

Abstract
A hallmark feature of prions, whether in mammals or yeast and fungi, is exponential growth associated with fission or autocatalysis of protein aggregates. We have employed a rigorous kinetic analysis to recent data from transgenic mice lacking a glycosylphosphatidylinositol membrane anchor to the normal cellular PrPC protein, which show that toxicity requires the membrane binding. We find as well that the membrane is necessary for exponential growth of prion aggregates; without it, the kinetics is simply the quadratic-in-time growth characteristic of linear elongation as observed frequently in in vitro amyloid growth experiments with other proteins. This requires both: i), a substantial intercellular concentration of anchorless PrPC, and ii), a concentration of small scrapies seeding aggregates from the inoculum, which remains relatively constant with time and exceeds the concentration of large polymeric aggregates. We also can explain via this analysis why mice heterozygous for the anchor-full/anchor-free PrPC proteins have more rapid incubation than mice heterozygous for anchor-full/null PrPC, and contrast the mammalian membrane associated fission or autocatalysis with the membrane free fission of yeast and fungal prions.
Prions are distinguished from other amyloid diseases both by their infectious character and the observed exponential growth of infectious material in vivo (1). There is a correspondence to this of prion-like proteins in yeast and fungi, for which spontaneous fission is reported in vitro (2). In fact, it has been argued that the replication necessary for infection in mammals and non-Mendelian inheritance in yeast/fungi requires the fission or autocatalysis that drives the exponential growth (1). It has been established for yeast prions that additional chaperone proteins most likely facilitate the fission of aggregates in living cells (1). It is an open question what mechanism drives the exponential growth in mammals. Here we show by a rigorous kinetic analysis of recent disease time course data that the exponential growth is tied to membrane anchoring of the prion protein, suggesting that either mechanical fission of areal prion aggregates or oligomeric autocatalysis of membrane bound prions explain the observed behavior.
Chesebro et al. (3) recently studied transgenic (Tg) mice lacking a GPI membrane anchor to the normal cellular PrPC protein and discovered that these mice grew infectious prions without suffering neuronal death. We denote these anchorless cellular prions as 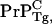 and anchor-full wild-type (WT) cellular prions by
and anchor-full wild-type (WT) cellular prions by 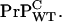 When inoculated with infectious scrapies prions (PrPSc) at a dose that induces clinical symptoms within 140–160 days for WT mice, the Tg mice were symptomless up to 400–600 days, even though proteinase resistant PrP-res, an indicator of infectivity, accumulated and surpassed the maximal WT levels.
When inoculated with infectious scrapies prions (PrPSc) at a dose that induces clinical symptoms within 140–160 days for WT mice, the Tg mice were symptomless up to 400–600 days, even though proteinase resistant PrP-res, an indicator of infectivity, accumulated and surpassed the maximal WT levels.
In Fig. 1 the Tg mice PrP-res concentration (crosses) of Chesebro et al. (3) are plotted versus the square of time, together with a linear regression fit (line) with a high regression coefficient (R = 0.97). This time dependence is consistent with short time kinetics described by linear polymer elongation via monomer addition without fission or autocatalysis (4), illustrated schematically in Fig. 2. Assuming the PrP-res concentration to be a proxy for the total protein content in aggregate, simple kinetic arguments predict a behavior, before monomer depletion and seed nucleus depletion, of
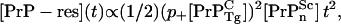 |
(1) |
with 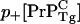 the elongation rate at intercellular monomer Tg prion concentration [
the elongation rate at intercellular monomer Tg prion concentration [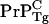 ], and [
], and [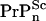 ] is the intercellular concentration of seeding nuclei from inoculated scrapies protein after initial hydrodynamic clearance. The validity of Eq. 1 at long times suggests that: i), there is a substantial homeostatic concentration of intercellular
] is the intercellular concentration of seeding nuclei from inoculated scrapies protein after initial hydrodynamic clearance. The validity of Eq. 1 at long times suggests that: i), there is a substantial homeostatic concentration of intercellular 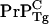 presumably due to slow clearance, and ii), [
presumably due to slow clearance, and ii), [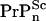 ] is hardly changed implying either that only a small fraction of seeds grow into large polymers or [
] is hardly changed implying either that only a small fraction of seeds grow into large polymers or [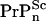 ] is maintained by steady proteolytic degradation of large remnant aggregates from the dose. Given a similar de novo production rate of
] is maintained by steady proteolytic degradation of large remnant aggregates from the dose. Given a similar de novo production rate of 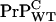 in inoculated WT mice, we speculate that the associated saturation of [PrP-res] arises from loss of
in inoculated WT mice, we speculate that the associated saturation of [PrP-res] arises from loss of 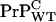 after cell death.
after cell death.
FIGURE 1 .

Quadratic-in-time fit to infectious prion time course data of Chesebro et al. (3).
FIGURE 2 .

Schematic model for linear elongation driven growth of infectious prion material from inoculated seeds and anchorless cellular prion proteins.
This elongation hypothesis is testable by: i), genetically engineering mice to overexpress 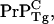 which will quadratically modulate the PrP-res concentration (4), and ii), by varying the initial dose of PrPSc, which will linearly modulate the PrP-res concentration.
which will quadratically modulate the PrP-res concentration (4), and ii), by varying the initial dose of PrPSc, which will linearly modulate the PrP-res concentration.
Another striking observation of Chesebro et al. (3) was that mice heterozygous for expression of 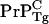 and
and 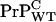 display shorter incubation time upon inoculation than mice with one
display shorter incubation time upon inoculation than mice with one 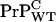 copy and one inactive fusion construct. We argue that this is due to an enhanced concentration for
copy and one inactive fusion construct. We argue that this is due to an enhanced concentration for 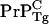 relative to
relative to 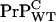 and that PrP-res obtained from
and that PrP-res obtained from 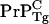 also templates
also templates 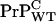 conversion. The latter is speculative but should be testable.
conversion. The latter is speculative but should be testable.
To establish the expectation that the 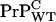 concentration is lower than
concentration is lower than 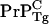 in the WT/Tg heterozygotes, it is sufficient to establish that
in the WT/Tg heterozygotes, it is sufficient to establish that 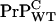 concentration in WT mice is lower than
concentration in WT mice is lower than 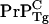 concentration in the anchorless Tg mice. To make this clear, we compare estimates for cellular prion concentrations in homozygous WT mice with homozygous Tg mice. Before the postulated cell-death driven saturation of infectious material, WT mice inoculated with a concentration [
concentration in the anchorless Tg mice. To make this clear, we compare estimates for cellular prion concentrations in homozygous WT mice with homozygous Tg mice. Before the postulated cell-death driven saturation of infectious material, WT mice inoculated with a concentration [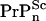 ] of scrapies seeds will have a time course of the general form (4)
] of scrapies seeds will have a time course of the general form (4)
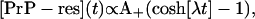 |
(2) |
where λ = ln(2)/t2 is the percentage growth rate and t2 is the doubling time. The coefficient A+ can be determined from the short time behavior of Eq. 2, which, like Eq. 1, is described by linear elongation given by
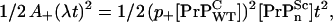 |
(3) |
where [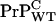 ] is the homeostatic concentration of membrane bound WT PrPC and we have assumed that the WT and Tg mice have the same PrP-res elongation coefficient p+.
] is the homeostatic concentration of membrane bound WT PrPC and we have assumed that the WT and Tg mice have the same PrP-res elongation coefficient p+.
On the other hand, at long times (but before saturation) Eq. 2 gives
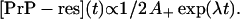 |
(4) |
Now, we set t+ = 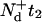 as the time it takes WT mice to reach clinically detectable levels of PrP-res concentration at the inoculum level generating a seed nuclei concentration [
as the time it takes WT mice to reach clinically detectable levels of PrP-res concentration at the inoculum level generating a seed nuclei concentration [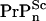 ], where
], where 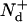 is the number of doublings experienced in that process, and t− is the time it takes Tg mice to reach the same clinical concentration of PrP-res for the same initial inoculation dose. By taking suitable ratios to eliminate A+, [
is the number of doublings experienced in that process, and t− is the time it takes Tg mice to reach the same clinical concentration of PrP-res for the same initial inoculation dose. By taking suitable ratios to eliminate A+, [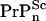 ], and p+, the ratio of homeostatic concentrations of cellular prions from the WT mice to the Tg mice is given by
], and p+, the ratio of homeostatic concentrations of cellular prions from the WT mice to the Tg mice is given by
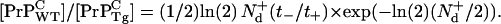 |
(5) |
From Chesebro et al. (3), t+ = 150 days, and t− = 400 days. A reasonable estimate (5) for the number of doublings is 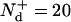 for the dose of Chien et al. (1). With these numbers, Eq. 5 gives a concentration ratio (and hence elongation rate ratio) for WT/Tg mice of 0.036. This is reasonable given that likely slower PrPC clearance in the Tg case will lead to a higher extracellular concentration of cellular prion protein. By employing the arguments of Chien et al. (1) we obtain elongation rate values of 0.13/day(Wt) and 3.5/day (Tg). The former is in good agreement with estimates made elsewhere for linear elongation based upon analysis of dose-incubation curves (6).
for the dose of Chien et al. (1). With these numbers, Eq. 5 gives a concentration ratio (and hence elongation rate ratio) for WT/Tg mice of 0.036. This is reasonable given that likely slower PrPC clearance in the Tg case will lead to a higher extracellular concentration of cellular prion protein. By employing the arguments of Chien et al. (1) we obtain elongation rate values of 0.13/day(Wt) and 3.5/day (Tg). The former is in good agreement with estimates made elsewhere for linear elongation based upon analysis of dose-incubation curves (6).
Hence, in the WT/Tg and WT/null heterozygotes explored in Chesebro et al. (3), we anticipate in each case the membrane bound 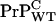 concentration to be about half that of the homozygous WT mice, whereas the intercellular
concentration to be about half that of the homozygous WT mice, whereas the intercellular 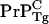 concentration should be about half that of the homozygous Tg mice. Because, as shown with infectious prions bound to electrodes (7), templating and conversion can be driven by scrapies material not bound to the membrane surface, we expect the incubation time of the WT/Tg heterozygotes to be significantly accelerated relative to the WT/null heterozygotes as is observed.
concentration should be about half that of the homozygous Tg mice. Because, as shown with infectious prions bound to electrodes (7), templating and conversion can be driven by scrapies material not bound to the membrane surface, we expect the incubation time of the WT/Tg heterozygotes to be significantly accelerated relative to the WT/null heterozygotes as is observed.
We note that membrane associated exponential growth might be due to: i), as yet undiscovered membrane specific enzymes splitting aggregates, in analogy to the role of Hp104a for yeast prions (1); ii), mechanical breakage of aggregates due to membrane curvature or membrane undulations (8); iii), oligomeric autocatalysis arising from interneuronal templating by infectious oligomeric seeds bound to one or the other membrane (9).
Acknowledgments
We are grateful for conversations with H. Levine, J. N. Onuchic, M. Oldstone, A. N. Parikh, and J. Weissman.
We acknowledge the support of the U.S. Army (Congressionally Directed Medical Research Program, grant NP020132) (D.L.C. and R.P.P.S), National Science Foundation grant PHY0216576 (D.L.C. and S.Y.), and the J. S. Guggenheim Memorial Foundation (D.L.C.).
References
- 1.Chien, P., J. S. Weissman, and A. H. DePace. 2004. Emerging principles of conformation based prion inheritance. Annu. Rev. Biochem. 73:617–656. [DOI] [PubMed] [Google Scholar]
- 2.Collins, S. R., A. Douglass, R. Vale, and J. S. Weissman. 2004. Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol. 2:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chesebro, B., M. Trifilo, R. Race, K. Meade-White, C. Teng, R. LaCasse, L. Raymond, C. Favara, G. Baron, S. Priola, B. Caughey, E. Masliah, and M. Oldstone. 2005. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 308:1435–1439. [DOI] [PubMed] [Google Scholar]
- 4.Ferrone, F. A. 1999. Analysis of protein aggregation kinetics. Methods Enzymol. 309:256–274. [DOI] [PubMed] [Google Scholar]
- 5.Carlson, G. A., C. Ebeling, S. L. Yang, G. Telling, M. Torchia, D. Groth, D. Westaway, S. J. DeArmond, and S. B. Prusiner. 1994. Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc. Natl. Acad. Sci. USA. 91:5690–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kulkarni, R. V., A. Slepoy, R. R. P. Singh, D. L. Cox, and F. Pazmandi. 2003. Theoretical modeling of prion disease incubation. Biophys. J. 85:707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flechsig, E., I. Hegyi, M. Enari, P. Schwarz, J. Collinge, and C. Weissmann. 2001. Transmission of scrapie by steel-surface-bound prions. Mol. Med. 7:679–684. [PMC free article] [PubMed] [Google Scholar]
- 8.Slepoy, A., R. R. P. Singh, F. Pazmandi, R. V. Kulkarni, and D. L. Cox. 2001. Statistical mechanics of prion disease. Phys. Rev. Lett. 87:058101. [DOI] [PubMed] [Google Scholar]
- 9.Yang, S. C., H. Levine, J. N. Onuchic, and D. L. Cox. 2005. Structure of infectious prions: stabilization by domain swapping. FASEB J. 19:1778–1782. [DOI] [PubMed] [Google Scholar]