

Abstract
Myosin-binding protein-C (MyBP-C) is a thick filament-associated protein that binds tightly to myosin. Given that cMyBP-C may act to modulate cooperative activation of the thin filament by constraining the availability of myosin cross-bridges for binding to actin, we investigated the role of MyBP-C in the regulation of cardiac muscle contraction. We assessed the Ca2+ sensitivity of force (pCa50) and the activation dependence of the rate of force redevelopment (ktr) in skinned myocardium isolated from wild-type (WT) and cMyBP-C null (cMyBP-C−/−) mice. Mechanical measurements were performed at 22°C in the absence and presence of a strong-binding, nonforce-generating analog of myosin subfragment-1 (NEM-S1). In the absence of NEM-S1, maximal force and ktr and the pCa50 of isometric force did not differ between WT and cMyBP-C−/− myocardium; however, ablation of cMyBP-C-accelerated ktr at each submaximal force. Treatment of WT and cMyBP-C−/− myocardium with 3 μM NEM-S1 elicited similar increases in pCa50, but the effects of NEM-S1 to increase ktr at submaximal forces and thereby markedly reduce the activation dependence of ktr occurred to a greater degree in cMyBP-C−/− myocardium. Together, these results support the idea that cMyBP-C normally acts to constrain the interaction between myosin and actin, which in turn limits steady-state force development and the kinetics of cross-bridge interaction.
INTRODUCTION
Cardiac muscle contraction is initially triggered by the binding of Ca2+ to troponin C (TnC), which activates the thin filament via a series of intermolecular events involving TnI, TnT, and tropomyosin (1). Although Ca2+ binding to TnC is a vital component of the regulation of muscle contraction, it is well established that binding of Ca2+ to TnC alone is unable to fully activate the thin filament. Instead the development of steady-state force and the kinetics of force development result from the synergistic actions of Ca2+ binding to TnC and strong binding of myosin cross-bridges to actin (2–4). This synergy is evident in the biphasic form of the force-pCa (pCa = −log[Ca2+]) relationship commonly seen in skinned myocardial preparations, which is steeper at low levels of Ca2+ activation (i.e., forces <0.50 Po) than at high. If force development was solely regulated by the noncooperative binding of Ca2+ to TnC, the steepness (i.e., Hill coefficient, nH) of the force-pCa relationship should be ∼1, since cardiac TnC contains a single low affinity Ca2+-specific binding site (5). However, the steepness of the force-pCa relationship in cardiac muscle is relatively high (i.e., nH > 4) (6–8), which strongly suggests the involvement of cooperative interactions in the Ca2+ activation process (4,9). Such cooperative processes may include the synergistic effects of strongly bound cross-bridges to enhance Ca2+ binding to TnC and to directly recruit additional cross-bridges to strong binding states, i.e., cross-bridge-induced cross-bridge binding (9).
With regard to cooperative activation of myocardium, most research to date has focused primarily on the roles played by thin filament regulatory proteins such as TnT (1,4), and comparatively little work has been done to determine the degree to which thick filament accessory proteins modulate cooperative activation of the thin filament. One prospective thick filament protein is myosin-binding protein-C (MyBP-C), which in skeletal muscle is localized in discrete bands (∼7–9) spaced at regular intervals (∼43 nm) along the central region of each half of the A-band (10,11). MyBP-C is thought to restrict cross-bridge binding to actin, possibly by tethering the myosin cross-bridge to the thick filament backbone (12–14) via interactions of two myosin-binding domains within MyBP-C to light meromyosin and subfragment 2 (15,16). These binding domains may act in concert (17) or independently (18,19) to tether the myosin heads to the thick filament. MyBP-C was originally proposed to form a trimeric collar around the thick filament (20), thereby providing a reasonable explanation for cross-bridge tethering, and recent work by Watkins and co-workers (14,21) has provided experimental support for this idea.
To investigate the role of MyBP-C in the regulation of myocardial contraction, in particular its effects on force and the kinetics of force development, we previously developed a cMyBP-C null mouse (cMyBP-C−/−) (22). Compared to age-matched wild-type (WT) controls, cMyBP-C ablation slightly reduced the Ca2+ sensitivity of force but did not affect maximum isometric force (19,22) and increased peak normalized power output (23). Ablation also had striking effects on in vivo left ventricular function, as manifested by reduced peak systolic elastance, a marked abbreviation of the time course of systolic ejection (24) and a prolonged duration of isovolumic relaxation (22). Thus, in normal myocardium cMyBP-C appears to contribute significantly to both systolic and diastolic function.
Since cMyBP-C is postulated to constrain the availability of myosin cross-bridges for binding to actin, we hypothesized that cMyBP-C normally acts to limit cooperative activation of the thin filament. In this model, cMyBP-C functions to attenuate steady-state force and repress the rate of force development during submaximal activations. Thus, ablation of cMyBP-C should increase steady-state isometric force and accelerate the rate of force redevelopment during submaximal activation due to greater cross-bridge availability to actin. Further, based on the premise that there is greater cooperative activation of the thin filament by endogenous cross-bridges in cMyBP-C−/− myocardium, we hypothesized that cMyBP-C−/− myocardium would exhibit altered responsiveness to NEM-S1, a strong-binding nonforce-generating derivative of myosin subfragment 1 (S1) that accelerates force development at submaximal Ca2+ concentrations in WT myocardium. These ideas are investigated here by characterizing the activation dependence of the rate of force development (ktr) in skinned preparations isolated from both WT and cMyBP-C−/− myocardium.
MATERIALS AND METHODS
Experimental solutions
All activating solutions were prepared using the computer program of Fabiato (25) with stability constants corrected to pH 7.0 and 22°C listed by Godt and Lindley (26). The composition of relaxing solution was as follows (in mM): 100 KCl, 20 imidazole, 4 MgATP, 2 EGTA, and 1 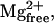 pH 7.0 at 22°C. All activating solutions contained (in mM): 100 N,N-bis[2-hydroxyethyl]-2-aminoethanesulfonic acid (BES), 15 creatine phosphate, and 5 1,4-dithiothreitol (DTT). Furthermore, pCa 9.0 solution contained (in mM): 7 EGTA, 5.49 MgCl2, 4.66 ATP, and 0.017 CaCl2; pCa 4.5 solution contained (in mM): 7 EGTA, 5.29 MgCl2, 4.72 ATP, and 7.01 CaCl2; and preactivating solution contained (in mM): 5.29 MgCl2, 4.67 ATP, and 0.07 EGTA. All activating solutions had an ionic strength of 180 mM (adjusted with potassium propionate) and a pH of 7.0 at 22°C. A range of pCa solutions containing differing concentrations of free Ca2+ (i.e., pCa 6.2–5.4) were prepared by mixing appropriate volumes of pCa 9.0 and pCa 4.5 solutions.
pH 7.0 at 22°C. All activating solutions contained (in mM): 100 N,N-bis[2-hydroxyethyl]-2-aminoethanesulfonic acid (BES), 15 creatine phosphate, and 5 1,4-dithiothreitol (DTT). Furthermore, pCa 9.0 solution contained (in mM): 7 EGTA, 5.49 MgCl2, 4.66 ATP, and 0.017 CaCl2; pCa 4.5 solution contained (in mM): 7 EGTA, 5.29 MgCl2, 4.72 ATP, and 7.01 CaCl2; and preactivating solution contained (in mM): 5.29 MgCl2, 4.67 ATP, and 0.07 EGTA. All activating solutions had an ionic strength of 180 mM (adjusted with potassium propionate) and a pH of 7.0 at 22°C. A range of pCa solutions containing differing concentrations of free Ca2+ (i.e., pCa 6.2–5.4) were prepared by mixing appropriate volumes of pCa 9.0 and pCa 4.5 solutions.
Experimental animals
Homozygous cardiac MyBP-C knockout mice (cMyBP-C−/−) were generated as described previously (22) and were maintained on an SV/129 background. Age-matched (i.e., 3–6 months) WT SV/129 mice were obtained from Taconic Farms (Germantown, NY). All animal usage was conducted under the strict guidelines established by the University of Wisconsin Animal Care and Use Committee.
Preparation of skinned myocardium
Chemically skinned myocardial preparations were obtained from cMyBP-C−/− and WT hearts as described previously (27). Briefly, both cMyBP-C−/−and WT mice were injected intraperitoneally with heparin (5,000 U heparin/kg body wt). After 20 min, the mice were placed in a glass bell jar and were anesthetized with inhaled isoflurane. After the establishment of deep anesthesia, confirmed by the loss of the pedal reflex and lack of muscular tension in the limbs, the mice were euthanized by inducing a pneumothorax. The heart was rapidly excised and the left and right ventricles were isolated in Ca2+-free Ringer's solution containing (in mM): 118 NaCl, 4.8 KCl, 2 NaH2PO4, 1.2 MgCl2, 25 HEPES, 11 glucose, and 0.5 CaCl2, pH 7.4 at 22°C. The ventricles were immediately frozen in liquid N2 for 10 min to improve the subsequent quality of mechanical preparations. The frozen ventricles were thawed and homogenized with a Polytron (Lucern, Switzerland) homogenizer for ∼2 s in ice-cold relaxing solution. The cellular homogenate was centrifuged at 120 × g for 1 min and resuspended in fresh ice-cold relaxing solution. After a second centrifugation, the pelleted myocardial preparations were resuspended in fresh relaxing solution containing 250 μg/ml saponin and 1% Triton X-100 for 30 min at 22°C. The chemically skinned preparations were washed three times with ice-cold relaxing solution and allowed to settle. After the final wash, the skinned myocardial preparations were dispersed in a glass petri dish with 50 ml of ice-cold fresh relaxing solution. The petri dish was kept on ice at all times until use. Mechanical experiments were always performed on cardiac preparations isolated on the day of the experiments. Despite exhibiting significant cardiac hypertrophy (22), skinned cardiac preparations from cMyBP-C−/− hearts did not show observable differences in ultrastructure or striation pattern compared with WT cardiac preparations.
Experimental apparatus
Skinned preparations with well-defined edges and no free ends evident in the middle region were transferred from the petri dish to a stainless steel experimental chamber (28) containing fresh relaxing solution. For mechanical measurements, skinned myocardial preparations (dimensions: ∼800 μm × 100–200 μm) were mounted between a force transducer (model 403A; Aurora Scientific; Aurora, Ontario, Canada) and a motor (model 312B, Aurora Scientific). A 500-μm long myocardial segment remained exposed to the solution between the force transducer and motor. Before mechanical measurements, the experimental apparatus was set on the stage of an inverted microscope (Olympus; Tokyo, Japan) fitted with a 40× objective and a closed-circuit television camera (model WV-BL600; Panasonic; Tokyo, Japan). Light from a halogen lamp was passed through a cut-off filter (transmission >620 nm) and was used to illuminate the skinned myocardium. Sarcomere length and myocardial dimensions were recorded during relaxation and activation using the video camera and a VHS recorder (model SVO-1420; Sony; Tokyo, Japan). Activated cardiac preparations that exhibited length changes >5% of initial sarcomere length were discarded and the data not used. Changes in force and motor position were sampled (16-bit resolution, DAP5216a; Microstar Laboratories; Bellevue, WA) at 2.0 kHz with SLControl software (29) and saved to computer files for later analysis. Changes in force were also recorded on a chart recorder using a slow time base. All experiments were performed at 22°C and at a sarcomere length of ∼2.20 μm measured in relaxing solution.
Preparation and use of NEM-S1
Myosin S1 was purified from rabbit fast-twitch skeletal muscle and modified with N-ethylmaleimide (NEM), as described previously (30). Although NEM-S1 significantly increases myosin ATPase activity in solutions containing myosin, regulated actin, and Ca2+ (31,32), it exhibits no ATPase activity of its own (31). NEM-S1 forms long-lasting complexes with actin in the presence or absence of Ca2+ and ATP (30). In this study, the concentration of NEM-S1 was estimated by absorbance at 280 nm (with light-scattering correction performed at 320 nm) using a mass absorptivity of 0.75 and a molecular mass of 118 kDa for myosin S1. A stock solution of NEM-S1 (i.e., 20–30 μM) was prepared by overnight dialysis against a solution of 20 mM imidazole, pH 7.0, and 1 mM DTT. A working solution of NEM-S1 (i.e., 10–15 μM) was prepared immediately before use by mixing equal volumes of 2× stock of pCa 9.0 solution and NEM-S1 stock. The concentration of NEM-S1 was adjusted by adding the appropriate amount of 1× pCa 9.0 solution. Before any mechanical measurements, each skinned myocardial preparation was incubated for 15 min at 22°C in solution of pCa 9.0 containing either 1 or 3 μM NEM-S1. Thereafter, for measurements of force or the rate of force development, the preparation was initially incubated with NEM-S1 in a solution of pCa 9.0 to allow for NEM-S1-binding to actin and was subsequently transferred to preactivating solution for 1 min and then into activating solutions of varying pCa (i.e., pCa 6.2–4.5) without NEM-S1 (30). After each mechanical measurement, the preparation was returned to solution of pCa 9.0 containing NEM-S1. Thus, the preparation was incubated in NEM-S1-free activating solutions for no more than 2 min, during which time negligible amounts of NEM-S1 would be debound (30).
Specific experimental protocols
Rate of tension development
The rate constant of force redevelopment (ktr) in skinned myocardium was assessed using a modification of the experimental protocol originally described by Brenner and Eisenberg (33). Measurement of ktr involves a mechanical slack-restretch maneuver to detach bound myosin cross-bridges from actin in steadily Ca2+-activated myocardium. Each skinned preparation was transferred from relaxing to activating solutions of varying free Ca2+ (i.e., pCa 6.2–4.5) and allowed to generate steady-state force. The myocardial preparation was rapidly (<2 ms) slackened by 20% of its original length, resulting in a rapid reduction of force to near zero (i.e., <5% of steady isometric force). This was followed by a brief period of unloaded shortening (i.e., 20 ms) after which the preparation was rapidly restretched to its original length. Force redevelopment after the slack-restretch maneuver and force recovery to the original steady-state value reflects the rate of myosin cross-bridge cycling between weakly bound and strongly bound, force-generating states (33). A ktr-pCa relationship was obtained by initially activating the skinned myocardium in solution of pCa 4.5 and then in a series of submaximally activating solutions between pCa 6.2 and 5.4. To assess any decline in the maximal rate of force redevelopment, the preparation was activated in a solution of pCa 4.5 at the end of each experimental protocol. The decline in maximum force at pCa 4.5 during the experiment was usually <10%, but preparations showing decreases >15% were discarded and the data not used. The reference value of maximal ktr for each activation was obtained by interpolation between the initial and final measurements of maximal ktr. The apparent rate constants of force redevelopment (ktr) were estimated by linear transformation of the half-time of force redevelopment, i.e., ktr = 0.693/t1/2, as described previously (34,35).
Force-pCa relationship
During measurements of force redevelopment, each preparation was to develop steady force in solutions of varying free Ca2+. The difference between steady-state force and the force baseline obtained after the 20% slack step was measured as the total force at that free [Ca2+]. Active force was then calculated by subtracting Ca2+-independent force in solution of pCa 9.0 from the total force and was normalized to the cross sectional area of the preparation, which was calculated from the width of the preparations assuming a cylindrical cross section. Force-pCa relationships were derived by expressing submaximal force (P) at each pCa as a fraction of maximal force (Po) determined at pCa 4.5, that is, P/Po. The apparent cooperativity in the activation of force development was inferred from the steepness of the force-pCa relationship and was quantified using a Hill plot transformation of the force-pCa data (36). The force-pCa data were fit using the equation P/Po = [Ca2+]n/(kn + [Ca2+]n), where n is the Hill coefficient and k is the [Ca2+] required for half-maximal activation (i.e., pCa50).
Statistics
All data are expressed as mean ± SE. Where appropriate, either a two-tailed t-test for independent samples or a paired t-test was used as a post hoc test of significance, with significance set at p < 0.05.
RESULTS
Steady-state mechanical properties in WT and cMyBP-C−/− skinned myocardium
Ablation of cMyBP-C had no significant effects on either Ca2+-independent force, measured in pCa 9.0, or maximal Ca2+-activated force (Table 1). Furthermore, we observed no significant difference in the Ca2+ sensitivity of force, that is, mean pCa50 was 5.73 ± 0.01 in WT myocardium and 5.72 ± 0.01 in cMyBP-C−/− myocardium. Although mean pCa50 was similar between groups, we observed a small, but significant increase in Ca2+-activated forces at low Ca2+ concentrations, i.e., pCa ≥ 5.9 in cMyBP-C−/− myocardium (Fig. 1 A). These results are consistent with the idea that elimination of cMyBP-C promotes increased binding of cross-bridges to actin. Consistent with the increase in Ca2+-activated force at low [Ca2+], we observed a significant reduction in the steepness of the force-pCa relationship for Ca2+-activated forces <0.50 Po in cMyBP-C−/− myocardium (n2 was 4.0 ± 0.2 vs. 5.6 ± 0.1 in WT myocardium; p < 0.05). These data indicate that ablation of cMyBP-C reduces the apparent cooperativity of force development.
TABLE 1.
Steady-state mechanical measurements in skinned preparations from WT and cMyBP-C−/− myocardium
| Group | Po (mN mm−2) | Prest (mN mm−2) | n2 | n1 | pCa50 |
|---|---|---|---|---|---|
| WT myocardium | |||||
| Control (12) | 20.7 ± 2.3 | 0.8 ± 0.1 | 5.6 ± 0.1 | 2.7 ± 0.1 | 5.73 ± 0.01 |
| 1 μM NEM-S1 (9) | 18.2 ± 1.7 | 1.6 ± 0.8* | 3.3 ± 0.1* | 2.6 ± 0.2 | 5.72 ± 0.01 |
| 3 μM NEM-S1 (12) | 15.2 ± 1.5 | 2.3 ± 0.4* | 2.6 ± 0.2* | 2.2 ± 0.2 | 5.79 ± 0.01* |
| cMyBP-C−/− myocardium | |||||
| Control (10) | 18.4 ± 2.3 | 0.8 ± 0.1 | 4.0 ± 0.2*† | 2.5 ± 0.5 | 5.72 ± 0.01 |
| 1 μM NEM-S1 (9) | 17.0 ± 1.9 | 1.9 ± 0.3* | 2.8 ± 0.2*† | 2.1 ± 0.1 | 5.72 ± 0.01 |
| 3 μM NEM-S1 (7) | 15.7 ± 1.6 | 3.7 ± 0.4*† | 1.5 ± 0.4*† | 2.1 ± 0.5 | 5.82 ± 0.02* |
All values are expressed as mean ± SE, with the number of experimental preparations given in parentheses.
Po, maximal Ca2+-activated force at pCa 4.5; Prest, Ca2+-independent force at pCa 9.0; n2, Hill coefficient for Ca2+-activated force <0.50 Po; n1, Hill coefficient for Ca2+-activated force >0.50 Po; pCa50, pCa required for half-maximal activation.
Significantly different from control (p < 0.05).
Significantly different from WT (p < 0.05).
FIGURE 1.

Effect of NEM-S1 on force-pCa relationships in skinned myocardium from WT and cMyBP-C−/−. (A) All values are mean ± SE. Smooth lines were generated by fitting the mean data to a Hill equation: P/Po = [Ca2+]n/(kn + [Ca2+]n), where n is the Hill coefficient and k is the [Ca2+] required for half-maximal activation (pCa50). The relationships between Ca2+-activated force and pCa were determined in the absence (open symbols) and presence (solid symbols) of 3 μM NEM-S1. pCa50 values for WT were control (○), 5.73 ± 0.01; 3 μM NEM-S1 (•), 5.79 ± 0.01, whereas pCa50 values for cMyBP-C−/− were control (Δ), 5.72 ± 0.01; 3 μM NEM-S1 (▴), 5.82 ± 0.01. (B) Hill plot transformations of the force-pCa data were generated using the following equation: log[Prel/(1 − Prel)] = n(log[Ca2+] + k), where Prel is force as a fraction of Po, n is the Hill coefficient, and k is the [Ca2+] required for half-maximal activation (pCa50). All values are mean ± SE. Fiber characteristics are listed in Table 1.
The effect of NEM-S1 on steady-state force
Our previous studies of cooperative mechanisms in skinned skeletal muscle fibers (30,35) and skinned myocardium (37) have used NEM-S1, a strong-binding, nonforce-generating derivative of myosin subfragment-1 (S1), to mimic the activating effects of endogenous strong-binding myosin cross-bridges. Those studies showed that 6–10 μM NEM-S1 is an upper limit in such experiments, since higher levels resulted in a marked decrease in maximal Ca2+-activated force (Po), presumably due to competitive inhibition of endogenous cross-bridge binding (30). In addition to its effects on steady-state force, treatment with 6 μM NEM-S1 nearly eliminated the activation dependence of the rate of force redevelopment and dramatically slowed the rate of force relaxation in WT skinned myocardium (37). Thus, to avoid saturating the cooperative mechanisms underlying activation of force in cardiac muscle, we used suboptimal concentrations of NEM-S1 (i.e., 1 μM or 3 μM) to examine the responsiveness of cMyBP-C−/− myocardium to the activating effects of strong-binding cross-bridges.
The effects of 1 and 3 μM NEM-S1 on Ca2+-independent force, maximal Ca2+-activated force, the Ca2+ sensitivity of force, and the steepness (i.e., n2 and n1) of the force-pCa relationship are summarized in Table 1. In both WT and cMyBP-C−/− myocardium, NEM-S1 increased both Ca2+-independent and submaximal Ca2+-activated forces (Table 1) in a concentration-dependent manner, i.e., the Ca2+-sensitivity of force (pCa50) increased significantly in both WT and cMyBP-C−/− myocardium. Furthermore, NEM-S1 significantly reduced the steepness (n2) of the force-pCa relationship for Ca2+-activated forces <0.50 Po in a concentration-dependent manner, an effect that was greater in cMyBP-C−/− myocardium (Fig. 1 B).
Acceleration of cross-bridge kinetics by NEM-S1
The rate constant of force redevelopment (ktr) after rapid release and restretch (33) provides an estimate of the rate of transition from weak-binding, nonforce-generating cross-bridges to strong-binding, force-generating cross-bridges in skeletal and cardiac muscles. Cooperative interactions within or along the thin filament, such as cross-bridge-induced cross-bridge binding, have been proposed to slow the rate of force redevelopment (38), thereby providing the basis for the steep activation dependence of ktr in cardiac muscle observed previously (27,37,39–41). If cMyBP-C constrains the availability of cross-bridges to actin, then removal of cMyBP-C should increase the rate of force redevelopment due, at least in part, to enhanced cross-bridge-induced binding of endogenous cross-bridges. Initial experiments were conducted in the absence of NEM-S1 to establish whether ablation of cMyBP-C accelerated the kinetics of cross-bridge interaction at all levels of submaximal activation. Experiments were then conducted in the presence of NEM-S1 to determine the effects of strong-binding cross-bridges on the activation dependence of the rate of force redevelopment in WT and cMyBP-C−/− myocardium.
Records of the time course of force redevelopment obtained in the absence and presence of NEM-S1 are shown in Fig. 2 for both WT and cMyBP-C−/− myocardium. Ablation of cMyBP-C accelerated the rate of force redevelopment as compared to WT at each submaximal [Ca2+] in both the absence (Fig. 2 A) and presence (Fig. 2 B) of NEM-S1. We observed a steep activation dependence of the rate of force redevelopment as the level of Ca2+ activation was varied from near threshold to maximal levels in both WT and cMyBP-C−/− myocardium. A summary of the ktr and relative ktr values for several levels of Ca2+ activation is presented in Table 2. The cumulative ktr-pCa relationships obtained in the absence and presence of 3 μM NEM-S1 are presented in Fig. 3. In the absence of NEM-S1, ktr varied with level of activation in both WT (1.4 ± 0.2 s−1 at pCa 6.0–30.0 ± 1.5 s−1 at pCa 4.5) and cMyBP-C−/− (4.2 ± 0.2 s−1 at pCa 6.1–30.6 ± 0.8 s−1 at pCa 4.5) myocardium. However, addition of 3 μM NEM-S1 significantly reduced the Ca2+ dependence of ktr in both WT (22.8 ± 0.8 s−1 at pCa 6.2–29.1 ± 1.0 s−1 at pCa 4.5) and cMyBP-C−/− (30.0 ± 1.1 s−1 at pCa 6.2–29.5 ± 0.8 s−1 at pCa 4.5) myocardium.
FIGURE 2.

Rate of force redevelopment in skinned myocardium from WT and cMyBP-C−/−. Differences in the rate constant of force redevelopment (ktr) were expressed relative to respective peak forces. (A) The rates of force redevelopment measured in the absence of NEM-S1 from two representative skinned myocardial preparations from either WT (left trace: pCa 4.5, P/Po = 1.0, ktr = 34.7 s−1; right trace: pCa 5.8, P/Po = 0.40, ktr = 5.1 s−1) or cMyBP-C−/− (middle trace: pCa 5.8, P/Po = 0.40, ktr = 11.4 s−1). (B) The rates of force redevelopment measured in the presence of NEM-S1 from two representative skinned myocardial preparations from either WT (left trace: pCa 4.5, P/Po = 1.0, ktr = 33.0 s−1; right trace: pCa 5.8, P/Po = 0.55, ktr = 16.1 s−1) or cMyBP-C−/− (middle trace: pCa 5.8, P/Po = 0.57, ktr = 23.5 s−1).
TABLE 2.
Summaries of ktr and Ca2+-activated force after treatment with NEM-S1
| WT
|
cMyBP-C−/−
|
|||||
|---|---|---|---|---|---|---|
| Control | 1 μM NEM-S1 | 3 μM NEM-S1 | Control | 1 μM NEM-S1 | 3 μM NEM-S1 | |
| pCa 4.5 | ||||||
| ktr (s−1) | 30.0 ± 1.5 | 29.6 ± 0.6 | 29.1 ± 1.0 | 30.6 ± 0.8 | 29.6 ± 0.7 | 29.5 ± 0.8 |
| pCa 6.0 | ||||||
| Force (P/Po) | 0.04 ± 0.01 | 0.12 ± 0.01 | 0.23 ± 0.02 | 0.07 ± 0.01 | 0.13 ± 0.02 | 0.29 ± 0.03 |
| ktr (s−1) | 1.4 ± 0.2 | 12.2 ± 1.1 | 18.2 ± 0.7 | 3.6 ± 0.2 | 15.6 ± 0.5 | 24.3 ± 1.1 |
| Relative ktr | 0.05 ± 0.01 | 0.41 ± 0.04 | 0.63 ± 0.03 | 0.12 ± 0.01 | 0.56 ± 0.04 | 0.83 ± 0.06 |
| pCa 5.8 | ||||||
| Force (P/Po) | 0.40 ± 0.02 | 0.38 ± 0.03 | 0.52 ± 0.02 | 0.38 ± 0.01 | 0.38 ± 0.02 | 0.53 ± 0.03 |
| ktr (s−1) | 3.3 ± 0.4 | 5.8 ± 0.3 | 11.1 ± 0.5 | 8.5 ± 0.3 | 10.4 ± 0.4 | 21.3 ± 0.9 |
| Relative ktr | 0.11 ± 0.01 | 0.20 ± 0.01 | 0.38 ± 0.01 | 0.28 ± 0.01 | 0.37 ± 0.03 | 0.73 ± 0.05 |
All values are expressed as mean ± SE. P/Po, relative Ca2+-activated tension; ktr, rate of tension redevelopment in s−1; and relative ktr, the relative rate of tension redevelopment. Both P/Po and relative ktr values obtained in pCa 6.0 and pCa 5.8 were normalized to maximal values obtained in pCa 4.5.
FIGURE 3.

Effect of NEM-S1 on the Ca2+ dependence of the rate of force redevelopment. Force redevelopment after rapid release and restretch was measured as a function of pCa in skinned myocardium from WT (○,•) and cMyBP-C−/− (Δ,▴) in the absence (open symbols) and presence (solid symbols) of 3 μM NEM-S1. All values are mean ± SE. Fiber characteristics are listed in Table 2.
The rate constant of force redevelopment was also plotted versus steady-state isometric force at each pCa to assess the variation of ktr as a function of the level of thin filament activation, induced by both Ca2+ and strong-binding cross-bridge (Fig. 4). In the absence of NEM-S1, ktr increased as a function of steady-state isometric force in WT myocardium, gradually at low forces and more steeply at high forces. However, in cMyBP-C−/− myocardium, we observed significantly faster rates of force redevelopment at all levels of submaximal activation compared to that observed in WT myocardium. These data are consistent with the idea that in normal myocardium, cMyBP-C constrains and thereby reduces the cooperative binding of endogenous cross-bridges to actin and in this way contributes to the activation dependence of the rate of force redevelopment. As seen in previous studies (37,39), the ktr-isometric force relationship was dramatically altered after NEM-S1 treatment. In WT myocardium, treatment with 3 μM NEM-S1 increased the rate of force redevelopment at low levels of activation to values near those obtained in maximally activated preparations. At intermediate levels of activation, NEM-S1 increased ktr in WT myocardium to values greater than those measured in control at similar relative isometric forces, but still less than the values obtained at both low and maximum Ca2+-activated forces. Treatment with NEM-S1 had qualitatively similar effects on the ktr-isometric force relationship in cMyBP-C−/− myocardium. But at all levels of submaximal activation, ktr values measured in cMyBP-C−/− myocardium were significantly faster than those measured in WT myocardium.
FIGURE 4.

Effect of NEM-S1 on the activation dependence of the rate of force redevelopment. Force redevelopment after rapid release and restretch was measured as a function of relative steady-state isometric force (P/Po) in skinned myocardium from WT (○,•) and cMyBP-C−/− (Δ,▴) in the absence (open symbols) and presence (solid symbols) of 3 μM NEM-S1. All values are mean ± SE. Fiber characteristics are listed in Table 2.
DISCUSSION
cMyBP-C is thought by many investigators to tether myosin cross-bridges to the thick filament, which would effectively control the availability of cross-bridges for binding to actin (12–14). Based on this premise, we investigated the idea that this constraint on cross-bridge availability would reduce the cooperative activation of force and the rate of force development by strong-binding cross-bridges during submaximal activations with Ca2+. We measured the activation dependence of isometric force and the rate constant of force development (ktr) in skinned myocardium from WT and cMyBP-C−/− mice. Experiments were done both in the absence and in the presence of NEM-S1 to determine the extent to which cMyBP-C modulates the responsiveness of the myocardial thin filament to the activating effects of strong-binding cross-bridges. In the absence of NEM-S1, WT and cMyBP-C−/− myocardium differed with regard to force generation. cMyBP-C−/− myocardium exhibited 1), a small, but significant increase in steady-state force at low levels of Ca2+ activation (Fig. 1 A), 2), a concomitant reduction in the steepness of the force-pCa relationship (Fig. 1 B), and 3), markedly greater rates of force development (ktr) during submaximal levels of Ca2+ activation (Figs. 3 and 4). Treatment of WT and cMyBP-C−/− myocardium with 3 μM NEM-S1 1) elicited significant increases in submaximal Ca2+-activated force (Fig. 1 A), with a corresponding increase in the Ca2+ sensitivity of force; and 2) markedly accelerated the rate of force development during submaximal levels of Ca2+ activation (Figs. 3 and 4), consistent with earlier results from WT myocardium (39). However, the activating effects of strong-binding cross-bridges were more pronounced in cMyBP-C−/− myocardium, suggesting that ablation of cMyBP-C further promotes cooperative interactions within the thin filament.
Steady-state mechanical properties of cMyBP-C−/− myocardium
Steady-state isometric force
Ablation of cMyBP-C did not affect either the Ca2+ sensitivity of force or maximal Ca2+-activated force (Table 1); however, there was a small but significant increase in steady-state force at low levels of Ca2+ activation compared to WT (Fig. 1 A), with a corresponding reduction in steepness of the force-pCa relationship for forces <0.50 Po (Fig. 1 B). The increase in submaximal force is consistent with the idea that cMyBP-C normally depresses the availability of myosin to actin, presumably via binding of the N-terminus of cMyBP-C to the S2 domain of myosin (42), i.e., ablation of cMyBP-C would eliminate this interaction and lead to greater probability of myosin binding to actin. The finding that there was no difference between cMyBP-C−/− and WT myocardium with respect to force at activation levels above half-maximal suggests that the activating effects of the small increase in bound cross-bridges with cMyBP-C ablation were obscured by the much greater activating effects of Ca2+ binding at higher levels of activation. Interestingly, in earlier studies, the acute extraction of cMyBP-C from rat myocardium resulted in increases in the Ca2+ sensitivity of force (12,13) and maximum force (13), which were not observed here (other than the increases in force at very low [Ca2+]). The differences in effects due to acute versus chronic ablation suggest that over time there are compensatory changes within the thick or thin filaments of cMyBP-C−/− myocardium that diminish or repress the effects on force due to cMyBP-C ablation. Although we haven't yet identified these compensatory changes, we have eliminated several possibilities such as myosin heavy chain expression (22) and troponin I phosphorylation (23), which are virtually unchanged in cMyBP-C−/− myocardium.
Rate of force development
Several investigators have reported that the rate of force development (ktr) in cardiac muscle is highly activation dependent (37,40,43,44), increasing ∼10-fold from low to high levels of activation. Typically, ktr is thought to be the sum of the forward (fapp) and reverse (gapp) rate constants describing the transition between force-generating and nonforce-generating states (33). A recent model by Campbell (38,45) predicts that activation dependence of ktr is a consequence of cooperativity in cross-bridge binding to the thin filament. At low levels of Ca2+, the number of noncycling cross-bridges is high, so that progressive recruitment of cross-bridges from this pool would act to slow the overall rate of force development. The first cross-bridges that bind recruit additional cross-bridges, which subsequently bind and recruit more cross-bridges and so forth, until force reaches a steady-state level. Thus, the rate of force development is slow at low Ca2+ due to progressive cooperative recruitment of cross-bridges to force-generating states. At higher levels of Ca2+, the cooperative slowing of force development is reduced because Ca2+-binding to TnC would immediately recruit most cross-bridges into the cycling pool, leaving relatively few noncycling cross-bridges available for cooperative recruitment.
In this study, ablation of cMyBP-C accelerated ktr measured during submaximal activations. MyBP-C is associated with the thick filaments, and based on studies of skeletal muscle (10) its location is presumably limited to a series of transverse stripes within the C-zones of the A-bands, such that the stoichiometric relationship between cMyBP-C and myosin cross-bridges is ∼1 cMyBP-C molecule for every 7–9 cross-bridges (10). This stoichiometry implies that cMyBP-C exerts its effects directly on only a small subset of cross-bridges, so that any effects of cMyBP-C on cross-bridge function are limited to a relatively small fraction of the total or are somehow communicated to other cross-bridges or to thin filament functional groups to alter contractile properties of the myofilament. Our results don't eliminate any of these possibilities but strongly support the idea that cMyBP-C modulation of cross-bridge availability influences the level of thin filament activation. Consistent with this idea, ablation of cMyBP-C resulted in a significant acceleration of the kinetics of force development at all levels of submaximal Ca2+ activation, but the most pronounced effects on the kinetics of force development occurred at low levels of activation where cooperative activation is most pronounced (9). The dramatic acceleration of cross-bridge kinetics observed in cMyBP-C−/− myocardium at low levels of activation is to be disproportionate with the modest increases in submaximal Ca2+-activated force in cMyBP-C−/− myocardium, which provides further support to the idea that the kinetics of force development in myocardium are sensitive to even small increases in numbers of cross-bridges (9).
Hofmann et al. (12) proposed that cMyBP-C tethers cross-bridges to the thick filament backbone and thereby represses their interaction with actin. Recent work has identified distinct structural domains within cMyBP-C that could contribute to formation of a collar around the thick filament, whereas other domains in N-terminal regions of cMyBP-C interact with the S2 domain of myosin (21). Eliminating cMyBP-C would also eliminate these interactions and presumably increase the mobility of cross-bridges and the likelihood of their interaction with actin, particularly at low levels of Ca2+. In cMyBP-C−/− myocardium, there is greater probability that the thin filament will be cooperatively activated by strong-binding cross-bridges at low levels of Ca2+ activation, which would reduce the slope of the force-pCa relationship and accelerate the rate of force development and in turn reduce the apparent cooperativity of the activation process. Consistent with this idea, the rate of force redevelopment was accelerated during submaximal Ca2+ activations of cMyBP-C−/− myocardium (Fig. 4). As observed previously (23), we did not observe any significant differences in the maximal rate of force redevelopment of cMyBP-C−/− and WT myocardium. This result is predictable given that at saturating levels of Ca2+ (pCa 4.5), there is little, if any, cooperative recruitment of cross-bridges.
Effects of strong-binding myosin cross-bridges
Force development in cardiac muscle appears to be very sensitive to the effects of even small numbers of strong-binding cross-bridges to cooperatively activate the myocardial thin filament (4,37,40,41). This feature of cardiac muscle suggested to us that ablation of cMyBP-C and the concomitant increase in endogenous strong-binding cross-bridges at submaximal concentrations of Ca2+ would alter the responsiveness of the cardiac thin filament to the activating effects of NEM-S1. In WT myocardium, the activating effects of strong-binding cross-bridges is particularly evident even at very low levels of Ca2+ (e.g., pCa 9.0), where virtually no Ca2+ is bound to TnC. Previous studies (37) showed that treatment of rat skinned myocardium with 6 μM NEM-S1 elicited a Ca2+-independent force that was ∼20% of maximal Ca2+-activated force. In this study, 3 μM NEM-S1 evoked Ca2+-independent forces that were 15% of maximal in WT myocardium and 24% of maximal in cMyBP-C−/− myocardium. Furthermore, whereas the Ca2+ sensitivity of force did not differ between WT and cMyBP-C−/− myocardium, we observe a greater NEM-S1-induced increase in submaximal force in cMyBP-C−/− myocardium compared to WT. The greater responsiveness of cMyBP-C−/− myocardium to 3 μM NEM-S1 supports the idea that ablation of cMyBP-C predisposes the thin filament to increased activation by strong-binding cross-bridges.
NEM-S1 treatment of WT and cMyBP-C−/− myocardium also accelerated the rate of force redevelopment at low Ca2+; however, as was seen in the effects of NEM-S1 on steady force, the rate of force redevelopment was increased to a much greater degree in cMyBP-C−/− myocardium. In fact, NEM-S1-treated cMyBP-C−/− myocardium exhibited rates of force redevelopment at low [Ca2+] (Fig. 4), similar to those observed during maximal activation. Thus, NEM-S1 significantly increased ktr in cMyBP-C−/− myocardium and thereby reduced the variation in ktr with level of activation, whereas in WT myocardium NEM-S1 evoked near-maximal ktr values only at very low levels of activation. As seen in previous studies (37,39) treatment with NEM-S1 did not alter the maximal rate of force redevelopment in WT or cMyBP-C−/− myocardium, again because the cooperative recruitment of cross-bridges is small or negligible during maximal Ca2+ activation (38).
Overall, we found that ablation of cMyBP-C led to marked acceleration in the rates of force redevelopment during submaximal activation and increased the responsiveness of the myocardial thin filament to the activating effects of strong-binding cross-bridges. Together, these results provide further support for the idea that cMyBP-C normally acts to modulate (repress) the interaction of myosin with actin, which in turn limits steady-state force development and the kinetics of cross-bridge binding to actin.
Acknowledgments
The authors thank Dr. R. J. Solaro and Mr. C. M. Warren (University of Illinois-Chicago) for the generous gift of the NEM-S1 used in these experiments.
This work was supported by grants PO1 HL47053, R01 HL82900, National Institutes of Health PAR-02-110 (to R.L.M.), and an American Heart Association postdoctoral fellowship (to J.E.S.).
References
- 1.Solaro, R. J., and H. M. Rarick. 1998. Troponin and tropomyosin: proteins that switch on and tune in the activity of cardiac myofilaments. Circ. Res. 83:471–480. [DOI] [PubMed] [Google Scholar]
- 2.Lehrer, S. S. 1994. The regulatory switch of the muscle thin filament: Ca2+ or myosin heads. J. Muscle Res. Cell Motil. 15:232–236. [DOI] [PubMed] [Google Scholar]
- 3.Swartz, D. R., R. L. Moss, and M. L. Greaser. 1996. Calcium alone does not fully activate the thin filament for S1 binding to rigor myofibrils. Biophys. J. 71:1891–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon, A. M., E. Homsher, and M. Regnier. 2000. Regulation of contraction in striated muscle. Physiol. Rev. 80:853–924. [DOI] [PubMed] [Google Scholar]
- 5.Johnson, J. D., J. H. Collins, S. P. Robertson, and J. D. Potter. 1980. A fluorescent probe study of Ca2+ binding to the Ca2+ specific sites of cardiac troponin and troponin C. J. Biol. Chem. 255:9635–9640. [PubMed] [Google Scholar]
- 6.Martin, A. F., R. M. Phillips, A. Kumar, K. Crawford, Z. Abbas, J. L. Lessard, P. deTombe, and R. J. Solaro. 2002. Ca2+ activation and tension cost in myofilaments from mouse hearts ectopically expressing enteric γ-actin. Am. J. Physiol. Heart Circ. Physiol. 283:H642–H649. [DOI] [PubMed] [Google Scholar]
- 7.Olsson, M. C., J. R. Patel, D. P. Fitzsimons, J. W. Walker, and R. L. Moss. 2004. Basal myosin light chain phosphorylation is a determinant of Ca2+ sensitivity of force and activation dependence of the kinetics of myocardial force development. Am. J. Physiol. Heart Circ. Physiol. 287:H2712–H2718. [DOI] [PubMed] [Google Scholar]
- 8.Rundell, V. L. M., V. Manaves, A. F. Martin, and P. P. deTombe. 2005. Impact of b-myosin heavy chain isoform expression on cross-bridge cycling kinetics. Am. J. Physiol. Heart Circ. Physiol. 288:H896–H903. [DOI] [PubMed] [Google Scholar]
- 9.Moss, R. L., M. Razumova, and D. P. Fitzsimons. 2004. Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circ. Res. 94:1290–1300. [DOI] [PubMed] [Google Scholar]
- 10.Craig, R., and G. Offer. 1976. The location of C-protein in rabbit skeletal muscle. Proc. R. Soc. Lond. B Biol. Sci. 192:451–461. [DOI] [PubMed] [Google Scholar]
- 11.Squire, J. M., P. K. Luther, and C. Knupp. 2003. Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J. Mol. Biol. 331:713–724. [DOI] [PubMed] [Google Scholar]
- 12.Hofmann, P. A., H. C. Hartzell, and R. L. Moss. 1991. Alterations in Ca2+ sensitive tension due to partial extraction of C-protein from rat skinned cardiac myocytes and rabbit skeletal muscle fibers. J. Gen. Physiol. 97:1141–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kulikovskaya, I., G. McClellan, R. Levine, and S. Winegrad. 2003. Effect of extraction of myosin binding protein C on contractility of rat heart. Am. J. Physiol. Heart Circ. Physiol. 285:H857–H865. [DOI] [PubMed] [Google Scholar]
- 14.Flashman, E., C. Redwood, J. Moolman-Smook, and H. Watkins. 2004. Cardiac myosin binding protein C: its role in physiology and disease. Circ. Res. 94:1279–1289. [DOI] [PubMed] [Google Scholar]
- 15.Starr, R., and G. Offer. 1978. The interaction of C-protein with heavy meromyosin and subfragment-2. Biochem. J. 171:813–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okagaki, T., F. E. Weber, D. A. Fischman, K. T. Vaughan, T. Mikawa, and F. C. Reinach. 1993. The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif. J. Cell Biol. 123:619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calaghan, S. C., J. Trinick, P. J. Knight, and E. White. 2000. A role for C-protein in the regulation of contraction and intracellular Ca2+ in intact rat ventricular myocytes. J. Physiol. 528:151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kunst, G., K. R. Kress, M. Gruen, D. Uttenweiler, M. Gautel, and R. H. A. Fink. 2000. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ. Res. 86:51–68. [DOI] [PubMed] [Google Scholar]
- 19.Harris, S. P., E. Rostkova, M. Gautel, and R. L. Moss. 2004. Binding of myosin binding protein-C to myosin subfragment S2 affects contractility independent of a tether mechanism. Circ. Res. 95:930–936. [DOI] [PubMed] [Google Scholar]
- 20.Offer, G., C. Moos, and R. Starr. 1973. A new protein of the thick filaments of vertebrate skeletal myofibrils: extractions, purification and characterization. J. Mol. Biol. 74:653–676. [DOI] [PubMed] [Google Scholar]
- 21.Moolman-Smook, J., E. Flashman, W. de Lange, Z. Li, V. Corfield, C. Redwood, and H. Watkins. 2002. Identification of novel interactions between domains of myosin binding protein-C that are modulated by hypertrophic cardiomyopathy missense mutations. Circ. Res. 91:704–711. [DOI] [PubMed] [Google Scholar]
- 22.Harris, S. P., C. R. Bartley, T. A. Hacker, K. S. McDonald, P. S. Douglas, M. L. Greaser, P. A. Powers, and R. L. Moss. 2002. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ. Res. 90:594–601. [DOI] [PubMed] [Google Scholar]
- 23.Korte, F. S., K. S. McDonald, S. P. Harris, and R. L. Moss. 2003. Loaded shortening, power output, and the rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ. Res. 93:752–758. [DOI] [PubMed] [Google Scholar]
- 24.Palmer, B. M., T. Noguchi, Y. Wang, J. R. Heim, N. A. Alpert, P. G. Burgon, C. E. Seidman, J. G. Seidman, D. W. Maughan, and M. M. LeWinter. 2004. Effect of cardiac myosin binding protein-C on mechanoenergetics in mouse myocardium. Circ. Res. 94:1615–1622. [DOI] [PubMed] [Google Scholar]
- 25.Fabiato, A. 1988. Computer programs for calculating total from specified free and free from specified total ionic concentrations in aqueous solutions containing multiple metals or ligands. Methods Enzymol. 157:378–417. [DOI] [PubMed] [Google Scholar]
- 26.Godt, R. E., and B. D. Lindley. 1982. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. J. Gen. Physiol. 80:279–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel, J. R., D. P. Fitzsimons, S. H. Buck, M. Muthuchamy, D. F. Wieczorek, and R. L. Moss. 2001. PKA accelerates rate of force development in murine skinned myocardium expressing α- or β-tropomyosin. Am. J. Physiol. Heart Circ. Physiol. 280:H2732–H2739. [DOI] [PubMed] [Google Scholar]
- 28.Moss, R. L. 1979. Sarcomere length-tension relations of frog skinned muscle fibers during calcium activation at short lengths. J. Physiol. 292:177–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campbell, K. S., and R. L. Moss. 2003. SLControl: PC-based data acquisition and analysis for muscle mechanics. Am. J. Physiol. Heart Circ. Physiol. 285:H2857–H2864. [DOI] [PubMed] [Google Scholar]
- 30.Swartz, D. R., and R. L. Moss. 1992. Influence of a strong-binding myosin analogue on calcium-sensitive mechanical properties of skinned skeletal muscle fibers. J. Biol. Chem. 267:20497–20506. [PubMed] [Google Scholar]
- 31.Williams, D. L., L. E. Greene, and E. Eisenberg. 1984. Comparison of effects of smooth and skeletal muscle tropomyosins on interactions of actin and myosin subfragment 1. Biochemistry. 23:4150–4155. [DOI] [PubMed] [Google Scholar]
- 32.Greene, L. E., D. L. Williams, and E. Eisenberg. 1987. Regulation of actomyosin ATPase activity by troponin-tropomyosin: effect of the binding of the myosin subfragment 1 (S-1)-ATP complex. Proc. Natl. Acad. Sci. USA. 84:3102–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brenner, B., and E. Eisenberg. 1986. Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl. Acad. Sci. USA. 83:3542–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Regnier, M., D. A. Martyn, and P. B. Chase. 1998. Calcium regulation of tension redevelopment kinetics with 2-deoxy-ATP or low [ATP] in rabbit skeletal muscle. Biophys. J. 74:2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fitzsimons, D. P., J. R. Patel, K. S. Campbell, and R. L. Moss. 2001. Cooperative mechanisms in the activation dependence of the rate of force development in rabbit skinned skeletal muscle fibers. J. Gen. Physiol. 117:133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strang, K. T., N. K. Sweitzer, M. L. Greaser, and R. L. Moss. 1994. Beta-adrenergic receptor stimulation increases unloaded shortening velocity of skinned single ventricular myocytes from rats. Circ. Res. 74:542–549. [DOI] [PubMed] [Google Scholar]
- 37.Fitzsimons, D. P., J. R. Patel, and R. L. Moss. 2001. Cross-bridge interaction kinetics in rat myocardium are accelerated by strong binding of myosin to the thin filament. J. Physiol. 530:263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell, K. 1997. Rate constant of muscle force redevelopment reflects cooperative activation as well as cross-bridge kinetics. Biophys. J. 72:254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stelzer, J. E., J. R. Patel, M. C. Olsson, D. P. Fitzsimons, L. A. Leinwand, and R. L. Moss. 2004. Expression of cardiac troponin T with COOH-terminal truncation accelerates cross-bridge interaction kinetics in mouse myocardium. Am. J. Physiol. Heart Circ. Physiol. 287:H1756–H1761. [DOI] [PubMed] [Google Scholar]
- 40.Regnier, M., A. J. Rivera, Y. Chen, and P. B. Chase. 2000. 2-deoxy-ATP enhances contractility of rat cardiac muscle. Circ. Res. 86:1211–1217. [DOI] [PubMed] [Google Scholar]
- 41.Regnier, M., H. Martin, R. J. Barsotti, A. J. Rivera, D. A. Martyn, and E. Clemmens. 2004. Cross-bridge versus thin filament contributions to the level and rate of force development in cardiac muscle. Biophys. J. 87:1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gruen, M., and M. Gautel. 1999. Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J. Mol. Biol. 286:933–949. [DOI] [PubMed] [Google Scholar]
- 43.Wolff, M. R., K. S. McDonald, and R. L. Moss. 1995. Rate of tension development in cardiac muscle varies with level of activator calcium. Circ. Res. 76:154–160. [DOI] [PubMed] [Google Scholar]
- 44.Palmer, S., and J. C. Kentish. 1998. Roles of Ca2+ and crossbridge kinetics in determining the maximum rates of Ca2+ activation and relaxation in rat and guinea pig skinned trabeculae. Circ. Res. 83:179–186. [DOI] [PubMed] [Google Scholar]
- 45.Razumova, M. V., A. E. Bukatina, and K. B. Campbell. 2000. Different myofilament nearest-neighbor interactions have distinctive effects on contractile behavior. Biophys. J. 78:3120–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]