

Abstract
Calmodulin is a small (148 residues), ubiquitous, highly-conserved Ca2+ binding protein serving as a modulator of many calcium-dependent processes. In this study, we followed, by means of molecular dynamics, the structural stability of the protein when one of its four bound Ca2+ ions is removed, and compared it to a simulation of the fully Ca2+ bound protein. We found that the removal of a single Ca2+ ion from the N-lobe of the protein, which has a lower affinity for the ion, is sufficient to initiate a considerable structural rearrangement. Although the overall structure of the fully 4 Ca2+ bound protein remained intact in the extended conformation, the Ca2+-removed protein changed its conformation into a compact state. The observation that the 3 Ca2+ loaded protein assumes a compacted solution state is in accord with experimental observation that the NSCP protein, which binds only three Ca2+ ions, is natively in a compact state. Examination of the folding dynamics reveals a cooperation between the C-lobe, N-lobe, and the interdomain helix that enable the conformation change. The forces driving this conformational change are discussed.
INTRODUCTION
The Ca2+ binding protein calmodulin (CaM) is a prevalent transducer of intracellular Ca2+ signals. The small protein (148 residues in vertebrates) is expressed in all eukaryotic cells, where it constitutes at least 0.1% of the total amount of proteins. This highly conserved protein participates in many signaling pathways that regulate crucial processes, such as growth, proliferation, and movement. The regulatory functions of CaM are controlled by intracellular Ca2+, which reacts with CaM at high affinity, and modulates its three-dimensional structure. As a result, the affinities of CaM to target proteins are affected, and CaM can bind, disengage, or wrap around them, causing their activation or deactivation (1).
CaM belongs to a family of proteins that are distinguishable by a structural motif known as the E-F hand. An E-F hand consists of an N-terminal helix, immediately followed by a Ca2+-coordinating loop and a C-terminal helix. CaM has four such motifs. The first two combine to form the globular N-terminal domain (the N-lobe), which is separated by a short flexible linker (interdomain helix) from the homologous C-terminal domain (the C-lobe), which consists of the two other E-F hands.
As calmodulin is the most studied E-F hand protein, a great deal of knowledge has been gathered about it. Ca2+ affinity to CaM (Kd = 5 × 10−7 M to 5 × 10−6 M) falls within the range of intracellular free Ca2+ concentrations (10−7M to 10−6M) of most cells, making it an ideal detector of Ca2+. The ion affinity of the C-lobe sites is three- to fivefold higher than that of the N-lobe, suggesting that the last Ca2+ to bind and the first to leave will do so to and from the N-lobe.
The vast majority of currently available crystallographic data of fully Ca2+ bound CaM (Holo-CaM) has shown that it adopts an extended dumbbell-like shape (2–10). The Ca2+ free (Apo-CaM) structure solved by NMR (11–13) and crystallography (14) has shown major conformational changes compared to these extended Holo-CaM structures. The most distinct difference between the two structures was in the shape of the central linker helix. Although its state seemed rigid in the Holo state, it becomes flexible in the Ca2+ free state. SAXS measurements have indicated that the binding of Ca2+ to the protein results in an enlargement of the gyration radius (15) and that Holo-CaM is found in an extended state in solution (16). This support the existence of Holo-CaM in and extended state in comparison to a more compact state assumed by Apo-CaM. Moreover, 4-ns molecular dynamics simulations, which were carried out on Apo-CaM and Holo-CaM (17), demonstrated the enhanced flexibility of Apo-CaM in comparison to Holo-CaM.
A number of other experiments, however, suggest the existence of a flexible Holo-CaM state. In a SAXS experiment performed by Heidorn and Trewhella (18) the measured pairwise distance distribution function suggests a flexible central helix (although the maximum vector length was in agreement with the extended crystal structures). Crystallographic structure of Holo-CaM in a globular compact conformation has recently been published (19), and NMR experiments on Holo-CaM also reveal a flexible central helix (20). Recent 20-ns molecular dynamics (MD) simulation has shown that the Holo-CaM can change its conformation from an extended to a compact state (21) and that this contraction started at the beginning of the simulation. Yet, it must be realized that this simulation was performed in negligible ionic strength. A different 11.5-ns MD simulation, also under low ionic strength due to addition of counter ions, showed unwinding of the central helix (22), whereas a 4-ns simulation of the Holo-CaM carried out in physiological salt solution revealed only minor conformational changes (23).
Although the experiments tend to agree that Apo-CaM exists in a compact conformation, different Holo-CaM experiments give different results as to the rigidity and conformation of the Holo-CaM. These contradictions may stem from the differences in the experimental conditions or simply by the fact that the proteins' conformation is at an equilibrium that includes the extended, compact, and other intermediary states. Under the latter assumption, the results seem to indicate that Holo-CaM's tendency to be in an extended conformation is much greater than Apo-CaM's. MD simulation can be used to evaluate what might be the preferred solution state of the Holo protein and to compare the stability and conformation tendency of the Holo and Apo CaM under the same conditions.
In this work we studied, via molecular dynamics simulations, the effect of removing a single Ca2+ ion from the low affinity domain on the structure of the protein. Accordingly, we compared a simulation of the Native 4 Ca2+ complex (Holo-CaM) to a simulation of the protein which was stripped of one Ca2+ ion at its N-terminal (N-CaM). The starting structure of the two simulations was the crystallographic structure of extended calmodulin with four bound Ca2+ ions (Protein Data Bank code 1CLL). The simulations were carried out in water supplemented with Na+, Ca2+, and Cl− ions to reach an ionic strength of I = 200 mM.
This study, which to our knowledge is the first to investigate the behavior of 3 Ca2+ bound CaM, reveals that there is a significant reduction in stability of the N-CaM protein compared to the Holo-CaM. This supports the findings of Komeiji et al. (17) that Holo-CaM is structurally more stable than Apo-CaM and gives a novel indication that even dissociation of a single Ca2+ ion from the protein significantly affects its structure. The Holo-CaM protein in our simulation remained in an extended conformation for the entire duration of the simulation, with some rotations of the lobes in relation to each other. On the other hand, the N-CaM had significant conformational changes, the most prominent of which occurred in the central helix, which bent at a certain point and allowed the two lobes to approach each other in a conformation quite similar to the compact structures seen in some crystals. Although the major conformational change itself occurred in a very short time span, the behavior of the N-CaM protein before the change was significantly different than the behavior of the Holo-CaM. This strengthens the notion that the removal of a Ca2+ ion led to increased instability which eventually drove to the compaction of the protein. Although it could be argued that the conformational transition between the extended and compact conformation may be much slower than the time span of the simulation, a previous independent simulation showed that structural collapse of the protein occurs at the ns timescale (21). In addition, a recent independent experimental study on a calmodulin-like protein containing three active Ca2+ binding sites, shows the 3Ca2+-bound protein to be in a compact conformation, consistent with our results (24).
MATERIALS AND METHODS
The MD simulations were performed using the GROMACS 3.21 package of programs (25–27) with the GROMOS96 force field (28). The calculations were carried out using the crystal structure of the fully Ca2+ bound calmodulin protein (Protein Data Bank code 1CLL) determined by x-ray crystallography at 1.7 Å (3), that was downloaded from the Protein Data Bank (29). The simulation was run twice, once with all four Ca2+ ions bound (Holo-CaM) and once with the Ca2+ ion between the helices A and B at the N-terminal end removed (N-CaM). Both proteins were embedded in a box containing the SPC water model (30), which extended to at least 12 Å between the protein and the edge of the box. Assuming normal charge states of ionizable groups corresponding to pH 7, the net charge of the protein plus the bound Ca2+ ions is −16e and −18e for the Holo-CaM and N-CaM, respectively. To neutralize the system and reach physiological ionic strength of 200mM, 34 sodium, 56 chloride, and 20 calcium ions were added for the Holo-CaM and 33 sodium, 57 chloride, and 20 calcium ions were added for the N-CaM, in random positions. Before the dynamics simulation, internal constraints were relaxed by energy minimization. After the minimization, an MD equilibration run was performed under position restraints for 20 ps followed by a 500-ps unrestrained equilibration run. After the equilibration, an MD production run was performed for a duration of 12.7 ns. During the MD run, the LINCS algorithm (31) was used to constrain the lengths of all bonds; the waters were restrained using the SETTLE algorithm (32). The time step for the simulation was 2 fs. The simulations were run under NPT conditions, using Berendsen's coupling algorithm for keeping the temperature and the pressure constant (P = 1 bar; τP = 0.5 ps; T = 300°K; τT = 0.1 ps) (33). VDW forces and short range electrostatic interactions were treated using a cutoff of 12 Å. Long-range electrostatic forces were treated using the particle mesh Ewald (PME) method (34). The coordinates were saved every 1 ps. All protein figures were created using the VMD computer program (35). The electrostatic analysis presented in Fig. 7 was recalculated using a cutoff of 2.4 nm. The solvent accessible surface area (SASA) was calculated using the g_sas program available in Gromacs (36).
FIGURE 7.

Electrostatic interactions up to a cutoff distance of 2.4 nm of Holo-CaM (solid line) and N-CaM (dashed line) averaged over 100 ps.
Interhelical angles were calculated using the definition of the dot product for two vectors: 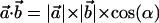 ,where each of the vectors
,where each of the vectors 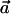 and
and 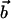 is taken as the vector from the average position of the six backbone atoms at the beginning of the helix to the average position of the six backbone atoms at the end of the helix, thus representing the direction of the helix.
is taken as the vector from the average position of the six backbone atoms at the beginning of the helix to the average position of the six backbone atoms at the end of the helix, thus representing the direction of the helix.  is the angle between the two vectors.
is the angle between the two vectors.
The entropy of the calmodulin molecule was calculated according to the quasi-harmonic assumption (37,38) by Eq. 1:
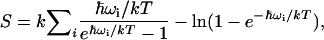 |
(1) |
where k is the Boltzmann constant, T is the absolute temperature, ωi are the quasiharmonic frequencies obtained from the diagonalized mass-weighed covariance matrix, and the summation is done on all eigenvalues. ωi is given by Eq. 2:
 |
(2) |
where λi are the eigenvalues of the diagonalized mass-weighted covariance matrix.
RESULTS AND DISCUSSION
The evolution of the calmodulin structure during the simulation
Two 12.7-ns simulations were performed: one with the CaM carrying 4 Ca2+ ions (Holo-CaM), and the other with a protein in which a single Ca2+ ion was removed out of the first ion binding site (between helices A and B) on the lower affinity N-lobe (N-CaM). The simulations were carried out in the presence of ample Ca2+ ions in the solution, so that the dynamics could detect both the structural effect of the ion stripping and a possible reoccupation of the site. The simulation of the Holo-CaM revealed a strong adherence to the initial structure, whereas the N-CaM protein, after intensive searching of the conformational space, suddenly collapsed upon itself to form a compact structure. This difference in shape and structural features is seen in Fig. 1. Frame A depicts the initial configuration of the Holo-CaM, as derived from the crystal structure. It has the typical dumbbell structure with a straight, extended interdomain helix. At the end of the simulation, the Holo-CaM (seen in frame B) retains the same general features, with minor rotations of the lobes, one with respect to the other, using the long axis of the interdomain helix as a pivot. In contrast to the Holo-CaM, the protein lacking a single Ca2+ ion from the low affinity site collapsed, exhibiting a new conformation where the interdomain helix lost its straight structure and both lobes established new contact areas with the interdomain helix and between themselves (compare frame C to A and B). The structural modulations, their dynamics and the forces which operate on the protein will be discussed below. To establish some reference for the evaluation of the gross structural modulation of the N-CaM protein, we shall first briefly discuss the results of the simulation carried out with the Holo-CaM protein.
FIGURE 1.

Structures of calmodulin. Frame A depicts the structure of the Holo-CaM as provided by the Protein Data Bank (PDB id: 1CLL). Frame B depicts the structure of Holo-CaM at the end of a 12.7-ns simulation, as described in Materials and Methods. Frame C presents the structure at the end of a 12.7-ns simulation of the protein with a single Ca2+ ion removed from the low-affinity, N-terminal site on the N-lobe. The N-lobe is colored in yellow, the central helix is in blue, and the C-lobe in cyan.
Holo-CaM protein
The root mean-square deviation (RMSD) value, calculated for the Holo-CaM protein backbone is presented in Fig. 2, bottom panel. The RMSD calculated for the whole protein increases to a maximum of ∼0.8 nm at t ∼ 4 ns and then decreases to a relatively stable value of ∼0.5 nm. This modulation of the RMSD values suggests that the protein samples the conformation space with no tendency to drift into new conformations that grossly differ from the initial one. To gain a better understanding of the states that the protein is sampling, we calculated the RMSD for the three structural elements of the protein; namely the N-lobe (residues 5–65), central helix (residues 66–92), and C-lobe (residues 93–147). These values are presented in Fig. 2, bottom panel, using red, green, and blue colors, respectively. It should be noted that although the total protein RMSD value appears to be quite large, the values calculated for the structural elements are much smaller. This is an indication that the lobes retain a rigid structure, while shifting their relative positions.
FIGURE 2.

Changes in structural parameters of Holo-CaM as a function of simulation time. (Top) The virtual dihedral angle for the Holo-CaM protein, The VDA is related to the right side ordinate. For more details, see Fig. 5. (Bottom) The RMSDs of backbone atoms are presented in solid lines for the backbone of the entire protein (black), or for defined domains: the N-lobe (red), the central helix (green), and the C-lobe (blue). The interhelical angle between helices A (residues 1–12) and B (residues 24–34) is presented as a dashed lined (black). The RMSDs are related to the left ordinate and the interhelical angle to the right one.
The RMSD fluctuations can be assigned to defined structural changes that are directly traced. Of the three structural elements, the interdomain helix is rigid with an RMSD of <0.2 nm, which is persistent during the whole length of the simulation. The RMSD of the C-lobe exhibits a minor increment during the first ∼1 ns of the simulation time, and then it maintains a constant value, in accordance with the general shape of the lobe as presented in Fig. 1, frames A and B. The only section of the lobe that exhibited an enhanced degree of freedom consisted of few residues located on the loop connecting helices F and G. The N-lobe had the largest RMSD value of all structural elements of the protein. During the first ∼2.5 ns the lobe seems very stable, but between 2.5 and 4.5 ns, there is a gradual structural transformation that increases the RMSD to ∼0.4 nm. Indeed, the N-lobe, as seen at the end of the simulation (Fig. 1, A and B), appears to be rotated with respect to its initial configuration. This rotation can be expressed by the interhelical angle between helices A and B, which shifts from ∼90° to ∼125° around this time as can be seen in Fig. 2, bottom panel, which shows a near perfect match between the change in the N-lobe RMSD (red line) and the interhelical angle (dashed line).
Examination of the RMSD value of the Holo-CaM reveals some features that appear to be a property of the whole structure, not assignable to any of the structural elements. These are represented by the relative rotation of the C- and N-lobes with respect to the interdomain helix, which acts as a pivot. This rotation is depicted in the top panel of Fig. 2, where the variation of the virtual dihedral angle (VDA, as defined in Fig. 5) is given as a function of time. Comparison of the VDA trace to the total RMSD of the protein (bottom panel, solid black line) clearly demonstrates that the peaks in RMSD value (at t ∼ 4000 and ∼ 7500 ps) are coupled with the increase of the VDA by ∼90°.
FIGURE 5.

Virtual dihedral angle between the planes defined by the two lobes and the central helix. Each plane was defined by the central helix (colored cyan) Cα atoms of residues Phe-92 and Pro-66 and by the Cα atom of residue Ala-46 in the N-lobe (blue) and the Cα atom of residue Glu-123 in the C-lobe (red). The atoms defining the planes are marked in yellow. The angle at t = 0 is illustrated in frame A. The angle of the N-CaM at t = 4 ns is presented in frame B.
Other parameters reflecting the conformational changes experienced by the Holo-CaM will be discussed during the analysis of the modifications the N-CaM protein has gone through, using the simulation of the Holo-CaM as a reference.
Our results are comparable to previous simulations of the Holo-CaM for 4 ns by Komeji et al. (17), 4 ns by Yang and co-workers (23), and 3 ns by Wriggers et al. (39). In those simulations too, the protein retained its dumbbell conformation with RMSD values of 0.4 to 0.6 nm, strengthening the impression of the relative stablility of the extended conformation of the Holo-CaM. The compaction of the Holo CaM, as simulated by (21) in the absence of screening electrolyte appeared as a rather slow process. The folding was initiated within the first nanosecond and proceeded for almost 5 ns. These dynamics differ markedly from that reported in this study and may be attributed to the absence of the screening electrolyte and their contribution to the electrostatic potential (40,41).
N-CaM protein
The RMSD of the N-CaM backbone, as it varies with time, is shown in Fig. 3, top panel. It can be seen that during the simulation, the N-CaM structure undergoes major conformational changes detected as a large increment of the RMSD at ∼7500 ps. During the first 7.5 ns of the simulation time, the protein remains rather stable, exhibiting a single reversible structural modulation during the ∼1.5–4.5 ns, where the RMSD was increased to ∼0.75 nm. This transient is reminiscent of the fluctuations detected for the Holo-CaM. After ∼7.5 ns, there is a fast fivefold increase in the RMSD, which reaches an apparent stable level within <0.5 ns. Examination of the lobes' RMSD values reveals that the C-lobe remained stable, with no increment at ∼7.5 ns (Fig. 3, top panel, blue trace). The N-lobe (red trace) exhibits a minor increase of the RMSD, which is delayed with respect to the total value calculated for the protein. As both lobes remain stable, the high RMSD reached by the whole protein is clearly the result of a structural change in the central helix (green trace). To ascertain that the constant RMSD after t = 8 ns does not conceal fluctuations between states with similar RMSD, the RMSD values were recalculated from that time point until the end of the run, using as a reference the atomic coordinates of the snapshot structure at 8000 ps. The RMSD values for the three structural domains are in the 0.2–0.35 nm range (data not shown), indicating that the structure gained at the end of the simulation is stable. The structural change that occurred at 7.5 ns will be termed, for reasons that will be discussed below, as the compaction event.
FIGURE 3.

Changes in structural parameters of Calmodulin with one Ca2+ ion removed from the N-lobe (N-CaM) as a function of simulation time. (Top) The RMSD value calculated for the heavy atoms of the protein's backbone. The color code refers to the RMSD of the total protein (black), N-lobe (red) interdomain helix (green), and the C-lobe (blue). (Middle) Quantitative description of the contraction of N-CaM molecule during the simulation; the traces correspond with the distance between the lobe's center-of-mass (black), the length of the interdomain helix (red), and the shortest distance between residues on C- and N-lobes (green). For details see Materials and Methods. (Bottom) The virtual dihedral angle (as defined in Fig. 5) of N-CaM at t < 7.5 ns (i.e., before the compaction event).
Since there are no previous simulations on N-CaM, we cannot directly compare our results to previous work. However, evidence of CaM destabilization upon removal of Ca2+ ions can be seen in the simulations of Apo-CaM carried out by Komeji and co-workers (17). In their 4-ns simulation, the Apo-CaM was much less stable than the Holo-CaM. The RMSDs of the Apo-CaM reached values of 1.1, 0.5, 0.4, and 0.3 nm for the total protein, N-lobe, central helix, and the C-lobe, respectively. These values are higher than the RMSD values calculated for the N-CaM in this simulation during the first 4 ns. This may hint for a correlation between the number of bound Ca2+ ions and the stability of the protein. However, to confirm this hypothesis, the simulations should be compared under the exact same conditions and with several repetitions.
The gyration radii of the proteins as a function of simulation time are shown in Fig. 4, bottom panel. During the simulation of the Holo-CaM, there was a slow decrease of the gyration radius from 2.2 nm (as calculated for the minimized crystal structure) down to 1.9 nm. This compaction appears to be a slow process and is attributed mostly to the change of the interhelical angle between helices A and B and the VDA between the lobes (see Fig. 2, bottom panel, dashed line, and top panel). The simulation of the N-CaM exhibits a completely different scenario; at t = 7500 ps the gyration radius drops rapidly to 1.7 nm and remains stable till the end of the run.
FIGURE 4.

Differences in structural parameters between Holo-CaM and N-CaM. (Top) The dynamics of the bending of the interdomain helix during the simulations of the Holo-CaM (solid line) and the N-CaM protein (dashed line). The interhelical angle is defined between the helix at the N-terminal side of the central helix (residues 61–72) and the helix at the C-terminal side of the central helix (residues 83–88). Angles were averaged over 100 ps to reduce their fluctuations. (Bottom) The variation of the gyration radius of the Holo protein (solid line) and of N-CaM (dashed line) during the simulation. The radii were averaged over 100 ps to reduce fluctuations.
An additional evaluation of the structural changes experienced by the N-CaM is presented in the middle panel of Fig. 3, by three structural features. The separation between centers of mass of the N-CaM protein (black trace) retains a constant value up to the moment of the compaction event (t = 7.5 ns), thus resembling the modulation of the radius of gyration (see Fig. 4, bottom panel). Similarly, the length of the interdomain helix (Fig. 3, red trace) is also constant until the point of compaction. On the other hand, the minimal distance between the lobes in N-CaM (green trace) shows great variability. It decreases gradually from 2.2 nm to 0.52 nm during the first 4 ns. Then, within a brief period (0.3 ns), it increases sharply to 2 nm, where it remains until 7 ns. It increases back to the original value just before the compacting event that finally reduces the value to ∼0.5 nm. Comparison of these variations before t = 7.5 ns with the stable pattern of the other two parameters presented in the panel, indicates some kind of conformational changes involving the lobes. The low RMSD values of the lobes imply that the observed approach between them must stem from a relative rotation, one toward the other, while maintaining a constant separation between their centers of mass.
This relative motion is expressed by the VDA between the lobes. This VDA is defined in Fig. 5: frames A and B present the protein at times 0 ns and 4 ns, respectively. It should be stressed that this VDA is meaningful only as long as the central helix retains its straight conformation. For this reason the VDA is drawn in the bottom panel of Fig. 3 only until t = 7.5 ns, the time point where the central helix is snapped into two helices (as discussed below). For the same reason, the VDA at t ∼ 6.5 (in which the central helix temporarily bent) does not represent the relative movement of the lobes.
The inspection of Fig. 5, frames A and B, reveals how the N-lobe rotates as a rigid unit, enabling its first helix (helix A) to approach the last helix (helix H) of the C-lobe. The variation of the VDA of the N-CaM protein as a function of the simulation time, up to the point when the massive compaction takes place, is detailed in the bottom panel of Fig. 3. The changes in the minimum distance between the lobes that occur up to t ∼ 4.5 ns, as presented in the middle panel of Fig. 3 (green tracing), can now be explained by a corresponding change in the dihedral angle up to this time.
At t = 7.5 ns the minimal distance between the lobes drops sharply to 0.6 nm (see Fig. 3, middle panel), where it remains until the rest of the simulation. At this time, the two helices, each from another lobe, are in physical contact (the contact is clearly identified by the variation with time of the Lennard Jones potential, as seen in bottom of Fig. 6). This reduction in the minimum distance between the lobes correlates with the drop in the separation between the edges of the central helix and between the mass centers of the lobes. This indicates that a major deformation of the central helix, which brings the two lobes closer to each other, has occurred. The deformation of the central helix can be clearly seen by visual inspection of the structures, where a breakage of the α-helix into two sections is clearly observed (compare Fig. 1, frame C, to Fig. 1, frames A and B). The angle between the two end sections of the interdomain helix, as measured for the Holo-CaM, is almost constant over the whole simulation time (Fig. 4, top panel), exhibiting only minor, rapid fluctuations that are consistent with the flexibility of the protein's structure. The N-CaM structure exhibits the same general features for the first 7.5 ns. However, at t = 7.5 ns, the angle between the two end sections of the interdomain helix increases, in <100 ps, from ∼15° to ∼150°. The time point of the interdomain helix bending matches the actual compaction event, which brings the two lobes close to each other.
FIGURE 6.

Interaction energies between the structural components of N-CaM. (Top) LJ energies (solid line) and the SR columbic interactions (dashed line) between the lobes and the interdomain helix averaged over 500 ps to reduce their fluctuations. (Bottom) LJ energies (solid line) and SR columbic interaction (dashed line) between the two lobes averaged over 100 ps.
The bending of the central helix in CaM was evidenced in some crystallographic structures and previous computational studies. In the crystal structures of both Apo-CaM (14) and Holo-CaM (19), the turn in the central helix occurs between residues 77 and 81. In the molecular dynamics simulation of the Holo-CaM at low ionic strength (21), the compaction of the protein stemmed from a loss of the helical structures between residues 75–79 and 82–86. In a simulation of the central helix without the lobes (42), the central helix formed a turn between residues 72 and 76 and in a simulation of the Holo protein (39) it bends locally near residue 74. The turn in the central helix of N-CaM at the end of our simulation consisted of residues 81–86. Apparently, the flexibility of the central helix in the N-CaM is an inherent property of the protein as the bending of the central helix has been witnessed twice in the simulation before the compaction event, at ∼2 ns and at 6.5 ns. Yet, in both cases, the deformations were not carried out to a sufficient extent to cause the major compaction seen at 7.5 ns. However, these bending events and the start of the compaction event all occur between Asp-78 and Glu-82 and between Lys-75 and Asp-78, respectively, which imply that this region is the weak spot of the helix. This is in agreement with the bending locations as seen in crystallographic data of compact calmodulin.
The effect of compaction on the solvent accessible surface area (SASA) of the protein was calculated for both the hydrophobic and the hydrophilic residues. Both parameters exhibited significant reduction. The hydrophobic SASA was reduced from 53 to 46 nm2 (18%) and the hydrophilic SASA dropped from 45 to 41 nm2 (11%). The reduction in SASA occurred at the beginning of the simulation and at t ∼ 7500 ps (which correspond to the compaction event) and at t ∼ 11,500 ps. The reduction of the total SASA of the protein resulted in a release of ∼35 water molecules to the bulk.
Energetic analysis of the compaction event
The compaction of the protein, initiated at ∼7500 ps, leads to the formation of a new conformation of the protein that exhibits no tendency to regain the initial state during the simulation. Considering the physiological behavior of calmodulin, shifting reversibly between the open and the closed states as a function of the intracellular Ca2+ concentration, one should not expect to find a high energy barrier, nor large differences in the free energy, between the initial and the compact states. Although MD simulations of the protein cannot yield thermodynamic parameters, one can directly calculate the short range Coulomb electrostatic (SR) and the Lennard Jones (LJ) components of the potential energy from the simulations. To analyze the factors which stabilize the compact state, the variations of these terms as a function of the simulation time are discussed below.
It must be recalled that to gain a comprehensive evaluation of the energetic aspects of the process, one needs to examine not only the protein but also its interaction with the solvent, and the solvent-solvent interactions. However, parameters such as the solvent's cavitation energy, solvent's entropy, solvation energy, and the long-range electrostatic energies in a periodic system pose both practical and conceptual difficulties when dealing with MD simulations. Therefore, we shall limit our discussion only to certain, well-understood aspects protein's energy, being aware that only part of the overall system is analyzed.
The intraprotein interactions were calculated between the three structural elements, the N- and C-lobes, and the interdomain helix. The lobes-central helix interaction energies (Fig. 6, top) clearly shows a decrease in the LJ interactions (solid line). Interestingly, this reduction is established in steps. The solid line reveals that the LJ stabilization, due to a better fitting of the contact between the structural elements, takes place both at the beginning of the simulation (during the first 3.5 ns), and later in two more phases. The first coincides with the compaction, whereas the second is near t = ∼10,500 ps time point, where no distinct structural change appears to take place.
The lobe-lobe LJ potential (bottom panel, solid line) is practically zero until the compaction event. Then, it decreases by as much as 40 kJ/mol. This interaction, however, does not stabilize in the scope of the simulation. Given that the LJ potential is a function of r−12, very small changes in the interatomic distances may suffice to drive sharp fluctuations in the LJ energy, making it harder for the potential to stabilize.
A similar analysis was carried out for the SR electrostatic interactions. However, it should be noted, that due to the nature of the electrostatic calculations in our analysis, the SR interactions should be considered only at a qualitative level. This is due to the fact that when computing the SR electrostatic energy between two groups in the protein, the screening effects by the solvent are not taken into account. Therefore, the absolute values of the electrostatic interaction may be equal to or lower than the results presented in Fig. 6. Nevertheless, the trends in the variation of the energy correctly represent the driving process in the simulation.
In contrast with the lobes-central-helix LJ interactions, the lobes-central-helix SR energy (top panel, dashed line), does not become more favorable before the compaction event. However, once the compaction began, the lobes-central-helix SR reduced substantially at t = 7500–10,500 ps. This appears to occur in two phases which correspond to the changes recorded for the LJ potential.
The lobe-lobe SR interactions (bottom panel, dashed line), in similarity with the LJ potential, has a zero value before the main compaction event. However, after some 3 ns of a relatively large range variations, the Coulomb potential manages to stabilize at t = ∼10,700 ps. It is of interest to point out that neither the RMSD values nor the interhelical angles reveal any significant changes at that time point. The ability of the SR to stabilize at the end of the simulation may be explained by fact that it is a function of r−1, which makes it less sensitive to distances than the LJ potential.
To ascertain that the difference in structural behavior between Holo-CaM and N-CaM is an inherent feature caused by the removal of the Ca2+ ion, we have examined the electrostatic interaction of the N-CaM before the compaction event and compared it to that of the Holo protein. As can be seen in Fig. 7, there is a significant difference between the variability of the electrostatic interactions between the simulations. Although the Holo-CaM electrostatic interactions seem to follow a well-defined average ∼−11,800 kJ/mol the N-CaM electrostatic interactions fluctuate over a much wider range.
In addition to the potential energy terms, one should also consider the entropic changes associated with the compaction of the protein. The enhanced association between the lobes reduces the freedom of motion of the interacting residues, thus affecting the entropy of the system. To estimate the contribution of this term, we calculated the entropy of the protein, before and after the compaction event. These calculations indicated that the entropy of the protein has decreased, upon compaction, from an average value of 18.7 ± 0.050 kJ/mol K (calculated in time frames of 1000 ps between 500 and 6500 ps) to 18.5 ± 0.099 kJ/mol K (calculated in time frames of 1000 ps between 8500 and 12,500 ps). This is equivalent to an entropic destabilization of the folded conformation by ∼52 kJ/mol (300 K). This decrease of the protein's entropy is due to formation of the new tight and compact conformation of the protein at the end of the simulation. However, the entropic gain for each water molecule released to the bulk during the compaction process, can be as high as 29 kJ/mol (43) which results in an upper bound of 1024 kJ/mol for the 35 water molecules released to the bulk. Of course, it is expected that the entropic gain will be much less since the released water are surface water, but this may still be sufficient to balance the entropic loss of the protein.
CONCLUSIONS
A 12.7-ns MD simulation of the Holo-CaM, under physiological conditions (I ≥ 100 mM, T ∼ 30°C), shows that the protein retains its dumbbell conformation in solution. Once one of the Ca2+ ions was removed (and we selected a Ca2+ ion located on the lower affinity N-lobe domain), the protein became less stable, which led to its rearrangement into a compact state. This transition is initiated by the random rotation of the lobes around the pivot made by the long axis of the interdomain helix. During these rotations, the interdomain helix, at its weak section (between residues 77 and 86), loses its helical form and snaps the central-helix into two helices. This conformational change is driven by the favorable electrostatic and LJ interactions between the lobes, and between each lobe and the interdomain helix. On the other hand, a loss of the protein's entropy reduces the favorability of this process. As a result, the protein may retain its capacity to recapture a Ca2+ ion and revert to the initial configuration without the necessity to have an external energy source to drive this conformation change.
Acknowledgments
The authors acknowledge the use of computer resources belonging to the High Performance Computing Unit, a division of the Inter-University Computing Center in Israel, and to the Bioinformatics Unit at the Tel Aviv University. R.F. acknowledges the Colton Foundation for its support through the Colton Scholarship.
This research is supported by the United States-Israel Binational Science Foundation (grant No. 2002129).
References
- 1.Chin, D., and A. R. Means. 2000. Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 10:322–328. [DOI] [PubMed] [Google Scholar]
- 2.Babu, Y. S., C. E. Bugg, and W. J. Cook. 1988. Structure of calmodulin refined at 2.2 Å resolution. J. Mol. Biol. 204:191–204. [DOI] [PubMed] [Google Scholar]
- 3.Chattopadhyaya, R., W. E. Meador, A. R. Means, and F. A. Quiocho. 1992. Calmodulin structure refined at 1.7 Å resolution. J. Mol. Biol. 228:1177–1192. [DOI] [PubMed] [Google Scholar]
- 4.Taylor, D. A., J. S. Sack, J. F. Maune, K. Beckingham, and F. A. Quiocho. 1991. Structure of a recombinant calmodulin from Drosophila melanogaster refined at 2.2-Å resolution. J. Biol. Chem. 266:21375–21380. [DOI] [PubMed] [Google Scholar]
- 5.Rao, S. T., S. Wu, K. A. Satyshur, K. Y. Ling, C. Kung, and M. Sundaralingam. 1993. Structure of Paramecium tetraurelia calmodulin at 1.8 Å resolution. Protein Sci. 2:436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabernero, L., D. A. Taylor, R. J. Chandross, M. F. VanBerkum, A. R. Means, F. A. Quiocho, and J. S. Sack. 1997. The structure of a calmodulin mutant with a deletion in the central helix: implications for molecular recognition and protein binding. Structure. 5:613–622. [DOI] [PubMed] [Google Scholar]
- 7.Wilson, M. A., and A. T. Brunger. 2000. The 1.0 Å crystal structure of Ca2+-bound calmodulin: an analysis of disorder and implications for functionally relevant plasticity. J. Mol. Biol. 301:1237–1256. [DOI] [PubMed] [Google Scholar]
- 8.Yun, C. H., J. Bai, D. Y. Sun, D. F. Cui, W. R. Chang, and D. C. Liang. 2004. Structure of potato calmodulin PCM6: the first report of the three-dimensional structure of a plant calmodulin. Acta Crystallogr. D Biol. Crystallogr. 60:1214–1219. [DOI] [PubMed] [Google Scholar]
- 9.Han, B. G., M. Han, H. Sui, P. Yaswen, P. J. Walian, and B. K. Jap. 2002. Crystal structure of human calmodulin-like protein: insights into its functional role. FEBS Lett. 521:24–30. [DOI] [PubMed] [Google Scholar]
- 10.Grabarek, Z. 2005. Structure of a trapped intermediate of calmodulin: calcium regulation of EF-hand proteins from a new perspective. J. Mol. Biol. 346:1351–1366. [DOI] [PubMed] [Google Scholar]
- 11.Kuboniwa, H., N. Tjandra, S. Grzesiek, H. Ren, C. B. Klee, and A. Bax. 1995. Solution structure of calcium-free calmodulin. Nat. Struct. Biol. 2:768–776. [DOI] [PubMed] [Google Scholar]
- 12.Zhang, M., T. Tanaka, and M. Ikura. 1995. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 2:758–767. [DOI] [PubMed] [Google Scholar]
- 13.Ishida, H., K. Nakashima, Y. Kumaki, M. Nakata, K. Hikichi, and M. Yazawa. 2002. The solution structure of apocalmodulin from Saccharomyces cerevisiae implies a mechanism for its unique Ca2+ binding property. Biochemistry. 41:15536–15542. [DOI] [PubMed] [Google Scholar]
- 14.Schumacher, M. A., M. Crum, and M. C. Miller. 2004. Crystal structures of apocalmodulin and an apocalmodulin/SK potassium channel gating domain complex. Structure. 12:849–860. [DOI] [PubMed] [Google Scholar]
- 15.Seaton, B. A., J. F. Head, D. M. Engelman, and F. M. Richards. 1985. Calcium-induced increase in the radius of gyration and maximum dimension of calmodulin measured by small-angle x-ray scattering. Biochemistry. 24:6740–6743. [DOI] [PubMed] [Google Scholar]
- 16.Kataoka, M., J. F. Head, A. Persechini, R. H. Kretsinger, and D. M. Engelman. 1991. Small-angle x-ray scattering studies of calmodulin mutants with deletions in the linker region of the central helix indicate that the linker region retains a predominantly alpha-helical conformation. Biochemistry. 30:1188–1192. [DOI] [PubMed] [Google Scholar]
- 17.Komeiji, Y., Y. Ueno, and M. Uebayasi. 2002. Molecular dynamics simulations revealed Ca2+-dependent conformational change of calmodulin. FEBS Lett. 521:133–139. [DOI] [PubMed] [Google Scholar]
- 18.Heidorn, D. B., and J. Trewhella. 1988. Comparison of the crystal and solution structures of calmodulin and troponin C. Biochemistry. 27:909–915. [DOI] [PubMed] [Google Scholar]
- 19.Fallon, J. L., and F. A. Quiocho. 2003. A closed compact structure of native Ca2+-calmodulin. Structure. 11:1303–1307. [DOI] [PubMed] [Google Scholar]
- 20.Barbato, G., M. Ikura, L. E. Kay, R. W. Pastor, and A. Bax. 1992. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry. 31:5269–5278. [DOI] [PubMed] [Google Scholar]
- 21.Shepherd, C. M., and H. J. Vogel. 2004. A molecular dynamics study of Ca2+-calmodulin: evidence of interdomain coupling and structural collapse on the nanosecond timescale. Biophys. J. 87:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiorin, G., R. R. Biekofsky, A. Pastore, and P. Carloni. 2005. Unwinding the helical linker of calcium-loaded calmodulin: a molecular dynamics study. Proteins. 61:829–839. [DOI] [PubMed] [Google Scholar]
- 23.Yang, C., G. S. Jas, and K. Kuczera. 2001. Structure and dynamics of calcium-activated calmodulin in solution. J. Biomol. Struct. Dyn. 19:247–271. [DOI] [PubMed] [Google Scholar]
- 24.Rabah, G., R. Popescu, J. A. Cox, Y. Engelborghs, and C. T. Craescu. 2005. Solution structure and internal dynamics of NSCP, a compact calcium-binding protein. FEBS J. 272:2022–2036. [DOI] [PubMed] [Google Scholar]
- 25.Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Comm. 91:43–56. [Google Scholar]
- 26.Lindahl, E., B. Hess, and E. Van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. (Online). 7:306–317. [Google Scholar]
- 27.van Der Spoel, D., E. Lindahl, B. Hess, A. R. van Buuren, E. Apol, P. J. Meulenhoff, D. P. Tieleman, A. L. T. M. Sijbers, A. K. Feenstra, R. van Drunen, and H. J. C. Berendsen. 2004. GROningen Machine for Molecular Simulations. Groningen, The Netherlands.
- 28.van Gunsteren, W. F., S. R. Billeter, A. A. Eising, P. H. Hunenberger, P. Kruger, A. E. Mark, W. R. P. Scott, and I. G. Tironi. 1996. Biomolecular Simulation: The GROMOS96 Manual and User Guide. Vdf Hochschulverlag AG an der ETH Zurich, Zurich:1–1024.
- 29.Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, and J. Hermans. 1969. Interaction models for water in relation to protein hydration. Nature. 224:175–177.5343518 [Google Scholar]
- 31.Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- 32.Miyamoto, S., and P. A. Kollman. 1992. SETTLE: an analytical version of the SHAKE and RATTLE algorithms for rigid water models. J. Comput. Chem. 13:952–962. [Google Scholar]
- 33.Berendsen, H. J. C., J. P. M. Postma, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 34.Essman, U., L. Perela, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pederse. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8592. [Google Scholar]
- 35.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: Visual Molecular Dynamics. J. Mol. Graph. 14:33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- 36.Eisenhaber, F., P. Lijnzaad, P. Argos, C. Sander, and M. Scharf. 1995. The double cube lattice method: efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J. Comput Chem. 16:273–284. [Google Scholar]
- 37.Levy, R. M., M. Karplus, J. Kushick, and D. Perahia. 1984. Evaluation of the configurational entropy for proteins: application to molecular-dynamics simulations of an alpha-helix. Macromolecules. 17:1370–1374. [Google Scholar]
- 38.Andricioaei, I., and M. Karplus. 2001. On the calculation of entropy from covariance matrices of the atomic fluctuations. J. Chem. Phys. 115:6289–6292. [Google Scholar]
- 39.Wriggers, W., E. Mehler, F. Pitici, H. Weinstein, and K. Schulten. 1998. Structure and dynamics of calmodulin in solution. Biophys. J. 74:1622–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson, J. D. 1975. Classical Electrodynamics. John Wiley & Sons, New York.
- 41.Solmajer, T., and E. L. Mehler. 1991. Electrostatic screening in molecular dynamics simulations. Protein Eng. 4:911–917. [DOI] [PubMed] [Google Scholar]
- 42.van der Spoel, D., B. L. de Groot, S. Hayward, H. J. Berendsen, and H. J. Vogel. 1996. Bending of the calmodulin central helix: a theoretical study. Protein Sci. 5:2044–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunitz, J. D. 1994. The entropic cost of bound water in crystals and biomolecules. Science. 264:670. [DOI] [PubMed] [Google Scholar]