenberg RD, Schrenzel M, Krieger M. Circ. Res. 2002;90:270–276. doi: 10.1161/hh0302.104462. [DOI] [PubMed] [Google Scholar]