

Abstract
The molecular mechanism leading to the cancer metastasis to bone is poorly understood but yet determines prognosis and therapy. Here, we define a new molecular pathway that may account for the extraordinarily high osteotropism of prostate cancer. By using SPARC (secreted protein, acidic and rich in cysteine)-deficient mice and recombinant SPARC, we demonstrated that SPARC selectively supports the migration of highly metastatic relative to less metastatic prostate cancer cell lines to bone. Increased migration to SPARC can be traced to the activation of integrins αvβ and αvβ5 on tumor cells. Such activation is induced by an autocrine vascular endothelial growth factor (VEGF)/VEGF receptor (VEGFR)-2 loop on the tumor cells, which also supports the growth and proliferation of prostate cancer cells. A consequence of SPARC recognition by αvβ5 is enhanced VEGF production. Thus, prostate cancer cells expressing VEGF/VEGFR-2 will activate αvβ5 and αvβ5 on their surface and use these integrins to migrate toward SPARC in bone. Within the bone environment, SPARC engagement of these integrins will stimulate growth of the tumor and further production of VEGF to support neoangiogenesis, thereby favoring the development of the metastatic tumor. Supporting this model, activated integrins were found to colocalize with VEGFR-2 in tissue samples of metastatic prostate tumors from patients.
If prostate cancer is locally confined, the 5-year survival rate is nearly 100%; however, if the cancer metastasizes to distant sites, the 5-year survival rate is only 31%. Bone is by far (80–85%) the primary site of prostate cancer metastasis. Despite the high incidence and serious consequences of skeletal metastasis of prostate cancer, the mechanisms underlying this osteotropism are poorly understood. However, it is clear that bone-specific matrix proteins and their receptors on the surface of prostate cancer cells play a pivotal role in the process (1-3). Recent analysis of bone extracts has revealed that a key factor that mediates prostate cancer cell invasion is the protein osteonectin or SPARC1 (secreted protein, acidic and rich in cysteine) (4). SPARC is a component of bone matrix that modulates cellular interactions with the extracellular matrix, exerting an anti-adhesive, promigratory effect on cells (5). SPARC is expressed by certain metastatic tumors, and its expression correlates with invasive activity. Therefore, in recent publications, SPARC has also been referred to as the “proinvasive” protein (6). Despite the obvious importance of this particular bone matrix protein in cancer development and metastasis, the mechanisms of its recognition by prostate cancer cells remain unclear.
Cell-surface receptors on prostate tumor cells that are particularly important for bone matrix recognition and that could contribute to the directed metastasis of prostate cancer cells to bone are members of the integrin family of heterodimeric adhesion receptors. In particular, the integrins that share a common αv subunit, αvβ3 and αvβ5 subunits, have been implicated in prostate cancer cell biology (7-11). For example, changes in αvβ3 expression correlate with the progression to androgen independence of prostate cancer (12) and with differences in the adhesive and migratory properties of different prostate cancer cell lines (13). Studies in vivo have demonstrated that tumor growth is inhibited by αvβ3-blocking antibodies (14). Central to the function of integrins is their capacity to be activated (15). Integrin activation can modulate ligand repertoire or can markedly enhance their apparent affinity for a particular ligand. In previous studies, we reported that the functional activity of αvβ3 on endothelial and tumor cells could be regulated by VEGF (16). VEGF has also been implicated in prostate carcinogenesis and metastasis as well as in angiogenesis. VEGF and VEGFRs are expressed by prostate carcinoma cells at a high level (17, 18), and their expression correlates with increasing grade, vascularity, and tumorigenicity (19, 20). These relationships have been observed in humans as well as in animal models of prostate cancer. High VEGF levels in prostate cancer are associated with poor prognosis (17). In addition, VEGF produced by tumor cells affects bone remodeling and might, therefore, facilitate nesting of metastatic cells in bone (21).
In the present study, we identify an interrelationship between SPARC, the αv integrins, and VEGF, which may define the mechanism responsible for the metastasis of prostate cancer to bone: (i) SPARC attracts and subsequently anchors metastatic tumor cells within the bone; (ii) cell-surface αv integrins mediate the migration of prostate tumor cells to bone and, subsequently, the retention and adaptation of the tumor cells within their new microenvironment; (iii) VEGF and its receptors regulate integrin activity to influence the recognition of the bone matrix; and finally, (iv) engagement of SPARC by the integrins induces VEGF production to support bone remodeling and neovascularization to nourish the metastatic tumor.
EXPERIMENTAL PROCEDURES
Cell Lines and Materials
The LNCaP lineage-derived human prostate cancer cell line, LNCaP-C4–2, is androgen-independent and highly tumorigenic, with a proclivity to metastasize to bone (33). All human prostate cancer cell lines LNCaP, LNCaP-C4–2, PC3, and lacZ-transfected CWR22R (H-clones), kindly provided by Dr. Lloyd A. Culp (Case Western Reserve University, Cleveland, OH), were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum. Recombinant SPARC was expressed in insect cells and purified as described previously (34). Platelet-derived purified SPARC was obtained from Hematologic Technologies, Inc. (Essex Junction, VT). Fibronectin was purchased from Roche Diagnostics, and collagen type I was obtained from Calbiochem. Antibodies against αvβ3 (LM609), αvβ5 (P1F6), α5β1 (JBS5), and α2β1 (BHA2.1) integrins were obtained from Chemicon (Temecula, CA). Anti-VEGF and anti-VEGFR-2 were obtained from R&D Systems (Minneapolis, MN). Anti-platelet-derived growth factor (PDGF) was obtained from Sigma, and WOW-1 was provided by Dr. Shattil, La Jolla, CA. cRGDfV peptide was obtained from Peninsula Laboratories Inc. (San Carlos, CA).
Preparation of Bone Extracts
SPARC-null and control mice of the same genetic background were generated as described previously (23). The SPARC-null mice do not contain detectable amounts of SPARC mRNA or protein. Bones excised from 6-week-old male wild-type and SPARC-null littermates (from pelvic region and limb) (1 g) were crushed and extracted for 24 h at 4 °C, as described previously (4). Extracts were centrifuged at 25,000 × g for 30 min, and the supernatant fractions were dialyzed against phosphate-buffered saline.
Cell Migration Assays
Cell migration assays on Transwell plates (8-μm pore size) were performed as described previously (27). Ligand (recombinant or purified SPARC or bone extract, fibronectin, and collagen) was diluted to a selected concentration, and 10 μl of this solution was placed on the lower surface of a polycarbonate filter and air-dried. Cell migration was performed in the presence of blocking antibodies against αvβ3, αvβ5, α5β1, and α2β1, neutralizing antibodies to VEGF, VEGFR-2, and PDGF (20 μg/ml each) or cRGDfV peptides (20 μm).
Flow Cytometry
WOW-1, a Fab fragment, reacts selectively with activated αvβ3 and αvβ5 (29). Its binding to cells was assessed by flow cytometry, as described previously (16). In selected experiments, cells were preincubated for 5 min in the presence of an inhibitor (anti-VEGF or anti-VEGFR-2 neutralizing antibodies or VEGFR2/Fc chimera; 20 μg/ml each). WOW-1 Fab was then added at a final concentration of 30 μg/ml, followed by the addition of Alexa 488 goat anti-mouse IgG (Molecular Probes, Eugene, OR) at 15 μg/ml. After 30 min, the cells were washed and analyzed by flow cytometry. Specific binding of WOW-1 Fab was defined as that which could be inhibited by 10 mm EDTA. Flow cytometry was performed with a FACScan instrument, and the data were analyzed with the CellQuest software program (version 1.2).
Proliferation Assays
These assays were performed as described (35). Briefly, cells were maintained in 1% serum for 20 h prior to experiments. Trypsinized cells were distributed into 96-well microtiter plates (2 × 105 cells/well) in the presence or absence of inhibitors. The cells were labeled with 1 μCi of [3H]thymidine per well. After 24–48 h, the cells were washed, and the radioactivity was precipitated with trichloroacetic acid and quantified by scintillation counting.
Soft Agar Assays
LNCaP-C4–2 cells were trypsinized, washed, and resuspended in 0.4% Bacto-agar (Difco, Detroit, MI) prepared in RPMI 1640 medium supplemented with 10% fetal bovine serum. Cells were plated on the top of 0.7% agar in the presence or absence of VEGFR-2/Fc chimera or anti-VEGFR-2 blocking antibodies (10 μg/ml each). After 5 days, the colonies were photographed, and the number of colonies per power field was quantified 7 days after plating.
Relative Quantitative Real Time PCR
Total RNA from two prostate cancer cell lines (LNCaP and LNCaP-C4–2) was prepared with RNeasy mini kits (Qiagen, Valencia, CA). Cells were plated in wells, which were left uncoated (control) or were coated with SPARC (200 ng/well), anti-αvβ5 integrin antibodies (P1F6), or control nonimmune IgG (10 μg/ml each). In selected experiments cRGDfV (20 μM) was added to the cells; after specific time periods of incubation at 37 °C, RNA was isolated. Real time PCR was performed using SYBR Green PCR core reagents (PerkinElmer Life Sciences) in an ABI Prism 7700 sequence detection system (PerkinElmer Life Sciences). The forward primer was 5′-GAGGAGTCCAACATCACCATGC-3′, located on exon 3; the reverse primer was 5′-CGTTTAACTCAAGCTGCCTCGCC-3′, located on exon 6. A comparative Ct (threshold cycle) method as described previously by Livak and Schmittgen (36) was used to detect relative gene expression where gene expression in treated samples was measured by the quantitation of cDNA converted from an mRNA. The mRNA corresponded to a VEGF gene relative to an untreated control sample, which served as a physiological reference. All quantitations of VEGF gene were normalized to an endogenous control, GAPDH. Relative quantitation of VEGF mRNA expression was performed by the comparative Ct method (36) with the formula 2−ΔΔCt, where ΔΔCt = [ΔCt VEGF (treated sample) – ΔCt GAPDH (treated sample)] – [ΔCt VEGF (calibrator/control sample) – ΔCt GAPDH (calibrator/control sample)]. ΔCt represents the mean Ct value of each sample, and GAPDH is the endogenous control used to normalize all the quantifications of VEGF gene. Alternatively, VEGF was measured by quantitative enzyme-linked immunosorbent assay (R&D Systems).
Immunohistochemistry
Formalin-fixed and paraffin-embedded tissue blocks of prostate cancer and bone metastasis were cut into 6-μm sections and stained with monoclonal antibodies against SPARC (Zymed Laboratories Inc.), CD31 (DAKO, Carpinteria, CA), αVβ3 integrin (Chemicon), and WOW-1 Fab and with antibody against VEGFR-2 (Santa Cruz Biotechnology, Santa Cruz, CA), using the antigen retrieval technique described previously (37). Sections were counter-stained with hematoxylin (Vector). A negative control was performed to ensure the specificity of peroxidase immunostaining by using nonimmune IgG. Sections were examined on an Olympus microscope, and representative areas were photographed using an Olympus digital camera. The quantitative data for the density of blood vessels and intensity of WOW-1 staining was obtained with the Image-Pro Plus program (version 4.5.1.22). Eight paired samples for primary prostate cancer and bone metastasis were analyzed. For each sample, five fields were quantified for the area of CD31-positive blood vessel and the density of WOW-1 staining. These two parameters were normalized to those of normal prostate tissue. The significance was calculated by using the paired t test in Excel.
RESULTS
SPARC Plays a Critical Role in Prostate Cancer Cell Migration to Bone
In a previous study, SPARC was isolated as a protein for bone extracts that supported the preferential migration of prostate cancer cells. The development of SPARC-null mice (22, 23) provides a means to critically address the role of SPARC in prostate tumor cell migration. Bone matrix proteins were extracted from wild-type (WT) and SPARC-null animals and were assessed for their ability to promote migration of prostate cancer cells that are known to metastasize to bone. In a Transwell system, migration of PC3 cells was directly proportional to the amount of bone extract and reached a maximum at 200 ng/well (Fig. 1A). Maximal migration of PC3 cells toward wild-type bone extract was about 4-fold higher than their migration toward SPARC-null bone extract. The deficient migration of PC3 cells toward the SPARC-deficient bone extracts was increased by 2.1-fold in the presence of recombinant SPARC at the concentration of 100 μm and completely restored by the addition of 300 μm SPARC (∼10-fold increase) (Fig. 1B). Addition of the most prominent cell matrix protein, fibronectin, also improved PC3 cell migration toward SPARC-null bone extract; however, the effect of fibronectin was relatively modest (∼2.5-fold in the presence of 300 μm) compared with SPARC at the same concentration (Fig. 1B). No significant improvement in PC3 cell migration was observed in the presence of collagen at the same concentrations (Fig. 1B). Similar results were obtained when SPARC purified from platelets was added (data not shown).
Fig. 1.
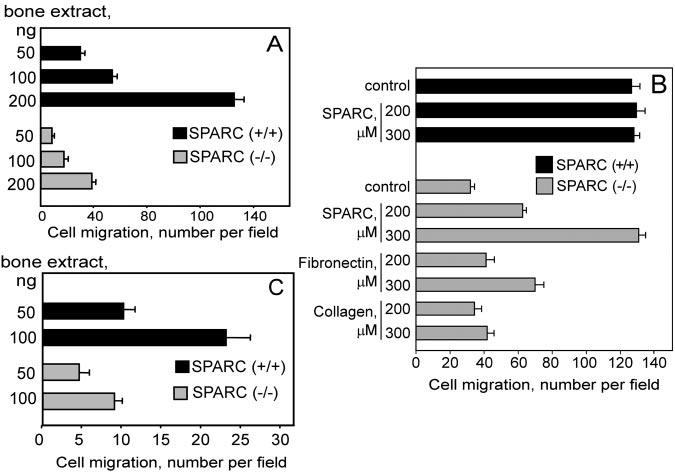
Migration of prostate cancer cells toward bone extracts from SPARC (+/+) and SPARC (−/−) mice. A and C, migration of PC3 and LNCaPC4–2 cells to bone extracts (50, 100, and 200 ng/well) of normal and SPARC (−/−) mice, respectively. Migration was quantified by performing microscopic counts of 10–12 random fields at 200× power. B, SPARC restores the migration of PC3 cells toward SPARC (−/−) bone extracts. Cells were placed in Transwell plates coated with bone extract (200 ng/well) and with or without recombinant SPARC, fibronectin, or collagen (200 and 300 μm). After 4 h of incubation, cell migration was quantified. The data shown are means ± S.D. of three separate experiments.
Metastatic Prostate Cancer Cells Exhibit Increased Migration to SPARC
Next we compared the migration of LNCaP cells to that of its highly metastatic variant, LNCaP-C4–2. The maximal migration of LNCaP-C4–2 cells was 1.85-fold higher than that of LNCaP cells (Fig. 2A). These data indicate a correlation between the metastatic potential of prostate cancer cells and their migration to SPARC. We obtained similar results with other representative metastatic prostate cancer cell lines, namely PC3 and CWR22Rv1-H (not shown). By using wild-type and SPARC-null bone extracts, we demonstrated that SPARC is a crucial factor for the migration of highly metastatic LNCaP-C4–2 cells. In these experiments, migration toward wild-type bone extract was optimal at 100 ng of bone extract per well and was 2.87-fold higher than migration toward SPARC-null bone extract (Fig. 1C).
Fig. 2.

A, migration of LNCaP and LNCaP-C4–2 cells toward recombinant SPARC at different concentrations (50, 100, and 200 ng/well). Integrins and VEGF regulate cell migration toward SPARC. B, cell migration to SPARC after treatment with or without anti-integrins αvβ3, anti-αvβ5, anti-α5β1, anti-α2β1 monoclonal antibody, and cRGDfV peptide. C, effect of neutralizing antibodies to VEGF, VEGFR-2, and PDGF on migration to SPARC.
Prostate Cancer Cell Migration to SPARC and Bone Matrix Proteins Is Mediated by Integrins and Controlled by VEGF
Because αvβ5 and αvβ3 have been implicated in the recognition of a number of bone-specific matrix proteins (24), we assessed the effects of specific anti-integrin blocking antibodies on prostate cancer cell migration to SPARC (Fig. 2). Blockade of αvβ3 and αvβ5 resulted in 35 and 70% inhibition of LNCaP-C4–2 cell migration to SPARC, respectively, indicating a role of both integrins in the response (Fig. 2B). In contrast, blockade of either α5β1 or α2β1 integrin had no effect on the migration of LNCaP-C4–2 cell to SPARC (Fig. 2B). Similar results were obtained with PC3 cells (data not shown). Significant inhibition with cyclic RGDfV peptide (70–80%), which is a relatively specific antagonist of αvβ5, substantiates a primary role for this integrin in migration to SPARC. Direct interaction of LNCaP and PC3 prostate cancer cells with SPARC and the cooperative roles of αvβ5 and αvβ3 were also demonstrable in adhesion assays (not shown). To our knowledge, this is the first demonstration that recognition of SPARC is mediated by these integrins.
In previous studies, we and others (16, 25) demonstrated that the adhesive and migratory phenotypes of certain tumor cells are sustained by an autocrine loop initiated by VEGF. Prostate cancer cells express high levels of VEGF (18), and VEGF production correlates with the metastatic potential of prostate cancer cells (26). The highly metastatic variant LNCaP-C4–2 produces higher amounts of VEGF compared with its nonmetastatic counterpart LNCaP (73.8 ± 8 pg/ml versus 19.5 ± 2.5 pg/ml after 72 h, respectively). Because prostate cancer cells not only produce VEGF but also express VEGF receptors (VEGFR-1 and VEGFR-2 by FACS), we postulated that the migration to SPARC could be controlled by a VEGF-dependent autocrine loop. Therefore, we assessed the effects of VEGF and VEGFR antagonists on the migration of prostate cancer cells to SPARC. As shown in Fig. 2C for LNCaP and LNCaP-C4–2, anti-VEGF- and anti-VEGFR-2-neutralizing antibodies antagonized the migration of prostate cancer cells to SPARC to a level similar to that obtained with anti-integrin-blocking antibodies and the cRGDfV peptide (∼80% inhibition). In contrast, anti-PDGF-neutralizing antibodies inhibited the migration rate by ∼35%. These results suggest that αvβ5 and αvβ3, together with VEGF/VEGFR-2, play a critical role in prostate cancer cell migration to SPARC. Similar results were obtained when bone extracts were substituted for SPARC (Fig. 3, A and B).
Fig. 3.
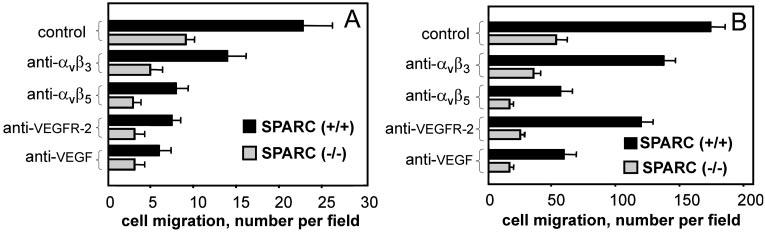
A, LNCaP-C4–2 cells were preincubated with or without anti-αVβ3, anti-αVβ5 monoclonal antibody, and anti-VEGF/VEGFR-2 and placed in bone extracts from SPARC (+/+) and SPARC (−/−) mice. B, migration of PC3 cells respectively to bone extracts from SPARC (+/+) and SPARC (−/−) mice after treatment with or without anti-αvβ3, anti-αvβ5 monoclonal antibody, and anti-VEGF/VEGFR-2 inhibitors. Cell migration was quantitated by performing microscopic counts of 10–20 random fields at 200X power. The data shown are means ± S.D. of three separate experiments.
In the absence of SPARC, migration was significantly lower but still depended on αvβ5 and αvβ3 integrins and VEGF. We attribute this residual activity to the presence of other matrix proteins (e.g. bone sialoprotein and osteopontin), which are known to interact with αvβ5 and αvβ3 (27, 28).
VEGF Supports Prostate Tumor Growth by Autocrine Stimulation of VEGFR-2
As shown in Fig. 4A, proliferation of LNCaP-C4–2 cells was inhibited by a blockade of VEGFR-2 by >70%. These data indicate that a VEGF-dependent autocrine loop, initiated by engagement of VEGFR-2, influences LNCaP-C4–2 growth. Because the anchorage-independent tumor growth is one of the defining characteristics of neoplastic transformation, we determined how the blockade of VEGF affects the growth of these prostate tumor cells in soft agar. When LNCaP-C4–2 cells were grown in the presence of anti-VEGF-neutralizing antibodies and VEGFR2/Fc, the number of colonies was significantly decreased (Fig. 4C). Of note, the size of colonies was also altered. In the presence of VEGF inhibitors, tumor colonies were significantly smaller (Fig. 4B).
Fig. 4.
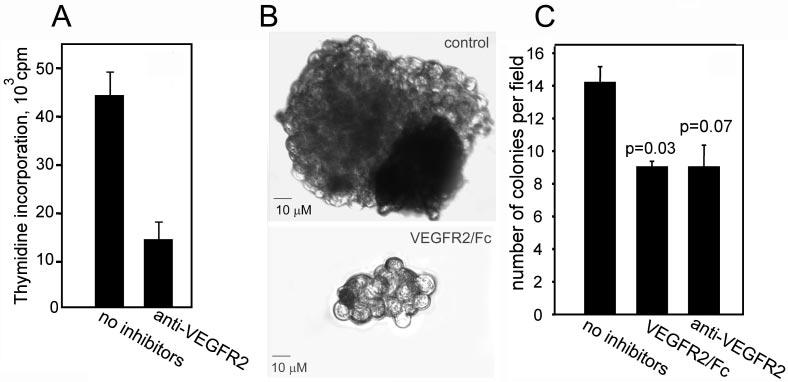
A, proliferative response of LNCaP-C4–2 cells was measured by [3H]thymidine incorporation in the presence of control or blocking anti-VEGFR-2 antibodies (10 μg/ml each). The data shown are means ± S.D. of quadruplicates in one experiment and are representative of three separate experiments. B and C, growth of LNCaP-C4–2 in soft agar. Cells were plated on top of the agarose base in the presence or absence of VEGFR-2/Fc chimera or anti-VEGFR-2 blocking antibodies. After 5 days, the colonies were photographed (B), and the number of colonies per high power field was quantified (C). The data shown are means ± S.D. of quadruplicates in one experiment and are representative of three separate experiments.
VEGF Activates Integrins on Metastatic Prostate Cancer Cells by an Autocrine Loop in Vitro and in Vivo
To assess the molecular basis for increased recognition of SPARC by integrins on metastatic prostate cancer cells, we analyzed the activation state of their αv integrins using WOW-1 Fab (29), which reacts selectively with activated αvβ3 and αvβ5 (29). As monitored by FACS (Fig. 5A), WOW-1 binding to LNCaP-C4–2 cells is 4.2 times higher than to LNCaP cells, despite the expression of similar levels of αVβ3 and αVβ5 by both cell lines (specific fluorescence intensity values were 15 ± 3 and 13 ± 5 for αVβ3 and 18 ± 4 and 20 ± 6 for αVβ5). These data indicate that the functional activity of αVβ3 and αVβ5 integrins is higher on the metastatic variant of prostate cells. In addition, Fig. 5A demonstrates that integrin activation can be inhibited by neutralization of VEGF, because anti-VEGFR-2 antibodies inhibited WOW-1 binding by >60%. We also analyzed two other metastatic prostate cancer cell lines, PC3 and CWR22Rv1. Integrin activation monitored by WOW-1 binding was inhibited by >50% when VEGF or VEGFR-2 were neutralized.
Fig. 5.
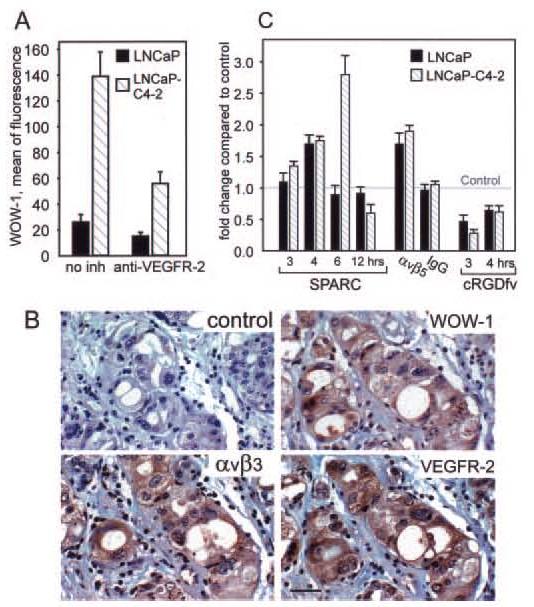
A, αVβ3 and αVβ5 integrin activation states on LNCaP and LNCaP-C4–2 cells were detected by WOW-1 binding. Cells were preincubated in the absence or presence of anti-VEGFR-2 blocking antibodies. WOW-1 Fab was then added, followed by the addition of Alexa 488 goat anti-mouse IgG. After 30 min, the cells were washed and analyzed by flow cytometry. B, adjacent tissue sections of metastatic prostate cancer were stained with control nonimmune antibodies (control), LM609 (αVβ3), WOW-1, and anti-VEGFR-2, followed by secondary antibodies (ABC kit from Vector). Bar, 50 μM. C, SPARC induces VEGF in prostate cancer cells. VEGF mRNA was quantified relative to control (assigned a value of 1). The data shown are means ± S.D. of triplicates in one experiment and are representative of three separate experiments.
αVβ3 and αVβ5 are present in an activated state on prostate tumor cells in vivo (Fig. 5B). Tumor areas were stained with WOW-1. The pattern of WOW-1 staining was similar but more restricted than that of LM609, the first demonstration of αVβ3 activation in vivo. VEGFR-2, which is critical for induction of the VEGF-dependent autocrine loop, colocalized with activated αVβ3 and αVβ5, supporting the concept that engagement of VEGFR-2 by VEGF induces integrin activation. Interestingly, in the sites of bone metastasis, but not in the primary prostate adenocarcinoma, SPARC expression within or around invading tumor follows WOW-1 expression, indicating colocalization and possible interaction (Fig. 6, A and C).
Fig. 6.
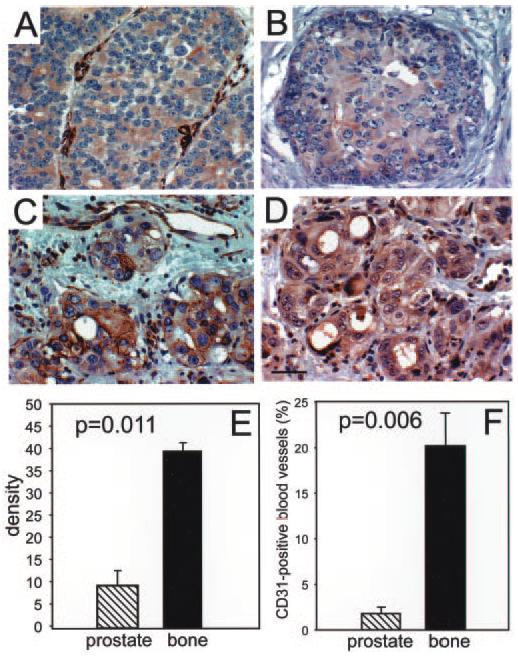
Paired tissue sections of prostate tumor (A and B) and bone metastasis (C and D) were stained with antibodies against SPARC (A and C) and WOW-1 (B and D), followed by secondary antibodies (ABC kit from Vector). D, bar, 50 μm. E, the mean intensity of WOW-1 expression was quantified with Image-Pro Plus software for four representative fields for each patient. The density of WOW-1 staining in normal prostate tissue was negligible, and it was subtracted from the values for primary tumor and bone metastasis. Bars, means ± S.E. for four patient samples in each primary tumor and bone metastasis. F, blood vessels were visualized by staining for CD31, an endothelial cell marker. The area of the CD31-positive blood vessels was measured with Image-Pro Plus software and is expressed as a percentage of the total area. Four representative fields were quantified for each patient. Bars, means ± S.E. for four patient samples for both primary tumor and bone metastasis.
SPARC Increases VEGF Levels via αvβ5 Integrin Ligation in Prostate Cancer Cells
Because VEGF affects SPARC engagement by integrins, we considered whether SPARC engagement might alter VEGF production and hence the autocrine loop. Accordingly, we assessed VEGF production in LNCaP and LNCaP-C4–2 cells plated on SPARC. After 3 or 4 h, the VEGF mRNA level in the LNCaP-C4–2 cells was increased 1.35- or 1.75-fold, respectively, and reached a maximum value (2.8-fold) after 6 h (Fig. 5C). The increase detected in metastatic LNCaP-C4–2 cells in VEGF mRNA after 6 h was 1.65-fold greater than in nonmetastatic LNCaP cells, and in all of these cases, the Ct value of GAPDH was ∼16. Quantitative enzyme-linked immunosorbent assay demonstrated that VEGF expression was also altered at the protein level and that blockade of αVβ5 with cRGDfV peptide suppressed VEGF production in LNCaP-C4–2 and LNCaP cells by 3.57 ± 0.16- and 2.1 ± 0.1-fold, respectively. Thus, ligation of αVβ5 by SPARC amplifies VEGF production and provides metastatic prostate cancer cells with a significant growth advantage in the bone matrix. In similar experiments using M21 melanoma cells that do not metastasize to bone but which express comparable amounts of αVβ3 and αVβ5 to LNCaP-C4–2 cells, we did not observe any increase in VEGF production at 3, 4, 6, or 12 h after plating onto SPARC. In fact, VEGF expression decreased by 40% compared with control (data not shown). We can tentatively conclude that the increased production of VEGF by SPARC via αVβ5 is a characteristic displayed by the prostate cancer cells tested.
Activation State of αVβ3 and αVβ5 Integrins Is Increased in Bone Metastasis
The intensity of WOW-1 staining in primary tumor located within the prostate and tumor that metastasized to bone (matched pairs of tissue samples obtained from the same patient) were compared. An example of such paired specimens is shown in Fig. 6, B and D. Quantitative aspects of WOW-1 staining in prostate in comparison to bone metastasis are shown in Fig. 6E. This analysis reveals that the intensity of WOW-1 staining was 4-fold higher at the sites of bone metastasis. As anticipated, vascular density was significantly (19-fold) higher at these sites compared with the tumor within the prostate itself (Fig. 6F). Our results indicate that αVβ3 is activated in prostate tumors in vivo. Moreover, the extent of activation is significantly increased in the sites of bone metastasis compared with the localized tumor.
DISCUSSION
Previous studies have implicated SPARC, integrins, and VEGF as major players in the pathogenesis of prostate cancer. Our study develops a cohesive model which may explain the interrelationship between these molecules and account for the high osteotropism of prostate cancer metastasis. The major elements of this model are shown in Fig. 7 and are supported by the following findings. (i) SPARC is a key protein that attracts prostate cancer cells to bone. SPARC-deficient bone extracts support minimal migration of prostate cancer cells, and the addition of purified SPARC restores the rate of cell migration. (ii) Recognition of SPARC correlates with the metastatic potential of prostate cancer cell lines. High metastatic variants exhibit increased migration to SPARC compared with nonmetastatic ones. (iii) SPARC recognition is mediated by αVβ3 and αVβ5 integrins and is controlled by an autocrine loop in which VEGF engages VEGFR-2. (iv) VEGF supports prostate cancer cell proliferation and anchorage-independent growth. (v) Increased migration of metastatic cells to SPARC can be traced to the activation of αVβ3 and αVβ5 as monitored by WOW-1. Activation of these integrins is substantially higher on metastatic versus nonmetastic prostate cancer cells. Moreover, activated integrins are expressed by prostate tumors in vivo and colocalize with VEGFR-2. (vi) A particularly relevant pathophysiological consequence of SPARC recognition by αVβ5 is the up-regulation of VEGF production, which provides prostate cancer cells with a significant growth advantage in bone tissue. (vii) At sites of metastasis characterized by abundant SPARC, high VEGF production, and enhanced neovascularization, αVβ3 and αVβ5 integrin activation is substantially higher in comparison to the tumor localized in the prostate.
Fig. 7.
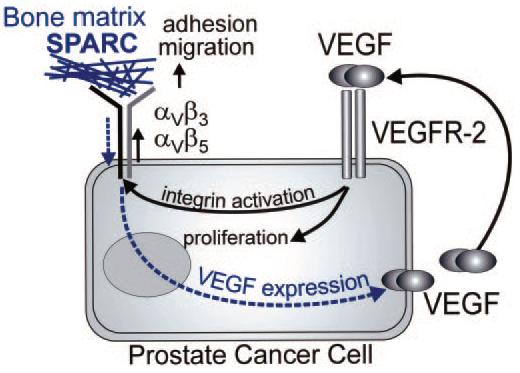
Mechanism of SPARC recognition by prostate cancer cells. The expression of VEGF and VEGFR2 on prostate tumor cells creates an autocrine loop that stimulates prostate cancer cell growth and proliferation and activates integrins αVβ and αVβ5. In turn, these activated integrins mediate enhanced adhesion and migration to SPARC and bone matrix. Engagement of the integrins by SPARC, in turn, enhances VEGF production.
Metastatic prostate cancer cells exhibit low migration to SPARC-deficient bone extracts, and the addition of recombinant SPARC completely rescues this response in a concentration-dependent manner. Although fibronectin was ∼5-fold less potent compared with SPARC, it also provided a certain increase in prostate cancer cell migration, which was due to the high level of expression of α5β1 (fibronectin receptor) on these cells. Our results strongly support the findings of Jacob et al. (4) who reported that extracts from bones but not from other tissues promoted a 4-fold increase in the invasive ability of human prostate carcinoma cells, with SPARC but not any other matrix protein as the factor responsible for this activity. In our studies, purified SPARC supported the enhanced migration of the metastatic prostate cancer cells LNCaP-C4–2 and PC3, compared with the nonmetastatic variant LNCaP. Thus, migration to SPARC positively correlates with a characterized metastatic potential of cell lines. We demonstrated that the cell adhesion receptors responsible for migration to SPARC are integrin αVβ5 and, to a lesser extent, αVβ3, but not α5β1 and α2β1. The differences in the migratory activity of metastatic versus non-metastatic cell lines can be explained by the differences in the activation state of the bone matrix receptors αVβ5 and αVβ3, as demonstrated by WOW-1 binding. Activated integrins are known to be responsible for increased cell migration and adhesion on a variety of cell types, including tumor cells. Thus, our results provide an explanation for the previously reported increased usage of αVβ3 by the metastatic subline LNCaP-C4–2 versus LNCaP cells in the migration to laminin (30). The integrin activation state and the recognition of SPARC are controlled by a VEGF-dependent autocrine loop, because the neutralization of VEGF or the inhibition of its receptor VEGFR-2 decreases WOW-1 binding and cell migration. Furthermore, activated αVβ3 colocalizes with VEGFR-2 on tumor cells in vivo. The existence of growth factor-dependent autocrine loop, which, in addition to the effect on the integrin functions, also supports tumor growth, is the most recently uncovered characteristic of malignant phenotype (16, 26).
Because of the increased VEGF production and autocrine stimulation, integrin activation is significantly higher on the metastatic prostate cancer cells compared with the nonmetastatic cells in vitro and, at sites of bone metastasis compared with the original prostate tumor, in vivo. Thus, at least two lines of evidence demonstrate that the integrin activation state controlled by VEGF contributes to the metastatic behavior of prostate cancer. These findings link together previously published observations that increased integrin function (31) as well as increased VEGF plasma levels in patients (32) are hallmarks of metastasis development.
We found that SPARC not only attracts prostate cancer cells to bone but also, by means of the ligation of integrin αVβ5, further increases VEGF production by metastatic cancer cells and provides an additional enhancement of the VEGF autocrine loop. In turn, VEGF facilitates the accommodation of tumor cells in the bone environment by stimulation of the growth of the tumor itself by a further increase of integrin activation on the tumor and by the induction of neovessel formation. Thus, high levels of SPARC at sites of bone metastasis lead to an increase in VEGF and integrin activation. Our results not only suggest a model to explain the osteotropism of prostate cancer metastasis but also to identify potential targets to prevent the process. For example, SPARC, activated integrins, VEGF, or VEGFR-2 would be candidate targets to interfere with metastatic process. Because prevention of bone metastasis is paramount to effective treatment of prostate cancer, these targets are particularly worthwhile pursuing.
Footnotes
This work was supported by CAPCURE Foundation and National Institutes of Health Grants DK60933 (to T. V. B.) and GM40711 (to E. H. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: SPARC, secreted protein, acidic and rich in cysteine; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor; LNCaP, lymph node carcinoma of prostate; PDGF, platelet-derived growth factor; cRGDfV, cyclic Arg-Gly-Asp-(d-Phe)-Val; Ct, threshold cycle; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; FACS, fluorescence-activated cell sorter.
REFERENCES
- 1.Smit JW, van der Pluijm G, Vloedgraven HJ, Lowik CW, Goslings BM. Thyroid. 1998;8:29–36. doi: 10.1089/thy.1998.8.29. [DOI] [PubMed] [Google Scholar]
- 2.Salih MA, Orhii PB, Chen C, Kalu DN. Mol. Cell. Endocrinol. 1999;147:149–159. doi: 10.1016/s0303-7207(98)00206-8. [DOI] [PubMed] [Google Scholar]
- 3.Fontana A, Delmas PD. Cancer. 2000;88:2952–2960. doi: 10.1002/1097-0142(20000615)88:12+<2952::aid-cncr11>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 4.Jacob K, Webber M, Benayahu D, Kleinman HK. Cancer Res. 1999;59:4453–4457. [PubMed] [Google Scholar]
- 5.Brekken RA, Sage EH. Matrix Biol. 2001;19:815–827. doi: 10.1016/s0945-053x(00)00133-5. [DOI] [PubMed] [Google Scholar]
- 6.Lane TF, Sage EH. FASEB J. 1994;8:163–173. [PubMed] [Google Scholar]
- 7.Rokhlin OW, Cohen MB. Prostate. 1995;26:205–212. doi: 10.1002/pros.2990260406. [DOI] [PubMed] [Google Scholar]
- 8.Varner JA, Cheresh DA. Curr. Opin. Cell Biol. 1996;8:724–730. doi: 10.1016/s0955-0674(96)80115-3. [DOI] [PubMed] [Google Scholar]
- 9.Lang SH, Clarke NW, George NJ, Testa NG. Clin. Exp. Metastasis. 1997;15:218–227. doi: 10.1023/a:1018465213641. [DOI] [PubMed] [Google Scholar]
- 10.van der Pluijm G, Vloedgraven HJ, Ivanov B, Robey FA, Grzesik WJ, Robey PG. Cancer Res. 1996;56:1948–1955. [PubMed] [Google Scholar]
- 11.Thalmann GN, Sikes RA, Devoll RE, Kiefer JA, Markwalder R, Klima I, Farach-Carson CM, Studer UE, Chung LW. Clin. Cancer Res. 1999;5:2271–2277. [PubMed] [Google Scholar]
- 12.Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, Pathak S, von Eschenbach AC, Chung LW. Cancer Res. 1994;54:2577–2581. [PubMed] [Google Scholar]
- 13.Zheng DQ, Woodard AS, Fornaro M, Tallini G, Languino LR. Cancer Res. 1999;59:1655–1664. [PubMed] [Google Scholar]
- 14.Sokoloff MH, Chung LW. Cancer Metastasis Rev. 1998;17:307–315. doi: 10.1023/a:1006170612253. [DOI] [PubMed] [Google Scholar]
- 15.Bazzoni G, Hemler ME. Trends Biochem. Sci. 1998;23:30–34. doi: 10.1016/s0968-0004(97)01141-9. [DOI] [PubMed] [Google Scholar]
- 16.Byzova TV, Goldman CK, Pampori N, Thomas KA, Bett A, Shattil SJ, Plow EF. Mol. Cell. 2000;6:851–860. [PubMed] [Google Scholar]
- 17.Ferrer FA, Miller LJ, Andrawis RI, Kurtzman SH, Albertsen PC, Laudone VP, Kreutzer DL. Urology. 1998;51:161–167. doi: 10.1016/s0090-4295(97)00491-3. [DOI] [PubMed] [Google Scholar]
- 18.Ferrer FA, Miller LJ, Lindquist R, Kowalczyk P, Laudone VP, Albertsen PC, Kreutzer DL. Urology. 1999;54:567–572. doi: 10.1016/s0090-4295(99)00156-9. [DOI] [PubMed] [Google Scholar]
- 19.Weidner N, Carroll PR, Flax J, Blumenfeld W, Folkman J. Am. J. Pathol. 1993;143:401–409. [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrer FA, Miller LJ, Andrawis RI, Kurtzman SH, Albertsen PC, Laudone VP, Kreutzer DL. J. Urol. 1997;157:2329–2333. [PubMed] [Google Scholar]
- 21.Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. Nat. Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 22.Sage EH. J. Histochem. Cytochem. 1999;47:1643A–1643. [PubMed] [Google Scholar]
- 23.Delany AM, Amling M, Priemel M, Howe C, Baron R, Canalis E. J. Clin. Invest. 2000;105:915–923. doi: 10.1172/JCI7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McHugh KP, Hodivala-Dilke K, Zheng MH, Namba N, Lam J, Novack D, Feng X, Ross FP, Hynes RO, Teitelbaum SL. J. Clin. Invest. 2000;105:433–440. doi: 10.1172/JCI8905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masood R, Cai J, Zheng T, Smith DL, Hinton DR, Gill PS. Blood. 2001;98:1904–1913. doi: 10.1182/blood.v98.6.1904. [DOI] [PubMed] [Google Scholar]
- 26.Soker S, Kaefer M, Johnson M, Kagsbrun M, Atala A, Freeman MR. Am. J. Pathol. 2001;159:651–659. doi: 10.1016/S0002-9440(10)61736-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Byzova TV, Kim W, Midura RJ, Plow EF. Exp. Cell Res. 2000;254:299–308. doi: 10.1006/excr.1999.4765. [DOI] [PubMed] [Google Scholar]
- 28.Zheng DQ, Woodard AS, Tallini G, Languino LR. J. Biol. Chem. 2000;275:24565–24574. doi: 10.1074/jbc.M002646200. [DOI] [PubMed] [Google Scholar]
- 29.Pampori N, Hato T, Stupack DG, Aidoudi S, Cheresh DA, Nemerow GR, Shattil SJ. J. Biol. Chem. 1999;274:21609–21616. doi: 10.1074/jbc.274.31.21609. [DOI] [PubMed] [Google Scholar]
- 30.Edlund M, Miyamoto T, Sikes RA, Ogle R, Laurie GW, Farach-Carson MC, Otey CA, Zhau HE, Chung LW. Cell Growth & Differ. 2001;12:99–107. [PubMed] [Google Scholar]
- 31.Pecheur I, Peyearuchaud O, Serre CM, Guglielmi J, Voland C, Bourre F, Margue C, Cohen-Solal M, Buffet A, Kieffer N, Clezardin P. FASEB J. 2002;16:1266–1268. doi: 10.1096/fj.01-0911fje. [DOI] [PubMed] [Google Scholar]
- 32.Jones A, Fujiyama C, Turner K, Fuggle S, Cranston D, Bicknell R, Harris AL. B. J. U. Int. 2000;85:276–280. doi: 10.1046/j.1464-410x.2000.00432.x. [DOI] [PubMed] [Google Scholar]
- 33.Thalmann GNN, Sikes RA, Wu TT, Degeorges A, Chang SM, Ozen M, Pathak S, Chung LW. Prostate. 2000;44:91–103. doi: 10.1002/1097-0045(20000701)44:2<91::aid-pros1>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 34.Bradshaw AD, Bassuk JA, Francki A, Sage EH. Mol. Cell. Biol. Res. Commun. 2000;3:345–351. doi: 10.1006/mcbr.2000.0237. [DOI] [PubMed] [Google Scholar]
- 35.Gardiner EE, D'Souza SE. J. Biol. Chem. 1997;272:15474–15480. doi: 10.1074/jbc.272.24.15474. [DOI] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Byzova TV, Goldman CK, Jankau J, Chen J, Cabrera G, Achen MG, Stacker SA, Carnevale KA, Siemionow M, Deitcher SR, DiCorleto PE. Blood. 2002;99:4434–4442. doi: 10.1182/blood.v99.12.4434. [DOI] [PubMed] [Google Scholar]