

Abstract
The rigid force fields currently used in molecular dynamics (MD) simulations of biomolecules are optimized for globular proteins. Whether they can also be used in MD simulations of membrane proteins is an important issue that needs to be resolved. Here we address this issue using the gramicidin A channel, which provides an ideal test case because of the simplicity of its structure and the availability of a wealth of functional data. Permeation properties of gramicidin A can be summarized as “it conducts monovalent cations, rejects anions, and binds divalent cations.” Hence, a comprehensive test should consider the energetics of permeation for all three types of ions. To that end, we construct the potential of mean force for K+, Cl−, and Ca2+ ions along the channel axis. For an independent check of the potential-of-mean-force results, we also calculate the free energy differences for these ions at the channel center and binding sites relative to bulk. We find that “rejection of anions” is satisfied but there are difficulties in accommodating the other two properties using the current MD force fields.
INTRODUCTION
A great deal of progress has been made in molecular dynamics (MD) simulations of biomolecules during the last two decades (for reviews see (1) and the accompanying articles in the special issue). MD simulations of realistic models of biomolecules are expected to contribute to our understanding of the structure-function relations in an essential way. Due to lack of structural information on membrane proteins, the initial efforts were mostly concentrated on simulations of globular proteins. An important exception here is the gramicidin A (gA) channel, whose structure has been known since 1971 (2). Thus starting with the work of Mackay et al. in 1984 (3), a growing number of MD simulations have been carried out for the gA system (for reviews see (4,5)). After the 1998 breakthrough of the MacKinnon group in determining the crystal structure of the KcsA potassium channel (6), there has been a great deal of interest in MD simulations of membrane proteins. The proliferation of relatively cheap computer clusters and the availability of user-friendly software have no doubt further fueled this interest. The literature on the subject is now growing at a rapid pace. For example, in a recent review article on simulations of membrane proteins (7), over 300 articles were cited, most of which were published in the last few years.
In most MD simulations of membrane proteins, rigid (i.e., nonpolarizable) force fields such as AMBER (8), CHARMM (9), or GROMACS (10) have been employed. These force fields are optimized under bulk conditions, and it is not clear from the outset that they will work for membrane proteins as well as globular ones. One worry is that in rigid force fields the polarization interaction is taken into account in a mean-field approximation by incorporating its effects in other interactions. While such an approximate treatment of polarization appears to have worked well in bulk water, there is no guarantee that the same force fields will work as well in a lipid environment because lipids have very different polarization characteristics to water. A recent semimicroscopic model calculation of the ionic free energies in the gA channel, which explicitly included polarization interaction, is very suggestive in this respect (11). Clearly one would like to ensure that the force fields employed in current MD simulations of membrane proteins produce reliable results before investing heavily in them. In terms of availability of both molecular structures and functional data, ion channels provide ideal testing grounds for this purpose. In particular, we favor the gA channel because it has one of the simplest structures in an open state and the amount of physiological data available for it is unmatched for any other channel.
A primary motivation for this study is that validation of a model should be based on as many measurable properties as possible. This point is particularly relevant for ion channels where there is an abundance of experimental data. In fact, in the case of the gA channel, the amount of available data is rather overwhelming (12–14). For our purposes, however, it will be sufficient to summarize the permeation properties of gA as “gA conducts monovalent cations, rejects anions and binds divalent cations.” As a pertinent example, we mention that there have been numerous model studies of the gA channel based on continuum electrostatics (15,16). Yet a full confrontation of such models with experimental data was attempted only recently (17,18). The results provided unequivocal evidence for the failure of continuum electrostatics in gA. Perhaps the clearest piece of evidence came from the inability of the model calculations to describe calcium binding and block of the gA channel regardless of the parameters employed. Calcium binding is one of the important descriptors of the gA function, but it was not considered in previous model studies using continuum electrostatics.
Inspection of the literature on MD simulations of the gA channel reveals a similar situation: almost all deal with monovalent cations and a few with anions, but there are no studies on binding of divalent cations (4,5). In summary, these MD studies show that rejection of anions can be accommodated within the current MD framework (19,20), but there are problems with the conductance of monovalent cations. For example, a recent potential of mean force (PMF) calculation employing the CHARMM force field found a 22 kT central barrier relative to the binding site and a −6 kT well-depth at the binding site for a K+ ion (21). The corresponding values obtained from the inversion of the available permeation data in gA using Brownian dynamics simulations are 5 kT for the barrier height and −8 kT for the well-depth (17). We emphasize that these values are well constrained because they are obtained using all the permeation data, not just a single conductance value (the other properties considered are the saturation of conductance with concentration and maintenance of a reasonable binding site). Extensive parameter studies in Edwards et al. (17) show that when only the conductance is considered, there is some variation in the allowed barrier and well values. But even then, the observed conductance value can be reproduced only when the barrier height is less than the absolute value of the well depth. This quantity corresponds to the stabilization energy of an ion calculated from the free energy difference when the ion is translocated from bulk to the channel center, and it must be negative. By that account, the error in the above K+ PMF is >16 kT, which translates to seven orders-of-magnitude error in conductance.
A more recent and improved calculation of the K+ PMF in gA—again obtained from MD simulations using the CHARMM force field—yielded 20 and −3 kT, respectively, for the barrier height and well depth, indicating similar errors for the stabilization energy and conductance (22). In Allen et al. (22), it was further estimated from continuum electrostatics calculations that inclusion of finite size effects and polarization of lipid molecules reduced the barrier height from 20 to 14 kT. The well-depth, however, remained at ∼−3 kT, which is somewhat shallow to explain the binding of K+ ions to gA and saturation of conductance with increasing concentration (17). Most importantly though, the stabilization energy of 11 kT is still too high and points to 4–5 orders-of-magnitude error in conductance. A much smaller discrepancy in conductance (only a factor of 30) was estimated in Allen et al. (22) using the one-dimensional Nernst-Planck equation. As pointed out by Levitt and others (23,24), the three-dimensional Brownian dynamics simulations provide a more reliable estimate of the channel conductance compared to the one-dimensional Nernst-Planck equation. Thus despite some improvements, there are still substantial errors in the calculated gA properties, and they need to be understood to ensure the reliability of MD simulations of membrane proteins.
One way to expedite the resolution of this debate is to consider further independent tests. As mentioned above, Ca2+ binding to gA has not been considered in MD simulations before, and hence its study would provide an additional probe for the force fields. For completeness, we present here a comprehensive study of the energetics of ion permeation that includes Cl− and K+ ions besides Ca2+, so that each ionic species with a distinct response to gA is represented under identical simulation protocols. We construct the PMF for each species of ions and compare the results with the observed behavior of these ions in gA as summarized above. We also consider the energetics for the binding of Ca2+ and Cl− ions as a pair to gA, which is suggested by some NMR experiments. For an independent validation of the PMF results, we calculate the free energy differences for a K+ ion at the channel center relative to bulk water using both the thermodynamic integration (TI) and free energy perturbation (FEP) methods. Similar calculations are carried out for a Cl− ion at the channel center and a Ca2+ ion at its binding site near the pore mouth.
METHODS
Model system and MD simulations
The simulation system is taken from a recent work, where the effects of peptide flexibility on ion permeation in gA were studied (25). Therefore, we give only a brief description of the system preparation and the MD protocols here. The model system is constructed using the VMD suite of software (26). The system consists of the gA dimer embedded in a bilayer of 96 dimyristoylphosphatidylcholine molecules and hydrated with ∼3200 water molecules. The 1MAG structure of Ketchem et al. (27) is used for the gA dimer. The initial structure is placed in an orthorhombic periodic box and equilibrated with surface-tension coupling until the surface area converged to the experimental lipid density of 60 Å2 per lipid (28). In the remaining simulations, the periodic box is fixed in the x and y directions at 60 and 52 Å, respectively, and a pressure coupling of 1 atm is applied in the z-direction, which results in an average box length of 64 Å in the z-direction. After lipid preparation, 24 water molecules in the reservoirs are replaced by 12 pairs of K+ and Cl− ions to create an electrolyte solution of ∼200 mM. This system is equilibrated for a total of 3 ns, where restraints applied to the gA atoms are gradually relaxed. For calcium simulations, two K+ ions in bulk water are replaced with a Ca2+ ion and a water molecule.
MD simulations are carried out using the NAMD code, version 2.5 (29) with the PARAM27 version of the CHARMM force field (9), which provides a complete set of parameters for all the atoms in the system. An NpT ensemble is used with periodic boundary conditions. Pressure is kept at 1 atm using the Langevin piston method with a damping coefficient of 5 ps−1 (30). Similarly, temperature is maintained at 298 K through Langevin damping with a coefficient of 5 ps−1. Electrostatic interactions are computed using the particle-mesh Ewald algorithm. The list of nonbonded interactions is truncated at 13.5 Å, and a switching cutoff distance of 10 Å is used for the Lennard-Jones interactions. A time-step of 2 fs is employed for all simulations. Trajectory data is written at 1-ps intervals during both equilibration and production runs.
Potential of mean force
The PMF of K+, Cl−, and Ca2+ ions along the gA channel axis are calculated using umbrella sampling (31), together with the weighted histogram analysis method (32,33). As the method was explained in Allen et al. (21) in some detail, we give a brief account here stressing only the differences in this work. Using an umbrella potential, we sample an ion's position at equal intervals along the channel axis during MD simulations of the system. The biased ion distributions obtained from the production runs are then unbiased and combined using the weighted histogram analysis method. In all cases, the ion coordinates are measured with respect to the center of mass of gA.
We employ umbrella potentials with a force constant of 12.5 kT/Å2 at 0.5 Å intervals. To avoid potential equilibration problems associated with dragging of an ion in the channel (21), here we have replaced individual water molecules in the pore with a K+ ion. This way we obtain 10 configurations with the ion placed at regular intervals along the channel axis. The K+ ion in each configuration then needs to be pushed by only 1 Å to either side to generate the full set of windows required in the PMF calculations. Outside the channel, where equilibration is not a problem, the ion is pushed along the central axis. A total of 81 windows covering the range [−20, 20] Å is employed for the K+ PMF. For Cl− and Ca2+ ions, the PMFs are found to rise steeply as the ions approach the pore mouth, indicating that sampling them inside the channel is not necessary. So for these ions, 23 windows covering the range [9, 20] Å are used in the construction of the PMF, including the one that considers the binding of Ca2+ and Cl− ions as a pair. In the case of Ca2+ ion, the PMF is calculated in two ways: first, with an additional biasing potential in the x and y directions to constrain it on the z axis; and second, without any radial restraints (all the umbrella potentials are unbiased during the final analysis).
For each window, the system is equilibrated for 200 ps and the trajectory data for ion positions is collected for 400 ps. To check the adequacy of this simulation time, we have performed a simple convergence test. We have divided the production data into four 100-ps sets and obtained separate PMFs from each set. All four partial PMFs are found to exhibit similar profiles, differing at most by a few kT from the total PMF. The observed deviations from the total PMF do not exhibit any definite trends, indicating that the system is equilibrated and exhibits statistical fluctuations around the average PMF profile.
The binding constant of an ion can be estimated from the integral of the one-dimensional PMF, W(z) (34,35),
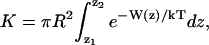 |
(1) |
where R is an effective radius for the pore, and the integration limits z1 and z2 are chosen in the bulk region where W vanishes. For the K+ PMF, adequate choices are R = 2 Å and z1, 2 = ± 15 Å.
Free energy difference calculations
Although the PMF calculations provide a detailed free energy profile for an ion permeating along the reaction coordinate, they are very time-consuming and require an inordinate amount of computing resources. If one is mainly interested in the height of the central barrier and the well-depth at the binding site for a given ion, these quantities can be computed with much less effort from free energy difference calculations. Naturally one needs to find the binding site first, but that can be achieved through a local simulation, i.e., a full PMF is not necessary. Because they require less time, the free energy difference calculations can be carried out for longer periods and hence offer a better handle for checking the convergence of the results.
Here we carry out free energy calculations for ions placed at the center of the channel and at the binding sites. They serve the dual purpose of providing an independent check on the PMF results and a tool for studying the convergence of free energy differences. We have used both the TI and the FEP methods in free energy calculations (36). In the former method, the free energy difference is obtained from
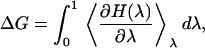 |
(2) |
where H(λ) = (1 − λ)H0 + λH1, with H0 and H1 representing the Hamiltonians of the initial and final states, respectively (e.g., if the initial state is an ion in the channel and a water molecule in bulk, in the final state these two are interchanged). The integral in Eq. 2 is performed using a Gaussian quadrature (37). We have experimented with various numbers of quadrature points (e.g., 3, 5, 7, and 12 points), and found that seven-point-quadrature provides sufficient accuracy for our purposes. This value is used in all subsequent calculations. The system is equilibrated for 200 ps for K+ and Cl− ions, and 500 ps for Ca2+ ion before production runs. In all three cases, the integrals are evaluated from 700 ps of production runs.
In the FEP method, the interval between λ = 0 and 1 is divided into k subintervals with {λi, i = 0, k}. For each λi value, the free energy difference is calculated from the ensemble average
 |
(3) |
The total free energy change is then obtained by summing the contributions from each subinterval, 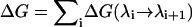 . The number of subintervals is chosen such that the free energy change at each step is at most 2 kT, otherwise the method loses its validity (36). For the K+ ion calculations described below, 40 subintervals have been used. Each window is equilibrated for 20 ps before a 30-ps production run. We have verified that doubling the simulation times (i.e., 40 ps equilibration and 60 ps production) results in a similar value—the difference is less than a kT, which is within the calculated statistical fluctuations.
. The number of subintervals is chosen such that the free energy change at each step is at most 2 kT, otherwise the method loses its validity (36). For the K+ ion calculations described below, 40 subintervals have been used. Each window is equilibrated for 20 ps before a 30-ps production run. We have verified that doubling the simulation times (i.e., 40 ps equilibration and 60 ps production) results in a similar value—the difference is less than a kT, which is within the calculated statistical fluctuations.
When a K+ ion is in the channel center, the dipoles of water molecules in the pore point away from the ion, whereas when there are only water molecules in the channel they all point in the same direction. Thus an alchemical transformation of a K+ ion in the channel center to a water molecule disrupts the orientation of one-half of the water molecules. The resulting fluctuations in the free energy calculations can be reduced by performing the transformation via an intermediate state with no charge, which we choose as a water molecule with the partial charges set to zero (denoted as W0). Thus we perform two calculations, ΔG(K+ → W0) and ΔG(W0 → W), whose sum gives the desired free energy change for the K+ → W transformation. Note that a similar transformation, W → W0 → K+, is carried out in bulk simultaneously to find the ionic free energy difference between the channel and bulk water.
RESULTS AND DISCUSSION
In the following, we present the ionic PMF and free energy differences obtained from MD simulations without applying any correction factors from continuum electrostatics. Our aim here is to test the predictive power of current MD force fields, and inclusion of correction factors from a lower-level theory that MD is supposed to replace would conflict with this aim. We note that the two main corrections suggested in Allen et al. (22) (i.e., finite system size and lipid polarizability) can be calculated within the MD framework. The first involves increasing the system size and monitoring the changes in the barrier height and well-depth for an ion from the free-energy calculations (work on this problem is in progress). The second includes corrections for the neglected polarization interaction, but only for the lipid molecules. As pointed out before (38), a self-consistent treatment of the polarization interaction is very important, and a partial incorporation of polarizability may introduce larger errors than would be found by completely neglecting it. Thus a fully polarizable MD force field is required for assessing the effect of the polarization interaction on the energetics of ion permeation.
Potential of mean force of ions
The results of the PMF calculations for K+, Cl−, and Ca2+ ions are shown in Fig. 1. The K+ PMF is similar to the previous MD results (21,22), when no correction factors are applied. The central barrier is 20 kT and the binding site has a depth of −7 kT. Thus the stabilization energy of the ion is 13 kT, which is smaller than the earlier results but still too large to describe the conductance data. While the barrier remains too large, the well-depth is close to the value of −8 kT obtained from the inversion of physiological data (17). Estimating the K+ binding constant from Eq. 1 yields K = 17.5 M−1, which is ∼3-times smaller than the experimental values obtained from NMR, e.g., K = 52.6 M−1 (39) and 60 M−1 (40). The exponential relationship between binding constant and the PMF suggests that the NMR results could be reproduced by a well-depth of −8 kT, consistent with the estimate obtained from the conductance data (17). The position of the binding site at 9.7 Å is in good agreement with the observed binding site for Tl+ at 9.6 Å (41), which is similar to K+ ion in size and hydration properties. In a similar PMF calculation that used the 1JNO structure for gA (42), the K+ binding site was found at 11.3 Å (22) with a depth of −3 kT. This suggests that 1MAG (27) may provide a better structure for describing the cation binding to gA.
FIGURE 1.

PMF profiles of K+, Cl−, and Ca2+ ions along the central axis of the gramicidin-A channel. In the calculation of the K+ PMF, the ion density is symmetrized around z = 0, so only the z > 0 portion is shown.
In the binding site, the K+ ion is coordinated by the carbonyl oxygens of Leu10, Leu12, Leu14, Trp13, Trp15, and two water molecules. To provide a better picture of the ion coordination, we show in Fig. 2 A the K+-O distance distribution for the Leu oxygens and the radial distribution function for the water oxygens (the latter quantity is chosen so as to keep the scales commensurate). The Trp oxygens are not shown to avoid cluttering of the figure but they are similar to those of Leu. It is clear from the distribution functions that the coordination shell is quite dynamic—only the Leu10 oxygen remains permanently in the first hydration shell, those of Leu12 and Leu14 make frequent excursions to the second hydration shell. This picture is consistent with the NMR experiments of Tian et al. (43,44), which show that the ion cannot interact with the three Leu oxygens simultaneously. On the other hand, the Leu10, Leu12, and Leu14 carbonyl dipoles deflect on average by 18°, 22°, and 27° upon K+ binding, which are substantially larger than the experimental results that limit such deflections to < 10° (43,44). The main reason for presenting the K+ ion results is to provide a reference point for the other ions, which is obtained under identical simulation conditions. So we do not dwell on the K+ ion results further here and turn to the PMFs of Cl− and Ca2+ ions, which have not been considered in MD simulations before.
FIGURE 2.

Distribution of ion-oxygen distances for the oxygens of water (solid line) and Leu10, Leu12, and Leu14 (dashed lines) when the K+ ion is in the binding site (A), and when the Ca2+ ion is in the binding site (B). To have similar scales with the Leu oxygens, the radial distribution function is shown for water oxygens (i.e., the distribution function is normalized by 4πr2Δr).
The profile for the Cl− PMF does not exhibit any special features. It steadily rises as the ion approaches the channel, reaching ∼20 kT at the pore mouth. The steep rise at 15 Å indicates that it is energetically unfavorable for the ion to lose any of its hydration waters to make contact with the peptide atoms. Overall, the PMF shows that Cl− ions have no chance of binding or entering the channel, and their rejection occurs outside the channel. Thus although gA is charge-neutral as a whole, the peptide charges are distributed such that it retains a significant valence selectivity due to the repulsive ion-dipole interactions. The most significant among these are the carbonyl groups of Leu10, Leu12, and Leu14 residues, whose oxygens point to the pore mouth, and as shown above play a critical role in the binding of a K+ ion.
The Ca2+ PMF exhibits a very weak binding site at ∼15 Å with a depth of 2 kT. MD simulations of a free Ca2+ ion in the binding site shows that it is slightly off-axis (1 ± 0.5 Å). To get a better view of the binding topology, we repeat the distribution function analysis in Fig. 2 A for a Ca2+ ion (Fig. 2 B). Integration of the radial distribution function in Fig. 2 B shows that the first hydration shell of Ca2+ contains, on average, 6.7 water molecules but a negligibly small number of Leu oxygens. The Leu oxygens are distributed near the second hydration shell and beyond, with the average distance of 6.8, 5.2, and 4.4 Å for Leu10, Leu12, and Leu14, respectively. Because the Ca2+ binding site is outside the channel, its binding causes negligible structural deformation on the peptide—the only notable change is that of Leu14 carbonyl, which deflects by 11° upon Ca2+ binding.
There are no direct experimental information on the binding site of a single Ca2+ ion, though indirect evidence from conductance measurements with mixed Cs+-Ca2+ solutions points to a binding site outside the channel (45,46). When symmetric solutions are used, increasing Ca2+ concentration reduces the Cs+ current, but even when Ca2+ is at 1 M level, the gA channel keeps conducting Cs+ ions (45). If a Ca2+ ion were to bind in the pore region, there would be an absolute block of the channel and no monovalent cations could pass through—a situation reminiscent of the Ca2+ channels (47). Similar experiments with asymmetric solutions that contain Ca2+ only on one side show that the Ca2+ block effect is observed on the side with Ca2+ ions as before, but there is no hindrance of Cs+ current from the other side that has no Ca2+ ions (46). Again binding of a Ca2+ ion in the pore would block the Cs+ current in both directions. These experiments are consistent with the structural information from x-ray and NMR experiments, which show that the divalent ions bind outside the gA channel (41,44,48). Therefore, to interpret the results we rely on those available for Ba2+ and Mn2+, whose properties bracket that of Ca2+ (e.g., the sizes of Mn, Ca, and Ba are 0.80, 0.99, and 1.35 Å, respectively, and the hydration energies are −437, −380, and −314 kcal/mol, respectively). The binding site of Ba2+ is found at 13 ± 0.2 Å (41), whereas the binding site of Mn2+ is specified by its distance from the Leu oxygens: 8.8, 6.4, and 8.6 Å for Leu10, Leu12, and Leu14, respectively, with an error of 2.1 Å (48). Also the Mn2+ ion is found to be off-axis by 3 ± 1.4. Å. Due to large errors it is difficult to estimate the precise position of the Mn2+ binding site but it is ∼18 Å. Thus the Ca2+ ion occupies an intermediate position between those of Ba2+ and Mn2+, as one would expect from their hydration energies. Because of its smaller hydration energy, it is easier for a Ba2+ ion to exchange one of its water molecules with a Leu oxygen, enabling it to come closer to the channel. Conversely, due to its higher hydration energy, such an exchange is not possible for a Mn2+ ion, which stays away from the channel. As seen in Fig. 2 B, the Ca2+-Leu14 oxygen distance fluctuates between the first and second shells, indicating that binding of water molecules to Ca2+ is intermediate between those of Ba2+ and Mn2+, consistent with their hydration energies.
The rapid rise in the PMF for z < 14 Å indicates that the Ca2+ ion would like to hold on to its first hydration shell. That is, it is energetically unfavorable for the ion to exchange the water in its hydration shell with the carbonyl oxygens of Leu10, Leu12, and Leu14 residues as in the case of monovalent cations. Instead, the water molecules in the hydration shell of the Ca2+ ion are observed to make hydrogen bonds with these residues. If held in this position, the Ca2+ ion can clearly block the channel and hinder permeation of monovalent ions as observed experimentally (45,46). However, the calculated binding energy of 2 kT is too small to bind a Ca2+ ion to gA, and more importantly, it is smaller than that for a K+ ion by 5 kT. To block the channel, the affinity of Ca2+ ions to gA must be much larger than that of K+ ions. This suggest that the error in the calculated binding energy of Ca2+ ion is much greater that 5 kT.
The Ca2+ PMF in the figure is obtained using a radial biasing potential, and hence it is along the central axis of gA. To make sure that there are no off-axis binding sites that are deeper than the central one, we have repeated the Ca2+ PMF calculation without a radial restraint. The resulting PMF is very similar to the one shown in Fig. 1, and does not exhibit any deeper binding sites. This shows that the Ca2+ PMF is rather flat in the radial direction.
While neither Cl− nor Ca2+ can bind to the pore region of gA individually, NMR experiments indicate that they may bind as a pair—Cl− near the carbonyl group of Trp11, and Ca2+ near the carbonyl group of Trp15 (49). In these positions, they would provide an absolute block of the channel preventing permeation of monovalent cations in conflict with the conductance measurements (45,46). Entropic considerations also make the prospect for such an ion pair binding very unlikely. Nevertheless, in the absence of any NMR experiments that directly contradict the results of Jing and Urry (49), we are compelled to consider the energetics of Ca2+-Cl− pair binding to gA. We construct the PMF for the pair of ions using two umbrella potentials separated by 2.56 Å, which is the contact distance of the ions found from the pair-distribution function. To mimic the experimental situation, the Cl− ion is placed closer to gA. During the production runs, the z1 and z2 coordinates of the two ions are sampled as before. The densities are unbiased using the center of the two-ion system, Z = (z1 + z2)/2, as the reaction coordinate. The resulting PMF for the binding of Ca2+-Cl− pair to gA is shown in Fig. 3. The PMF rises earlier and steeper than the PMFs of individual ions, and clearly shows that such a binding of Ca2+ and Cl− ions as a pair to gA is extremely unlikely.
FIGURE 3.

The PMF profile for the binding of Ca2+-Cl− pair to gA plotted against the center of the two-ion system, Z = (z1 + z2)/2.
Free energy differences of ions
The purpose of the free energy calculations is to provide an independent check on the PMF results as well as to study the convergence properties of the free energy simulations. Because the energetics of K+ ion permeation in MD simulations is fairly well established, we discuss it first. The results of the free energy differences for transferring a K+ ion from bulk to the channel center and the reverse process are presented in Table 1. Both the TI and FEP methods have been employed. As mentioned in Methods, the calculations are performed in two steps using a water molecule with no charges as an intermediate state. The difference between the TI and FEP calculations arises mainly from the free energy difference for the W → W0 transformation, which is found to be very small in the former and ∼1.6 kT in the latter. Statistical errors in the TI calculations are determined by dividing the data into 50-ps blocks and finding the standard deviation in the sample. For the K → W0 transformation, the error in both directions is found to be ∼1 kT, and for W0 → W, ∼0.5 kT. The total statistical error of 1.5 kT is consistent with the hysteresis effect observed in the forward and backward transformations.
TABLE 1.
Free energy differences for translocating a K+ ion from bulk to the channel center (ΔG+), negative of the reverse transfer (−ΔG–), and their average (ΔGav)
| ΔG+ | −ΔG– | ΔGav | |
|---|---|---|---|
| TI | 11.2 | 12.2 | 11.7 |
| FEP | 13.2 | 14.0 | 13.6 |
| PMF | — | — | 13.0 |
The TI and FEP results are compared to the free energy difference obtained from the K+ PMF (Fig. 1) in the last column. All free energies are given in units of kT.
The average free energy difference, ΔGav, quoted in the last column of Table 1 corresponds to the barrier-well difference in the PMF (Fig. 1), which is listed in the last row. We see that both the TI and FEP methods yield free energy differences that are consistent with the PMF result within the statistical errors. For computational convenience, we use the TI method in the remaining free energy calculations.
The above free energy difference was also calculated in Allen et al. (22) using the FEP method as 16.6 kT, which is 3 kT higher than our value. A similar difference is found for the stabilization energy in the PMF (17 vs. 13 kT here), suggesting a systematic difference between the two simulations. We note that the final value quoted for this free energy difference in Allen et al. (22) was 14.6 kT, which included a correction term that took into account the different density of water in the pore compared to bulk: kT ln [ρw(bulk)/ρw(pore]. This correction was calculated as −2 kT from the estimated pore water density of 7.5 times the bulk density (22). We remark that water is practically incompressible and such a large increase in water density in the pore is not possible. The free energy difference without this correction (16.6 kT) is, in fact, more consistent with the corresponding PMF value of 17 kT (22). Recent semimicroscopic calculations that include polarization interaction explicitly (11), give 6 kT for the K+ free energy difference, which is substantially lower than any of the MD simulation results.
As an example of the convergence of the free energy differences obtained with the TI method, we show in Fig. 4 the running averages of ΔG+ (solid line) and – ΔG– (dashed line), which are described in Table 1. The two legs of the transformation, K+ → W0 and W0 → W, are shown separately and labeled by K and W, respectively. The values quoted in Table 1 correspond to the sum of the final points in the K and W curves in Fig. 4. There is minimal hysteresis between the forward and backward directions (∼1 kT), and the running averages remain rather flat. The slight increase in the forward K result is caused by a larger than average fluctuation. We have followed this simulation for a further 200 ps and observed that the direction is reversed, i.e., it is a fluctuation, not a definite trend. Thus the free energy differences appear to have converged to reasonably well-defined values.
FIGURE 4.

Running averages of the free energy differences ΔG+ (solid lines) and −ΔG– (dashed lines) for transferring a K+ ion from bulk to the channel center and the reverse process, respectively. The curve labeled K shows the K+ → W0 leg of the transformation, and W shows the W0 → W leg. The total ΔG is obtained from the average of the two curves.
Having established the validity of the free energy calculations for K+ ions using the TI method, we next apply it to Cl− and Ca2+ ions in the gA system. As a Cl− ion has no binding site, the only relevant point for calculating the free energy difference is the channel center. In Fig. 5, we show the results of the free energy calculations for translocating a Cl− ion from bulk to the channel center (solid line) and the negative of the reverse transformation (dashed line). As before, the transformation is done in two steps but only the Cl− → W0 step is shown in Fig. 5. The W0 → W leg is identical to that shown in Fig. 4, and therefore is not duplicated here. In any case it is practically zero on the scale of Fig. 5. From the average of the forward and backward transformations, the free energy difference is obtained as ΔGav = 63.5 ± 2 kT. The statistical error is estimated from 50-ps blocks of data as in the case of K+ ion. Comments similar to K+ ion can also be made for Cl− ion about the consistency of the statistical fluctuations and hysteresis effects, and the convergence properties of the calculated free energy differences. Note that the same energy scale is used in Figs. 4–6 to make the comparison of the hysteresis effects between different ions easier. In an earlier MD simulation of gA (20), the quantity ΔΔG(K+ → Cl−) was calculated as 100 kT, which is much higher than our result: 63 − 13 = 50 kT. The recent semimicroscopic Monte Carlo calculations (11), on the other hand, give 20 kT for ΔG, which is much smaller than the 63 kT calculated here.
FIGURE 5.

Same as Fig. 4 but for a Cl− ion. The W0 → W leg of the free energy difference is the same as in Fig. 4, and therefore is not shown.
FIGURE 6.

Similar to Fig. 4 but for a Ca2+ ion at z = 14.5 Å on the channel axis. The W0 → W leg of the free energy difference is negligibly small and therefore not shown, to avoid cluttering of the Ca2+ ion result.
For a Ca2+ ion, the binding site at z = 14.5 Å is clearly the most relevant point for calculating the free energy difference. The results of the free-energy difference calculations for translocating a Ca2+ ion from bulk to the binding site (solid line) and the negative of the reverse transformation (dashed line) are shown in Fig. 6. Only the Ca2+ → W0 leg of the transformation is shown in the figure because the W0 → W leg yields a negligibly small free energy difference, which would overlap and obscure the calcium results. The binding site is sufficiently far from the channel to exhibit bulklike properties for uncharged species (and even for a K+ ion; see Fig. 1). The average binding energy obtained from the forward and backward transformations is ΔGav = – 1 ± 2 kT. The hysteresis effect is larger than that for the K+ ion, which is presumably due to the quadrupling of the ion-water interactions for a Ca2+ ion. Otherwise, it is within the calculated statistical fluctuations. The running averages for the free energy differences remain flat after 300 ps, indicating that the results have converged reasonably well. Overall, the calculated free-energy difference for the binding energy of a Ca2+ ion is consistent with that obtained from the PMF and provides an independent check of that result.
Because the calcium binding site is outside the channel, it is relatively easier to locate the problem with the energetics of binding compared to that of potassium. We recall that the effect of the finite size and lipid polarizability corrections on K+ ion was to lower the central barrier inside but they had virtually no effect on the binding energy at the pore mouth (22). Thus for a Ca2+ ion outside the channel, we expect these corrections to go in the opposite direction and reduce the magnitude of the binding energy even further. This suggests that description of calcium binding to the gA channel may provide an even stronger case for including polarization interaction in the MD force fields. To check the viability of this proposition, we have estimated the interaction energies of a K+ and a Ca2+ ion in their respective binding sites with the water column inside the channel. The results obtained from the average of 100 ps of MD data are −46 kT for a K+ and −133 kT for a Ca2+ ion. (The reason for more than doubling of the energy is better alignment of water dipoles with the stronger electric field of a Ca2+ ion.) Thus the ion-channel water interactions contribute to the stabilization energy of a Ca2+ ion relative to K+ by a ratio of 3:1. In a polarizable model, the dipole moment of water increases from the rigid value of 2.35 Debye (D) to ∼2.7 D (50)—a 15% increase. Even larger values are predicted from ab initio MD calculations (51). Assuming that increasing the dipole moment of water by 15% would roughly boost the stabilization energy of a Ca2+ relative to K+ by the same 3:1 ratio, we estimate the polarization effects to contribute to the relative stabilization energy by (−133 + 46) × 0.15 = −13 kT. This would change the binding energy of a Ca2+ ion relative to K+ from +5 kT to −8 kT, which is sufficient to explain the observed calcium binding and block data. Although this is a fairly simple estimate, it nevertheless shows that inclusion of polarization goes in the right direction toward the resolution of this problem. Naturally, one needs to repeat the above PMF calculations for potassium and calcium ions using a polarizable force field to show that inclusion of polarizability indeed fixes the problems pointed out in this article.
CONCLUSIONS
Molecular dynamics simulations of membrane proteins have been growing rapidly during the last decade. The majority of groups involved in such work use commonly available force fields such as AMBER, CHARMM, or GROMACS. These force fields have been optimized for globular proteins and their applicability to membrane proteins has not been well tested. A first test of these rigid force fields in the gA channel indicated serious problems in an MD description of K+ permeation through this channel (21). However, in a more recent work, the suggested discrepancy in K+ conductance was minimized by using correction factors, selective data, and the one-dimensional Nernst-Planck equation (22). These later results were interpreted in a recent review article (7) as “good agreement with experimental conductance data on gramicidin A was obtained.” As pointed out here, despite inclusion of some correction factors from continuum electrostatics, the situation with regard to K+ permeation is not that good—if one takes into account all the permeation data and uses the more realistic three-dimensional Brownian dynamics simulations, the discrepancy in the K+ conductance remains at ∼4–5 orders of magnitude.
In this article, we have carried out a more comprehensive test of the nonpolarizable CHARMM force field in the gA channel by studying the energetics of K+, Cl−, and Ca2+ ions. The potential of mean force along the channel axis is determined for each ion type, the latter two being the first such PMF calculations. We have also carried out free energy difference calculations at the channel center and binding site, which have provided an independent check on the discrepancies found in the PMF results, as well as a tool for studying the convergence properties of the free energy simulations. Convergence of the results are demonstrated in Figs. 4–6: the running averages of free energy differences remain flat and the hysteresis effects are within the expected statistical fluctuations.
Our results confirm those obtained from previous studies for K+ ion and extend them to Ca2+ ion—namely, that the rigid force fields can account for the binding configurations of monovalent and divalent cations but have difficulties in explaining the energetics of permeation of monovalent cations, and binding and block of divalent cations. The inability of the nonpolarizable force fields to describe conductance of monovalent cations was noted earlier and despite some recent improvements, this problem has not been resolved satisfactorily. This work shows that they also fail to describe the binding of divalent cations to gA and the ensuing block of the channel. Because the calcium-binding site is outside the pore, we believe that this result is more robust against simulation artifacts compared to that for a potassium ion, and hence makes a stronger case for the inclusion of the polarization interaction. We hope that these results will further stimulate construction of polarizable force fields for MD simulations of membrane proteins.
Acknowledgments
Calculations were carried out using the Barossa cluster at the Australian Center for Advanced Computing and Communications, and Compaq Alphaserver at the ANU Supercomputer Facility. We thank T. Cross, J. Hinton, P. Kuchel, and F. Separovic for useful comments on the interpretation of the NMR data on ion binding.
This work was supported by grants from the Australian Research Council.
References
- 1.Karplus, M. 2002. Molecular dynamics simulations of biomolecules. Acc. Chem. Res. 35:321–323. [DOI] [PubMed] [Google Scholar]
- 2.Urry, D. W. 1971. The gramicidin A transmembrane channel: a proposed πLD helix. Proc. Natl. Acad. Sci. USA. 68:672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackay, D. H. J., P. H. Berens, K. R. Wilson, and A. T. Hagler. 1984. Structure and dynamics of ion transport through gramicidin A. Biophys. J. 46:229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roux, B., and M. Karplus. 1994. Molecular dynamics simulations of the gramicidin channel. Annu. Rev. Biophys. Biomol. Struct. 23:731–761. [DOI] [PubMed] [Google Scholar]
- 5.Roux, B. 2002. Computational studies of the gramicidin A channel. Acc. Chem. Res. 35:366–375. [DOI] [PubMed] [Google Scholar]
- 6.Doyle, D. A., J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- 7.Ash, W. L., M. R. Zlomislic, E. O. Oloo, and D. P. Tieleman. 2004. Computer simulations of membrane proteins. Biochim. Biophys. Acta. 1666:158–189. [DOI] [PubMed] [Google Scholar]
- 8.Pearlman, D. A., D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham, S. DeBolt, D. Ferguson, G. Seibel, and P. Kollman. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics, and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Comm. 91:1–41. [Google Scholar]
- 9.MacKerell, A. D., Jr., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. D. Evanseck, M. J. Field, S. Fisher, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher III, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 10.Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Comm. 91:43–56. [Google Scholar]
- 11.Dorman, V. L., and P. C. Jordan. 2004. Ionic permeation free energy in gramicidin: a semimicroscopic perspective. Biophys. J. 86:3529–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersen, O. S., and R. E. Koeppe. 1992. Molecular determinants of channel function. Physiol. Rev. 72:89–158. [DOI] [PubMed] [Google Scholar]
- 13.Busath, D. D. 1993. The use of physical methods in determining gramicidin structure and function. Annu. Rev. Physiol. 55:473–501. [DOI] [PubMed] [Google Scholar]
- 14.Hille, B. 2001. Ionic Channels of Excitable Membranes, 3rd Ed. Sinauer Associates, Sunderland, MA.
- 15.Partenskii, M. B., and P. C. Jordan. 1992. Theoretical perspectives on ion-channel electrostatics: continuum and microscopic approaches. Q. Rev. Biophys. 25:477–510. [DOI] [PubMed] [Google Scholar]
- 16.Tieleman, D. P., P. C. Biggin, G. R. Smith, and M. S. P. Sansom. 2001. Simulation approaches to ion channel structure-function relationships. Q. Rev. Biophys. 34:473–561. [DOI] [PubMed] [Google Scholar]
- 17.Edwards, S., B. Corry, S. Kuyucak, and S. H. Chung. 2002. Continuum electrostatics fails to describe ion permeation in the gramicidin channel. Biophys. J. 83:1348–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jordan, P. C. 2002. Trial by ordeal: ionic free energies in gramicidin. Biophys. J. 83:1235–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorman, V., M. B. Partenskii, and P. C. Jordan. 1996. A semi-microscopic Monte Carlo study of permeation energetics in a gramicidin-like channel: the origin of cation selectivity. Biophys. J. 70:121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roux, B. 1996. Valence selectivity of the gramicidin channel: a molecular dynamics free energy perturbation study. Biophys. J. 71:3177–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen, T. W., T. Bastug, S. Kuyucak, and S. H. Chung. 2003. Gramicidin A as a test ground for molecular dynamics force fields. Biophys. J. 84:2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allen, T. W., O. S. Andersen, and B. Roux. 2004. Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. USA. 101:117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levitt, D. G. 1999. Modeling of ion channels. J. Gen. Physiol. 113:789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuyucak, S., O. S. Andersen, and S. H. Chung. 2001. Models of permeation in ion channels. Rep. Prog. Phys. 64:1427–1472. [Google Scholar]
- 25.Bastug, T., A. Gray-Weale, S. M. Patra, and S. Kuyucak. 2006. Role of protein flexibility in ion permeation: a case study in gramicidin A. Biophys. J. 90:2285–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 27.Ketchem, R. R., B. Roux, and T. A. Cross. 1997. High-resolution polypeptide structure in a lamellar phase lipid environment from solid state NMR derived orientational constraints. Structure. 5:1655–1669. [DOI] [PubMed] [Google Scholar]
- 28.Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kale, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- 30.Feller, S., Y. Zhang, R. Pastor, and B. Brooks. 1995. Constant pressure molecular dynamics: the Langevin piston method. J. Chem. Phys. 103:4613–4621. [Google Scholar]
- 31.Torrie, G. M., and J. P. Valleau. 1977. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 23:187–199. [Google Scholar]
- 32.Kumar, S., D. Bouzida, R. H. Swensen, P. A. Kollman, and J. M. Rosenberg. 1992. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 13:1011–1021. [Google Scholar]
- 33.Souaille, M., and B. Roux. 2001. Extension to the weighted histogram analysis method: combining umbrella sampling with free energy calculations. Comput. Phys. Commun. 135:40–57. [Google Scholar]
- 34.Levitt, D. G. 1986. Interpretation of biological ion channel flux data—reaction-rate versus continuum theory. Annu. Rev. Biophys. Biophys. Chem. 15:29–57. [DOI] [PubMed] [Google Scholar]
- 35.Roux, B., T. Allen, S. Bernèche, and W. Im. 2004. Theoretical and computational models of biological ion channels. Q. Rev. Biophys. 37:15–103. [DOI] [PubMed] [Google Scholar]
- 36.Beveridge, D. L., and F. M. DiCapua. 1989. Free energy via molecular simulation: applications to chemical and biomolecular systems. Annu. Rev. Biophys. Biophys. Chem. 18:431–492. [DOI] [PubMed] [Google Scholar]
- 37.Press, W. H., B. P. Flannery, S. A. Teukolsky, and W. T. Vetterling. 1989. Numerical Recipes. Cambridge University Press, Cambridge, UK.
- 38.Duca, K. A., and P. C. Jordan. 1998. Comparison of selectively polarizable force fields for ion-water-peptide interactions: ion translocation in a gramicidin-like channel. J. Phys. Chem. 102:9127–9138. [Google Scholar]
- 39.Hinton, J. F., W. L. Whaley, D. Shungu, R. E. Koeppe, and F. S. Millett. 1986. Equilibrium binding constants for the group I metal cations with gramicidin-A determined by competition studies and Tl+-205 nuclear magnetic resonance spectroscopy. Biophys. J. 50:539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jing, N. J., K. U. Prasad, and D. W. Urry. 1995. The determination of binding constants of micellar-packaged gramicidin A by 13C and 23Na-NMR. Biochim. Biophys. Acta. 1238:1–11. [DOI] [PubMed] [Google Scholar]
- 41.Olah, G. A., H. W. Huang, W. Liu, and Y. Wu. 1991. Location of ion-binding sites in the gramicidin channel by x-ray diffraction. J. Mol. Biol. 218:847–858. [DOI] [PubMed] [Google Scholar]
- 42.Townsley, L. E., A. W. Tucker, S. Sham, and J. F. Hinton. 2001. Structures of gramicidin A, B and C incorporated into sodium dodecyl sulfate micelles. Biochemistry. 40:11676–11686. [DOI] [PubMed] [Google Scholar]
- 43.Tian, F., K. C. Lee, W. Hu, and T. A. Cross. 1996. Monovalent cation transport: lack of structural deformation upon cation binding. Biochemistry. 35:11959–11966. [DOI] [PubMed] [Google Scholar]
- 44.Tian, F., and T. A. Cross. 1999. Cation transport: an example of structural based selectivity. J. Mol. Biol. 285:1993–2003. [DOI] [PubMed] [Google Scholar]
- 45.Bamberg, E., and P. Laeuger. 1977. Blocking of the gramicidin channel by divalent cations. J. Membr. Biol. 35:351–375. [Google Scholar]
- 46.Heitz, F., and C. Gavach. 1983. Ca2+-gramicidin interaction and blocking effects on the ionic channel. Biophys. Chem. 18:153–163. [DOI] [PubMed] [Google Scholar]
- 47.Corry, B., T. W. Allen, S. Kuyucak, and S. H. Chung. 2001. Mechanisms of permeation and selectivity in calcium channels. Biophys. J. 80:195–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Golovanov, A. P., I. L. Barsukov, A. S. Arseniev, V. F. Bystrov, S. V. Sukhanov, and L. I. Barsukov. 1991. The divalent cation-binding sites of gramicidin A transmembrane ion-channel. Biopolymers. 31:425–434. [DOI] [PubMed] [Google Scholar]
- 49.Jing, N., and D. W. Urry. 1995. Ion pair binding of Ca2+ and Cl− ions in micellar-packaged gramicidin A. Biochim. Biophys. Acta. 1238:12–21. [DOI] [PubMed] [Google Scholar]
- 50.Wallqvist, A., and R. D. Mountain. 1999. Molecular models of water: derivation and description. Rev. Comput. Chem. 13:183–247. [Google Scholar]
- 51.Silvestrelli, P. L., and M. Parrinello. 1999. Structural, electronic, and bonding properties of liquid water from first principles. J. Chem. Phys. 111:3572–3580. [Google Scholar]