

Abstract
Oncolytic herpes simplex virus-1 (HSV-1) mutants selectively replicate in and lyse tumor cells. Viral replication is dependent on the cellular proliferative mechanism. Estrogen increases cellular proliferation and decreases apoptosis in estrogen receptor positive (ER+) human breast cancer cells. We hypothesize that the cellular changes produced by estrogen may enhance oncolytic viral replication and improve treatment of ER+ breast cancer cells.
Estrogen increased proliferation and replication of the HSV-1 mutant NV1066 in ER+ breast cancer cells. Additionally, cells grown with estrogen had lower rates of apoptosis and higher bcl-2 levels at baseline and after infection. Estrogen enhanced the oncolytic effect of NV1066, with cell kills of 95% and 97% at MOIs of 0.1 and 0.5, compared to 53% and 87% respectively without estrogen (p < 0.001).
Therapy of ER+ human breast cancer cells with a replication-competent HSV-1 mutant is improved in the presence of estrogen, in contrast to more standard therapies, such as chemotherapy and radiation, which demonstrate decreased efficacy in similar conditions. These data provide the mechanistic basis for the use of an oncolytic HSV-1 in patients with hormone receptor positive breast cancer, particularly if disease progresses with conventional therapies.
Keywords: Apoptosis, Bcl-2, Herpes simplex, Hormonal therapy, Replication-competent viruses
Introduction
Breast cancer is the most commonly diagnosed new cancer in women, excluding cancers of the skin. In 2004, breast cancer alone accounted for approximately one-third of new cancer cases and 15% of cancer deaths in women in the United States(1). Breast cancer cells are often responsive to hormones, with estrogen and its metabolites being the most widely studied effectors. The cellular effects of estrogen have been implicated as possible factors in the induction and promotion of carcinogenesis(2). Additionally, estrogenic effects on cells have been demonstrated to decrease the efficacy of various chemotherapeutics and radiation therapy in vitro(3–6). Although early stage cancers may often be effectively treated with combination therapeutic regimens, the 5-year survival rates for stage III and IV breast cancer are only 46% and 12%, respectively(7). New treatment strategies are needed, particularly for patients with late stage disease and for those with tumors resistant to standard treatment protocols.
Multimutated replication-competent herpes simplex viruses (HSV) are an effective therapy against a number of malignancies in experimental models(8–13). Oncolytic HSV are genetically engineered to selectively replicate within tumor cells, sparing normal tissues. HSV-1 viral mutants are known to cause both lysis and apoptosis in targeted tumor cells(14–16). The antitumor efficacy of HSV-1 mutants depends upon their ability to infect and propagate within the tumor and upon host cell defense mechanisms that limit viral replication. Previous studies have demonstrated the effectiveness of HSV-1 mutants against both estrogen receptor positive (MCF-7 and T47D) and negative (MDA-MB-231 and MDA-MB-435) breast cancer cell lines(8;17). Given the established role of tamoxifen and of newer selective estrogen receptor modulators (SERMs) in the treatment of breast cancer, the interaction between hormonal therapy and oncolytic HSV-1 therapy deserves examination.
We hypothesize that in contrast to other in vitro therapies, estrogen will improve treatment of ER+ human breast tumors with an HSV-1 mutant due to estrogenic effects on cells, namely increasing cellular proliferation and decreasing apoptosis. These effects may create an environment favorable to viral replication and proliferation. We therefore designed this study to explore the effect of hormonal milieu on the efficacy of an HSV-1 mutant, NV1066, in the treatment of breast cancer cells. Specifically, the purpose of this study was to determine whether the virus is effective in the treatment of both hormone sensitive and hormone independent breast cancer cell lines, to assess the effects of estrogen on viral replication and efficacy, and to examine the mechanisms by which estrogen may affect viral therapy.
Materials and Methods
Cells and cell culture
MCF7, T-47D, and HCC38 cell lines were obtained from the American Type Culture Collection (ATCC®, Rockville, MD). MCF7 and T-47D cells are ER+, while HCC38 cells are estrogen receptor negative (ER−). MCF7 cells were grown in phenol red free MEM with 2 mM L-glutamine, 1.0 mM sodium pyruvate, 0.1 mM nonessential amino acids, 1.5 g/L sodium bicarbonate, 100 U/ml penicillin and 100 mg/ml streptomycin, and 10% charcoal stripped FCS (Atlanta Biologicals, Norcross, GA). T-47D and HCC38 cells were grown in phenol red free RPMI 1640 with 2 mM L-glutamine, 10 mM HEPES, 1.0 mM sodium pyruvate, 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, 100 U/ml penicillin and 100mg/ml streptomycin, and 10% charcoal stripped FCS. The serum contained less than 5 pg/ml of 17 β-estradiol and estrone per the manufacturer’s instructions. SKBr3, MDA-MB-435, and HCC1954 cells were similarly obtained from the ATCC® and grown in the recommended media. Cells were passaged either in the presence of 10 nM β-estradiol in DMSO vehicle or with vehicle alone. After one week in culture, distinct populations of cells were plated for experiments with either 10 nM β-estradiol or vehicle alone, added at a 1/1000 volume ratio to the media.
Virus
NV1066 is a replication-competent attenuated herpes simplex-1 mutant virus described previously(18). Briefly, the virus lacks single copies of the ICP-4, ICP-0, and γ134.5 genes (Figure 1). The ICP-4 and γ134.5 protein products have important anti-apoptotic roles in wild-type HSV-1(19;20). The virus additionally contains the enhanced green fluorescent protein (EGFP) sequence under the control of a cytomegalovirus (CMV) promoter. Viral stocks were generated from Vero cells using standard techniques.
Figure 1.
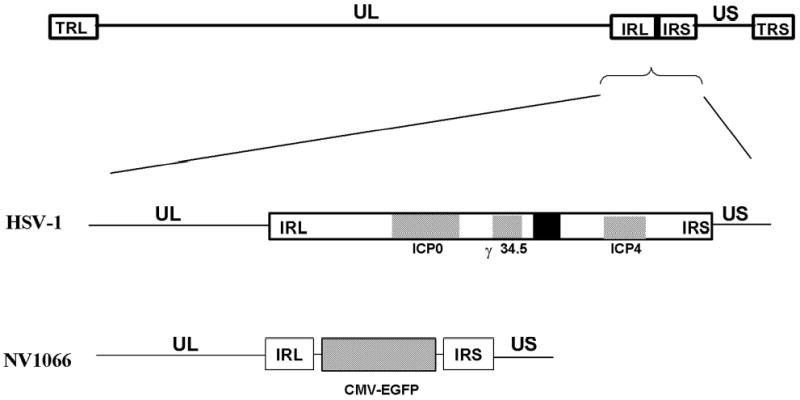
Molecular structure of NV1066. The wild-type HSV-1 genome consists of the unique long (UL) and unique short (US) sequences, flanked by inverted repeats, terminal and internal repeats long (TRL and IRL) and terminal and internal repeats short (TRS and IRS). NV1066 has deletions from the internal repeat sequences, with loss of one copy each of the ICP-0, ICP-4, and γ134.5 genes. The ICP-4 and γ134.5 genes have important anti-apoptotic functions in wild-type virus. A sequence for enhanced green fluorescent protein (EGFP) has been inserted into the viral backbone under the control of a constitutive cytomegalovirus promoter.
Determination of cellular proliferation
MCF7 cells were plated in 6-well plates (Costar, Corning Inc., Corning, NY) for assessment of cellular proliferation. Either 10 nM β-estradiol or vehicle was added to the cell suspension prior to plating. Each condition was assessed in triplicate. Cells were harvested with 0.25% trypsin in 0.02% EDTA and live cells were counted with trypan blue exclusion of dead cells.
Cell cycle analysis
Cell cycle distribution of MCF7 cells was measured using flow cytometry. Cells were harvested with 0.25% trypsin in 0.02% EDTA and centrifuged. Cells grown with either 10 nM β-estradiol or with vehicle alone were suspended in NP-40 solution (10 mM NaCl, 3.4 mM sodium citrate, 0.03% NP-40, 63 μM ethidium bromide, 10 μg/ml RNase) and incubated (1 hr) with gentle shaking. An equal volume of high sucrose solution (0.25 M sucrose, 78 mM citric acid, 100 μM ethidium bromide) was added, cells were centrifuged, and the pellet was re suspended in 0.5 ml of the combined solution. DNA content of ethidium bromide-stained nuclei was determined on a FACS Calibur machine equipped with CellQuest software (Becton Dickinson, San Jose, CA).
TUNEL Staining
MCF7 cells were plated in 4-well chamber slides (Lab-Tek, San Diego, CA) either with 10 nM β-estradiol or vehicle prior to TUNEL staining. Cells were incubated (37°C, 48 hr), then harvested, air-dried, and fixed (4% paraformaldehyde, 12 min). Cells were treated with 20 μg/ml proteinase K (2 min), then fixed again (4% paraformaldehyde, 2 min). After three phosphate buffered saline (PBS) washes, endogenase peroxidase was quenched with 0.1% H2O2 (15 min). After a dH2O rinse, slides were immersed in terminal deoxynucleotidyl transferase (TdT) buffer (3 mM Tris, pH 7.2; 14 mM sodium cacodylate; 1 mM Cobalt Chloride, 10 min). Next, cells were exposed to 30 units of TdT (Roche Applied Science, Indianapolis, IN) and 5μM biotin-dUTP per slide at 37°C in a moist chamber (1 hr). The reaction was stopped with 2X SSC and cells were blocked with 2% BSA in PBS to eliminate nonspecific reactivity. Slides were then exposed to the secondary stain (1 hr), washed in PBS and treated with 0.5% Triton-X100 (2 min). Slides were stained with a filtered diaminobenzidine (DAB) and H2O2 solution (0.0012%), counterstained with Harris Hematoxylin, washed in dH20 and mounted with mounting media. Several fields were inspected for TUNEL positivity under 40X magnification in bright-field mode using a Zeiss Axiophot-2 microscope (Carl Zeiss, Inc., Germany). The image-capture system consisted of a Retiga EX digital CCD camera (Qimaging, Burnaby, BC, Canada). At least 200 cells were examined. The same experiment was performed, infecting cells with a multiplicity of infection (MOI: number of viral plaque forming units per tumor cell) of 1.0 for the last 24 hours of the incubation.
Hoechst staining
MCF7 cells were plated in 4-well chamber slides either with 10nM β-estradiol or vehicle as previously described. Cells were incubated (37°C, 72 hr), then harvested, air-dried and fixed (4% paraformaldehyde, 12 min). After two PBS washes, cells were stained with 1 μg/ml DNA specific fluorochrome, Hoechst 33342 (NPE Systems, Inc., Pembroke Pines, FL: 10 min). Slides were washed in PBS twice and mounted with fluorescent mounting media. Several fields were examined until at least 200 cells were counted. Cells were first examined under a DAPI fluorescent filter to assess nuclear morphology. Cells were determined to be apoptotic based on accepted criteria for nuclear morphology including nuclear pyknosis, chromatin condensation, nuclear blebbing and fragmentation, and the formation of apoptotic bodies(21). Cells were also examined following infection with NV1066 at an MOI of 1.0 for the last 48 hrs of the incubation. Again, microscopic fields were examined for nuclear morphology using a DAPI fluorescent filter. The same microscopic fields were examined after changing the fluorescent filter to a GFP filter (fixed to allow passage of 470 ± 40 nm wavelength light). DAPI and GFP images were taken again using a Retiga EX digital CCD camera. Two scientists blinded to the treatment groups evaluated the specimens.
Quantitation of bcl-2
MCF7 cells were plated in 6-well flat-bottom plates with either 10 nM β-estradiol or vehicle. Cells were then infected with NV1066 at MOIs of 10.0 or 1.0 and incubated (37°C, 16 hr). Uninfected cells served as controls. Cells were lysed, proteins extracted with M-PER protein extraction reagent (Pierce, Rockford, IL), and bcl-2 assessed by standard ELISA (Oncogene Research Products, San Diego, CA). All conditions were performed in triplicate. Bcl-2 levels were standardized per total protein levels as measured by Bradford protein assay, using the total protein in the uninfected cells grown in the presence of estrogen as the control. For the manufacturer’s assay standard, one unit of bcl-2 equals the amount in the lysate of 5.6 x 104 HL60 cells.
Flow cytometry for EGFP
MCF7 cells were plated in 12-well plates with either 10 nM β-estradiol or vehicle and subsequently infected with NV1066 at MOIs of 0.1 or 0.5. Untreated cells served as negative controls. Cells were harvested with 0.25% trypsin in 0.02% EDTA, combined with the supernatant fraction, centrifuged, washed in PBS, and brought up in 100 μl PBS. Five μl of 7-amino-actinomycin (7-AAD, BD Pharmingen, San Diego, CA) was added as an exclusion dye for cell viability. Data for EGFP expression from 1x104 cells was acquired on a FACS Calibur machine and analyzed with Cell Quest software.
Viral Titering
MCF7 were plated in 6-well plates with either 10nM β-estradiol or vehicle and subsequently infected with NV1066 at MOIs of 0.1 or 0.5. Cells were harvested by cell scraping, combined with supernatant, and then subjected to three cycles of freeze-thaw lysis. Serial dilutions were made of the suspension and viral plaques were grown and counted on confluent Vero cells in a standard viral plaque assay. All samples were tested in triplicate.
Cytotoxicity assay
MCF7, T-47D, or HCC38 cells were plated in 12-well flat-bottom plates with either 10 nM β-estradiol or vehicle and subsequently infected with NV1066 at MOIs of 0.1 or 0.5. Every other day after infection, cells were washed in PBS and lysed (1.35% Triton-X) to release intracellular lactate dehydrogenase (LDH). The cytotoxic effect of viral infection on tumor cells was determined by comparison of levels of LDH in the uninfected and infected cellular lysates. LDH was quantified using a Cytotox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI) that measures the conversion of a tetrazolium salt into a formazan product. Absorbance was measured at 450 nm using a microplate reader (EL 312e: Bio-Tek Instruments, Winooski, VT) Results are expressed as the surviving fraction, based on the percentage of the LDH release compared to that of untreated, control cells. All samples were tested in triplicate.
Statistics
All pair wise comparisons were performed using the exact permutation distribution of the Wilcoxon test. To compare viral plaque forming units (PFU) over time, area under each viral-time curve was obtained and then compared using the Wilcoxon test.
Results
Effect of estrogen on cellular proliferation and cell cycle
To confirm estrogenic effects on ER+ human breast cancer cells, MCF7 cells were grown either in the presence of 10 nM β-estradiol or vehicle. Cells grown in 10 nM β-estradiol proliferated more rapidly than those grown without estrogen. After 10 days in culture (Figure 2a), there was a 2 fold increase in cell number in the cells grown in 10nM β-estradiol, compared to cells grown without estrogen (p < 0.05). To determine whether this observed effect correlated with population distribution, cell cycle analysis was performed on MCF7 cells (Figure 2b). 10nM β-estradiol increased the fraction of cells in S + G2 phases from 34% (± 0.4 %) to 44% (± 0.0%), while it decreased the fraction of cells in the resting G1 phase from 66% (± 0.4%) to 56% (± 0.0%). This relationship was maintained after 7 days in culture, although as cells became more confluent, the fraction of cells in the resting phase increased in both groups, 82% (± 0.2%) vs. 64% (± 0.1%) for no estrogen and 10nM β-estradiol respectively, with a corresponding decrease in percentage of cells in the S + G2 phase (p = 0.05).
Figure 2.
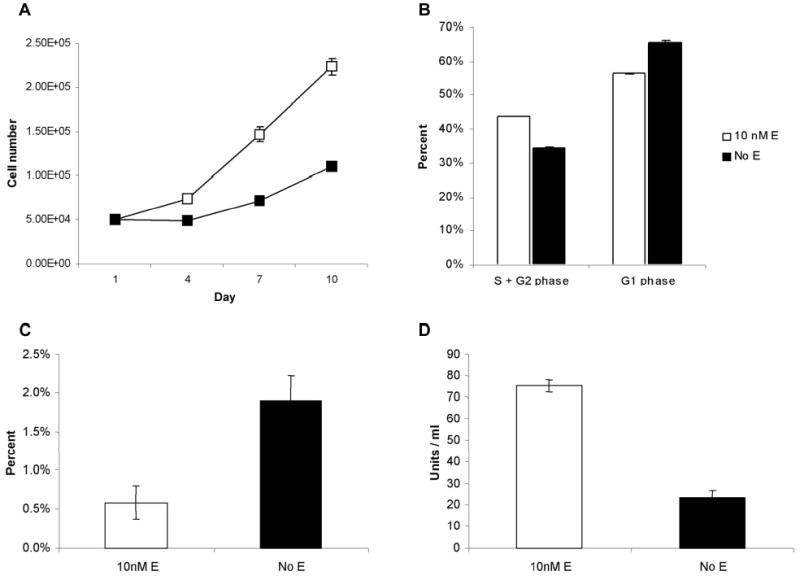
Confirmation of estrogenic effects on MCF7 cells. (a) Cellular proliferation with 10nM estrogen and with vehicle alone. There is a 2-fold increase in cell growth of MCF-7 cells in the presence of estrogen after 10 days (p < .02). (b) Cell cycle analysis. Estrogen increases the fraction of cells in S+G2 phases and decreases the fraction of cells in the resting G1 phase as measured by flow cytometry. (c) Baseline apoptotic rates. The absence of estrogen significantly increases baseline apoptotic rates in MCF7 cells. (d) Bcl-2 ELISA. Cells grown in estrogen have a 3.2 fold higher level of bcl-2 expression.
Determination of apoptotic index and bcl-2 levels
To determine baseline apoptotic rates within MCF7 cell populations, TUNEL staining, a nuclear stain that binds labeled uridine bases to the DNA nicks characteristic of apoptotic cells was performed and cells were counted under 40X magnification. The percentage of apoptotic cells was determined to be 0.59% (± 0.21%) in cells grown in 10 nM β-estradiol and 1.90% (± 0.31%) in cells grown in the absence of estrogen in non-confluent populations (Figure 2c, p = 0.05). This three-fold decrease in the percentages of apoptotic cells when grown with 10 nM β-estradiol was confirmed by examining cells stained with Hoescht nuclear stain under a DAPI filter (0.40 ± 0.07% vs. 1.35 ± 0.29%).
Because apoptotic rates have been demonstrated to inversely correlate with intracellular bcl-2 protein levels(22), we examined bcl-2 production using an ELISA to compare levels of this anti-apoptotic protein within the different cell populations. MCF7 cells were cultured either in the presence or absence of 10 nM β-estradiol for one week. The ELISA was performed with bcl-2 levels standardized to total protein levels. Cells grown in 10 nM β-estradiol had an approximate 3.2-fold increase in bcl-2 levels compared to cells grown without estrogen, with the cellular lysate containing 75 (± 3) units/ml of bcl-2 as compared to 24 (± 3) units/ml in cells grown without estrogen (Figure 2d, p = 0.05).
Viral proliferation
The EGFP transgene carried by NV1066 was used as a marker of infection, the protein product of which was detected by flow cytometry. EGFP is expressed 4–6 hours after viral entry into the cell and can localize functional virus. EGFP expression was used to infer viral proliferation, or percentage of live cells actively infected over time, among cell populations treated with either estrogen or no estrogen and subsequently with NV1066. With continued viral replication, all cell populations trended toward 100% infection after 5 days (Figure 3a). Cell-to-cell spread of virus, however, was significantly faster in cell populations grown in estrogen, particularly at the lower MOI, with differences as high as 20–25% in the first 2 or 3 days. It is striking that by day 5 after infection, an MOI of 0.1 in the cell population grown in 10 nM β-estradiol resulted in a higher percentage of infected cells than an MOI of 0.5 in cells grown without estrogen (Figure 3a, p = 0.05).
Figure 3.
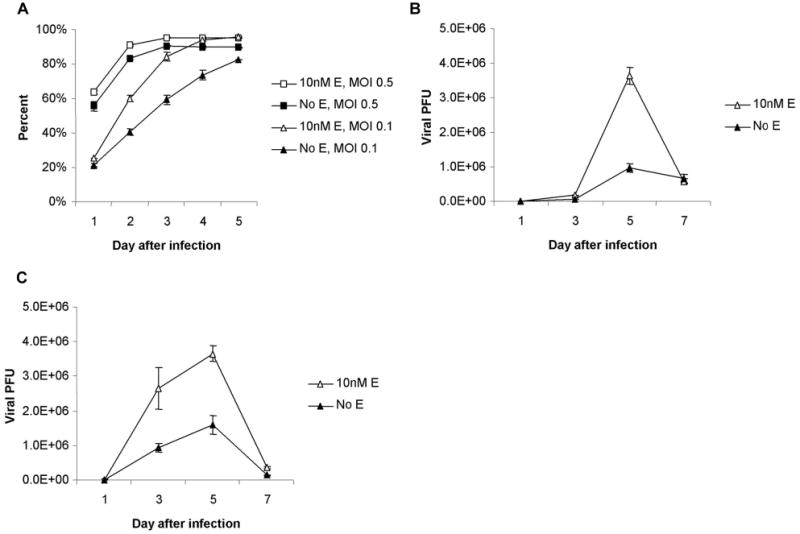
EGFP expression and viral replication in MCF-7 cells grown with or without 10nM β-estradiol. (a) Growth of cells in estrogen leads to significantly greater viral proliferation among live cells as measured by FACS analysis of EGFP expression. Even at a five-fold lower dose of virus (MOI 0.1 vs. MOI 0.5), significantly more EGFP expression is present in cells grown with estrogen by day 5 (p < .002). (b and c) Greater replication of NV1066 occurs in the presence of estrogen at MOIs of both 0.1 (b) and 0.5 (c). Peak viral titers occur on day 5, thereafter titers decrease as cell death limits further replication. Viral growth was 2.9 and 2.5 fold higher in MCF-7 cells grown in the presence of 10nM B-estradiol infected at MOIs of 0.1 and 0.5 respectively. More virus was produced at an MOI of 0.1 in cells infected in the presence of estrogen than at an MOI of 0.5 in cells infected without estrogen.
Viral replication
EGFP expression was measured at several time points in the first 18 hrs after infection with NV1066, with no significant differences in expression between cells grown with and without estrogen (data not shown). This suggested that there was no difference in initial viral binding and entry into cells in the two populations. To compare the amount of viral replication in the different treatment conditions, standard plaque assays were performed. Peak viral titers were achieved on day 5 with and without estrogen, at both MOIs of 0.1 and 0.5 (Figures 3b and 3c). Thereafter, titers decrease as cell death limits further viral replication. Importantly, NV1066 was noted to effectively replicate with or without estrogen. Cells grown and infected in the presence of 10 nM β-estradiol had significantly higher viral titers as compared to cells grown without estrogen at both MOIs. Treated with an initial MOI of 0.1, peak viral titers were 36.4 x 105 PFU/ml (± 2.35 x 105) and 9.83 x 105 PFU/ml (± 1.21 x 105), respectively (p = .05). This represents a 730- and 200-fold increase of the initial infecting dose, respectively. At an MOI of 0.5, peak titers were 36.6 x 105 PFU/ml (± 2.14 x 105) and 16 x 105 PFU/ml (± 2.65 x 105), respectively (p < .05). By estimating the area under the viral growth curves, the total virus produced in the presence of estrogen was 2.9- and 2.5-fold higher for the respective MOIs. Even when comparing an MOI of 0.1 in the estrogen group to an MOI of 0.5 in the no estrogen group, there is roughly a 1.6-fold increase in total virus in the estrogen group despite an initial MOI that is 5 times lower (p = 0.05).
Effect of viral infection on apoptotic fraction and bcl-2 levels
To assess the ability of NV1066 to induce apoptosis in human breast cancer cells, MCF7 cells were grown on chamber slides either with or without estrogen and then infected with NV1066 at an MOI of 1.0. A higher MOI was used to mimic conditions in culture after viral infection and subsequent replication have led to higher viral titers. Performing the experiments in the first 48 hours allowed comparison before large differences in cell numbers and viral titers arose in the two populations. TUNEL and Hoescht staining were performed in separate experiments. Representative microscopic fields from the cells infected either in the presence of 10 nM β-estradiol or without estrogen and then stained with the TUNEL stain are shown in Figure 4a and 4b, respectively. Apoptotic cells are identified by uptake of the brown stain. After 24 hours, 1.61 ± 0.15% of cells grown in the presence of estrogen were apoptotic, compared to 2.81 ± 0.36% of cells infected in the absence of estrogen, suggesting a protective effect of estrogen against early viral-induced apoptosis (p = 0.05).
Figure 4.
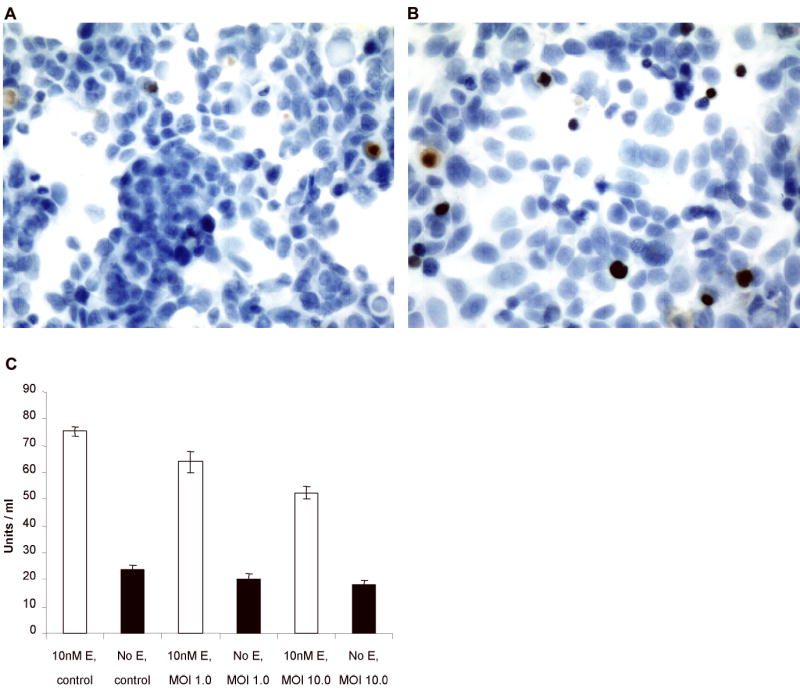
Effect of viral infection on apoptotic fractions and bcl-2 levels. (a and b) Representative fields of TUNEL staining examined by bright field microscopy at 40x magnification, grown either in 10nM β-estradiol (a) or no estrogen (b) demonstrating several brown stained apoptotic nuclei. Apoptotic fractions after 24 hours of infection with NV1066 were significantly lower in MCF-7 cells grown in estrogen (p < .01, t-test). (c) Lower rates of apoptosis in cells grown with estrogen were associated with higher levels of bcl-2 in both uninfected and infected cell populations. Infection with NV1066 for 16 hours significantly decreased bcl-2 levels in the cell population grown in estrogen, while it also decreased the already low bcl-2 levels in cells grown without β-estradiol.
To determine whether bcl-2 levels were affected by infection with NV1066, protein was harvested from separate cell populations infected either in the presence or absence of 10 nM β-estradiol. Bcl-2 levels were significantly lower in all of the groups grown in the absence of estrogen when compared with groups grown in 10 nM β-estradiol (Figure 4c). In the presence of 10 nM β-estradiol, infection with NV1066 at MOIs of 1.0 and 10.0 for 16 hours decreased bcl-2 levels by 15% and 30%, respectively as compared to control (p = 0.05). In the absence of estrogen, bcl-2 levels were decreased by similar percentages, although this did not reach statistical significance given the low baseline levels of expression (p = 0.2, p = 0.1, respectively). MOIs of 10.0, although higher than those initially used in other experiments, are reached in culture after viral replication has occurred.
To confirm these results, separate populations of cells were stained with a nuclear Hoescht stain. Two distinct mechanisms of cell death were noted to occur in the cell populations, apoptosis and oncolysis, which could be easily distinguished at 40x magnification, using a DAPI fluorescent filter. By viewing the cells concomitantly through a GFP filter, EGFP expression could be assessed and related to apoptosis (Figure 5a). To determine apoptotic rates after 48 hrs of NV1066 infection, MCF7 cells were grown either with or without estrogen, incubated for 48 hrs with NV1066, and fixed with Hoescht stain. A significant difference again existed between cells grown either in the presence or the absence of estrogen (Figure 5b). With an MOI of 1.0, apoptotic fractions were 7.86 ± 1.48% and 11.36 ± 1.33% (p = 0.05) for cells grown with and without β-estradiol, respectively. With a GFP filter, identical microscopic fields were examined to determine EGFP expression in relation to apoptosis. No apoptotic cells strongly expressed EGFP.
Figure 5.
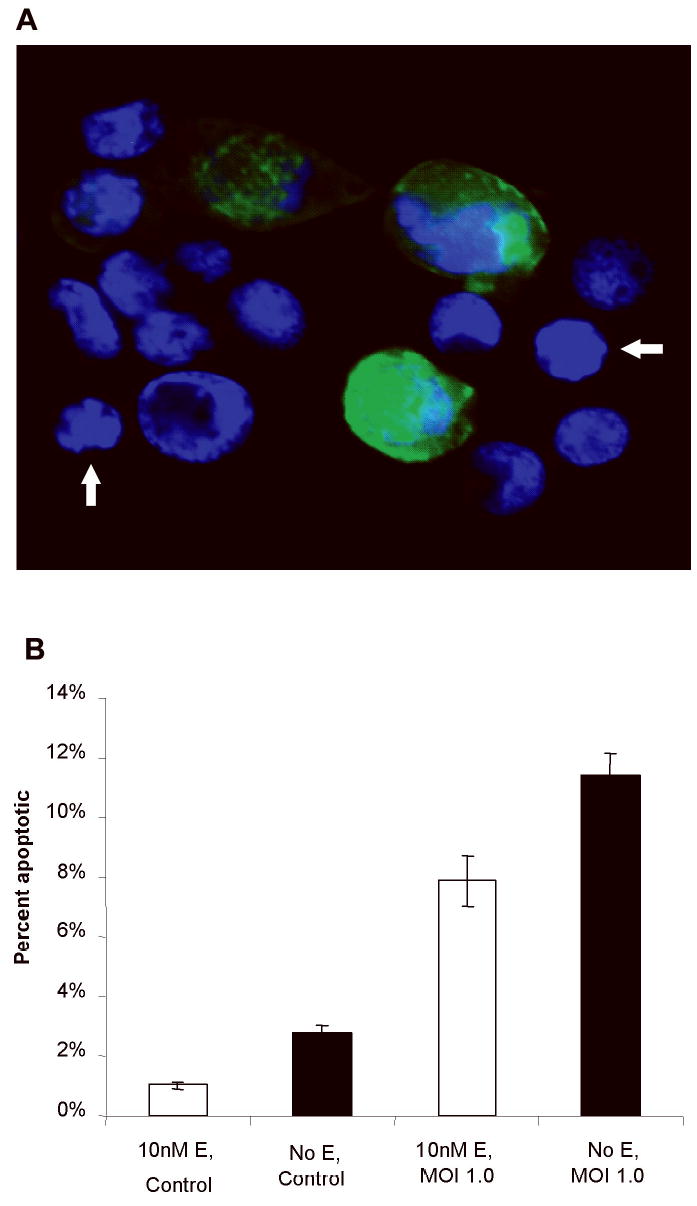
Hoescht staining of infected cells. (a) Overlay photo created by merging photographs taken through DAPI and GFP filters. Infected cell populations demonstrate two mechanisms of cell death, oncolysis and apoptosis. Oncolysis is a consequence of the virus overtaking the host cell machinery to reproduce, while apoptosis is a cellular defense mechanism to prevent reproduction and spread of the virus. Cells strongly expressing EGFP have cytoplasmic swelling and nuclear dispersion, consistent with oncolysis. Neighboring cells (arrows) display nuclear condensation and blebbing of the nuclear membrane, morphology consistent with apoptosis. (b) Apoptotic fractions following 48 hours of infection with NV1066 were significantly higher in cells grown in the absence of estrogen.
Viral cytopathic effects in vitro
To examine the effect of estrogen on oncolysis, cells were again plated either with 10 nM β-estradiol or vehicle alone, then subsequently infected with NV1066 at MOIs of 0.1 and 0.5. Results are reported as surviving fraction of untreated control cells. NV1066 is cytotoxic to MCF7 cells in a dose dependent manner (Figure 6a and 6b). There is a significantly greater cytopathic effect in cells treated in the presence of estrogen, particularly at the lower dose of virus, which is more dependent upon viral replication and proliferation for cell kill. By day 7, only 5.1% (± 0.64%) of cells are still alive in the group grown with estrogen, treated at an MOI of 0.1, compared to 47.5% (± 3.23%) of cells surviving in the no estrogen group treated with the same dose of NV1066 (p = 0.05). Significant differences are also present at the higher MOI of 0.5, with a 10–20% advantage to cell kill in the presence of estrogen on days 3 through 7. An MOI of 0.1 is more cytotoxic to MCF-7 cells grown in estrogen than is an MOI of 0.5 to cells grown without estrogen (12.7% cell survival, ± 1.39%), by day 7 (p = .05).
Figure 6.
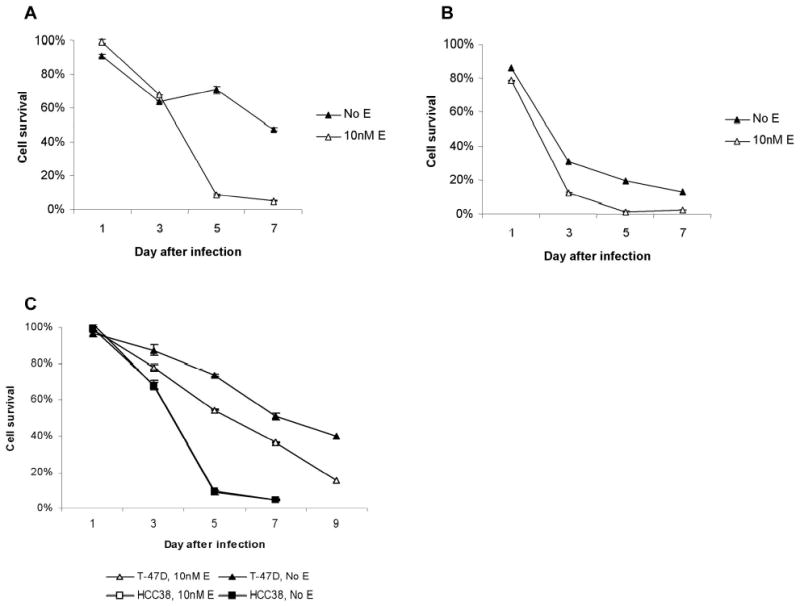
Cell survival following infection with NV1066 either in the presence of 10nM β-estradiol or vehicle alone. (a) MCF7 cells, MOI 0.1. Significantly less cell survival is demonstrated by day 7 following treatment with NV1066 in the presence of β-estradiol at an MOI of 0.1. Only 5.1% of cells survive compared to 47.5% without estrogen. (b). MCF7 cells, MOI 0.5. There was a 10–20% advantage to cell kill in the presence of estrogen on days 3 through 7. An MOI of 0.1 in estrogen is more cytotoxic to MCF7 cells than is an MOI of 0.5 to cells without estrogen. (c) Cytotoxicity of NV1066 to T47D and HCC38 cells. NV1066 at an MOI of 0.1 effectively killed both cell lines. In T-47D cells (ER+), a significantly greater percentage of cell kill was again noted in the presence of estrogen. HCC38 cells (ER−) were very sensitive to viral therapy with NV1066, however sensitivity was essentially identical with or without estrogen, with the survival curves overlapping.
The same experiment was performed using two other human breast cancer cell lines, T-47D and HCC38. T-47D cells are also ER+ and demonstrate increased proliferation and decreased apoptotic fractions in the presence of estrogen, similar to MCF7 cells (data not shown). In contrast, HCC38 cells are ER− and do not appear to be responsive to estrogenic effects in vitro, based upon cellular proliferation studies (data not shown). Although the cytotoxic effect of NV1066 was more delayed in the slower growing T-47D cells, a significantly greater percentage of cell kill was again noted in the presence of estrogen, particularly at the lower MOI of 0.1 (Figure 6c). By day 9, 39.9% (± 0.5%) of cells in the no estrogen group survived, compared to only 15.6% (± 0.3%) of cells treated with NV1066 in the presence of estrogen. This difference was not noted in HCC38 cells, although the cells were also quite sensitive to viral therapy, more so than the T-47D cells. Cytotoxicity was essentially identical with or without estrogen, with less than 5% survival in both treatment groups by day 7 (Figure 6c).
In addition, NV1066 was effective against multiple breast cancer cells even in the absence of estrogen (Figure 7). In vitro, at an MOI of 1, more than 90% of cells were killed by day 7 in all cell lines. This is observed across a wide range of breast cancer cells irrespective of phenotype. Cells tested are estrogen receptor positive (MCF-7, T47D) or negative (HCC38, SKBr3, MDA-MB-435s, HCC 1954), progesterone receptor positive (T47D) or negative (MDA-MB-435s, HCC 1954, HCC38), p53 positive (MCF-7, T47D, SKBr3), and HER2/c-erb-2 overexpressing (SKBr3, HCC1954).
Figure 7.
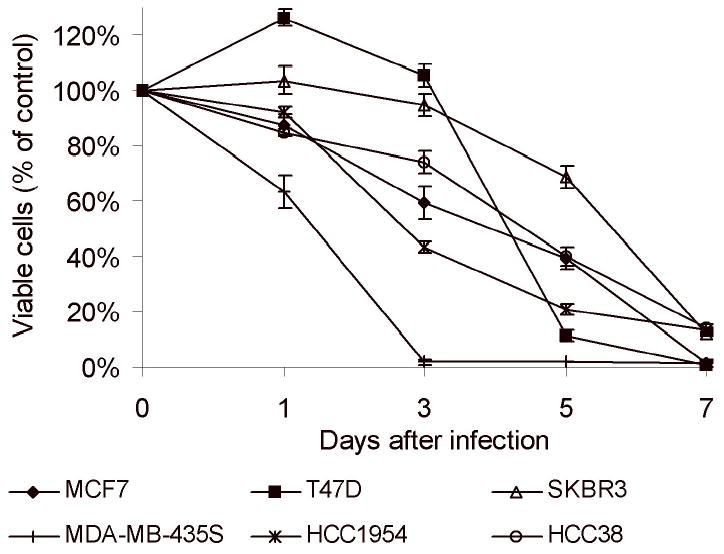
Cell survival following infection with NV1066 in multiple breast cancer cell lines at MOI of 1. NV1066 effectively killed more than 90% of all cells at an MOI of 1 by day 7. MDA-MB-435s and HCC1954 cells were exquisitively sensitive to NV1066. Nearly 60–90% of the cells were killed by day 3.
In contrast to the increased efficacy of NV1066 in the presence of estrogen, the decreased efficacy of chemotherapeutics and radiation therapy in the treatment of ER+ human breast cancer cells in the presence of estrogen has been demonstrated in numerous in vitro studies. In our experiments, presence of estrogen reduced cell kill in vitro in both estrogen receptor positive cell lines MCF-7 (51 ± 4% to 16 ± 2%) and T47D (26 ± 3% to 13 ± 1%) when exposed to taxol at a concentration of 5 μM. Similar results were observed at varying concentrations of taxol 0.5 and 50 μM (data not shown).
Discussion
Herpes simplex viral mutants such as NV1066 infect, replicate in, and lyse a variety of malignant tumor cells(8–13). Replication competent HSV-1 mutants are currently in clinical trials for recurrent malignant gliomas and for unresectable colorectal metastases to the liver(23;24). Phase I studies have demonstrated favorable safety profiles(23–25). Animal studies have demonstrated the efficacy of HSV-1 mutants in the treatment of metastatic sites relevant to breast cancer, including metastatic models of the lymph nodes, brain, liver, and pleural cavity(8;11;18;26). Despite the large body of literature concerning herpes viruses, little has been published regarding the interaction between viral therapy and hormonal milieu. Oncolytic therapy with replication competent HSV-1 mutants has previously been demonstrated to be effective against human breast cancer cell lines, without controlling for in vitro or in vivo estrogen levels(8;17). Using the HSV-1 mutant G207, Toda et al found a significant in vitro cytopathic effect in three of four human breast adenocarcinoma cell lines with an estrogen receptor negative cell line being least susceptible(8). In the same study, this group demonstrated efficacy of the virus in vivo in the treatment of subcutaneous tumors and in a metastatic brain tumor model. Subsequently, the same group reported on the successful use of G207 in purging occult breast tumor cells from human bone marrow cells in vitro(17). G207 was not cytotoxic to human bone marrow cells themselves, and in fact stimulated hematopoietic cell proliferation.
In our study, HSV was effective against multiple breast cancer cells irrespective of receptor status. In all cell lines tested, NV1066 killed greater than 90% of cells by day 7 after infection. Given the apparent efficacy of HSV-1 mutants against human breast cancer cells and the established role of hormonal therapy in the treatment of breast cancer, we sought to determine in this study whether the hormonal environment affects HSV-1 oncolytic therapy.
HSV-1 mutants are genetically altered for selective replication in rapidly dividing cells. Our laboratory has demonstrated that tumor cells with higher S-phase fractions and shorter doubling times are generally more sensitive to oncolytic HSV-1 viral therapy(12;27). Metabolically active tumor cells provide mutant viruses with an environment in which host cell machinery can be appropriated for viral replication. In contrast, viruses replicate poorly in quiescent cells. This biological phenomenon has important implications in the use of viral mutants in hormone sensitive tumors. In the present study, we demonstrate that ER+ human breast cancer cells have more rapid growth rates and higher S phase fractions when grown in the presence of estrogen. We believe that this plays a significant role in the increased replication and proliferation of NV1066 in cell populations grown in the presence of estrogen.
With regard to ER− tumors, the effects of estrogen on cellular proliferation are less clear. However, ER− breast cells have been demonstrated to have significantly higher proliferative indices than ER+ cells in several studies(28;29), which should promote robust viral replication and oncolysis. As demonstrated in this study, these tumors are also highly sensitive to viral therapy. Doses as low as one viral PFU per 10 tumor cells killed over 95% of ER− cell populations after one week in culture. It is particularly interesting that some of these lines appear to be exquisitely sensitive to viral therapy (MDA-MB-435s and HCC 1954 cell lines, 60–90% cell kill by day 3 with NV1066 at an MOI of 1). We believe that our data provide preliminary evidence to postulate that oncolytic viral therapy holds promise for ER− breast cancer. Factors that are present in ER− cells that enhance proliferation or inhibit apoptosis may favor replication competent viral vectors. Because ER− negativity may be associated with a poor prognosis following other forms of adjuvant therapy(30), oncolytic viral therapy holds promise in such a clinical setting.
Returning to ER+ breast cancer cells, part of estrogen’s ability to act as a growth and survival factor also occurs through the prevention of apoptosis(4;31). Previous studies have demonstrated that in the absence of exogenously provided estrogen, tumors of ER+ human breast cancer cells may regress with evidence for apoptotic cell death(32;33). Tamoxifen and other SERMs have also been demonstrated to cause apoptotic cell death(32–35). Wild-type HSV-1 has evolved mechanisms for suppressing apoptotic DNA fragmentation and cell death(14;36). HSV-1 mutants, including NV1066(15), lack functional viral proteins that have been demonstrated to contribute to this anti-apoptotic effect(16;19;20;37). Accordingly, previous reports have demonstrated that such HSV-1 mutants induce morphologic and biochemical changes consistent with apoptosis, whereas wild-type viruses do not(16;38). Apoptosis in both infected and in uninfected bystander tumor cells has been shown to limit replication of viral progeny and to decrease efficacy of viral-based therapy(39;40).
NV1066 lacks single copies of the viral genes ICP-4 and γ134.5. The ICP-4 protein product acts as a transcription factor to transactivate viral genes and plays a role in inhibition of apoptosis in infected cells(15). The γ134.5 gene encodes for the protein ICP34.5, which functions to inhibit the ability of infected cells to shut down protein synthesis and undergo apoptosis in the face of viral infection(15). Loss of these viral genes significantly attenuates NV1066 and renders infected cell populations susceptible to virally induced apoptosis. Apoptosis in infected cells and bystander cells may be considered a host cell defense mechanism to minimize the spread of viral progeny by limiting cellular production of virus. In the present study we demonstrate that estrogen led to decreased apoptotic fractions in infected cells and to subsequent higher viral titers and improved oncolysis. In contrast to these findings, similar differences in apoptotic fractions have been suggested to decrease the efficacy of taxol and of UV irradiation in MCF7 cells grown in the presence of estrogen(4). While a difference of 1–2% in apoptosis may not lead to a large cumulative difference in outcome using standard treatment regimens such as chemotherapy or radiotherapy, it can have a significant impact on replication-competent biological therapies. For example, consider a clinical scenario in which the virus infects two groups of 100,000 cells each. If one population of cells undergoes a 1% higher rate of apoptosis in response to viral infection, 1000 fewer cells go on to actively replicate virus. Assuming that each lysed tumor cell releases 100–1000 viral progeny, an extra 1000 cells may increase the effective dose of the virus by 100 000–1 000 000 PFU with only one life cycle. Experiments conducted in other cancer cell lines from our laboratory have shown that such inhibition of apoptosis resulted in significant advantage of viral replication and propagation.
Decreased efficacy of chemotherapeutics and of radiation therapy in the treatment of ER+ human breast cancer cells in the presence of estrogen has been demonstrated in several in vitro studies(3–6). Taxol and UV-irradiation are known to induce apoptosis in human breast cancer cells. Estrogen is thought to cause resistance to taxol and UV-light induced apoptosis in ER+ cells by increasing expression of anti-apoptotic genes and by increasing proliferation signals in the cells. These mechanisms are activated via specific steroid receptors.
Regarding apoptosis, the relationship between the hormonal environment and oncolytic viral therapy with regard to their effect on bcl-2 levels is of particular interest. Bcl-2 acts at the level of the mitochondria to block apoptosis(22). It is a member of a family of proteins that includes both prosurvival proteins and proapoptotic proteins. Although the anti-apoptotic effects of estrogen are incompletely understood, the bcl-2 pathway is thought to play a significant role(3;4;39;41). In the presence of estrogen, cell survival has been shown to correlate with increased expression of bcl-2(6). Recent studies have demonstrated that overexpression of the bcl-2 gene significantly inhibits apoptotic cell death and cytotoxicity induced by various chemotherapeutics and radiotherapy in vitro in human breast cancer cells(3–5). It has been suggested that drug sensitivity is directly related to the amount of bcl-2 expressed by the cell(42). Additionally, cells that overexpress bcl-2 demonstrate higher metastatic potentials in vivo in mouse models of metastatic breast cancer(38). Clinically, although it is difficult to assess the true prognostic meaning of bcl-2 overexpression due to its close association with estrogen receptor positivity, high bcl-2 levels have been associated with resistance to chemotherapy in advanced breast cancer(40). Treatment with bcl-2 antisense oligonucleotides in vitro has been demonstrated to sensitize ER+ breast cancer cells to treatment with anti-cancer drugs, including doxorubicin, paclitaxel, and cyclophosphamide(41). Reduction of bcl-2 levels by antisense oligonucleotides has also been demonstrated to be effective in facilitating apoptosis in ER− breast cancer cells with low baseline bcl-2 levels, suggesting that the relative reduction is perhaps more important than the absolute reduction(43).
Studies in the virology literature have suggested that specific HSV-1 mutants, including ICP4-null mutants, decrease bcl-2 levels by decreasing mRNA levels and by protein destabilization(38). This reduction of bcl-2 levels is thought to be a key event during apoptosis induced by these viruses and correlates with decreased viral yields after infection. In the present study we demonstrate that estrogen increases bcl-2 levels in ER+ breast cancer cells. This likely helps to protect the cells from early apoptosis following viral infection, which allows for subsequent replication and propagation of the virus. This early cytoprotective effect ultimately leads to increased cytotoxicity, as significantly more oncolytically active virus is produced. Furthermore, the ability of HSV-1 mutants to decrease bcl-2 levels, as confirmed in this study, may sensitize cells to other therapies, including chemotherapy and radiotherapy. This may have important implications in clinical trials in which virus is given concomitantly or prior to other therapies.
In summary, we believe that the effects of endogenous estrogen on ER+ human breast cancer cells establish an environment favorable to treatment with replication competent HSV-1 mutants. These oncolytic viruses are also quite effective against hormone receptor negative cell lines. Thus, oncolytic viral therapy may be an especially attractive option for patients who fail SERM therapy or who have hormone-insensitive tumors. Hormonal therapy with tamoxifen or other SERMs is given to most patients with ER+ tumors. However, approximately one-third to one-half of ER+ breast cancers fail to respond to tamoxifen initially and up to 90% develop resistance within one year(44;45). Most breast tumors with acquired SERM resistance continue to express ER of normal size and DNA-binding ability(46). Additionally, tumors that initially respond to SERMs may develop resistance to adjuvant chemotherapy by over expression of antiapoptotic proteins such as bcl-2 in residual cells(47). In vitro, high intracellular levels of bcl-2 have been shown to confer resistance against a variety of anti-cancer drugs and radiotherapy. In therapy with replication competent HSV-1 mutants, increased bcl-2 levels and a decreased rate of apoptosis should correlate with enhanced viral proliferation, replication, and persistence, leading to an improved oncolytic effect. NV1066 is therefore an attractive treatment strategy for breast cancers that may be resistant to other forms of therapy.
Further studies need to be completed to better define how HSV-based therapy would integrate with current breast cancer therapies. The results of this study suggest that HSV-1 mutants should not be used concurrently with tamoxifen or other SERMs. Estrogenic effects on cells create an environment favorable to viral replication and proliferation. The improved efficacy of replication competent viral vectors in this setting is unique. Combination therapy using HSV-1 mutants and either chemotherapy or radiation therapy also holds promise. Since genetically engineered herpes simplex viruses kill cancer cells by oncolytic mechanisms differing from standard anticancer therapies, there is an opportunity that synergistic interactions with other therapies might be found with the use of combination therapy. Improved understanding of the oncolytic herpes simplex viral life cycle resulted in an exploration of multiple such interactions between viral and cellular gene products. Our laboratory has demonstrated that such interactions are beneficial to reduce the dosage of individual therapies without compromising cell kill in multiple cancers(48;49). One possible mechanism of this synergy which has not been previously explored is the reduction of anti-apoptotic proteins caused by viral infection. In this paper, we confirm the results from previous studies that viral infection causes significant decreases in bcl-2 expression in infected cells. Overexpression of bcl-2 has been shown to inhibit cell death caused by various chemotherapeutics and radiation therapy. This raises the intriguing possibility that pretreatment with HSV-1 mutants may sensitize cells to these other forms of therapy. Further investigations are warranted as oncolytic HSV-1 based therapy is utilized clinically.
Acknowledgments
The authors thank Brian Horsburgh, Ph.D. and Medigene, Inc. for constructing and providing us with the NV1066 virus. We also thank Liza Marsh of the Department of Surgery at Memorial Sloan-Kettering Cancer Center for her editorial assistance with this manuscript.
Footnotes
Supported in part by training grant T32 CA09501 (B.M.S.), AACR-AstraZeneca Cancer Research and Prevention grant (P.S.A), grants RO1 CA 76416 and RO1 CA/DK80982 (Y.F.) from the National Institutes of Health, grant MBC-99366 (Y.F.) from the American Cancer Society, grant BC024118 from the US Army (Y.F.), grant IMG0402501 from the Susan G. Komen Breast Cancer foundation (Y.F. and P.S.A.) and grant 032047 from Flight Attendant Medical Research Institute (Y.F. and P.S.A.)
Study presented at ‘The American Society for Clinical Investigation’, 2002 in Chicago.
References
- 1.Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ. Cancer statistics. Ca: a Cancer Journal for Clinicians. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Lippert TH, Seeger H, Mueck AO. The impact of endogenous estradiol metabolites on carcinogenesis. Steroids. 2000;65:357–69. doi: 10.1016/s0039-128x(00)00101-x. [DOI] [PubMed] [Google Scholar]
- 3.Huang Y, Ray S, Reed JC, Ibrado AM, Tang C, Nawabi A, Bhalla K. Estrogen increases intracellular p26Bcl-2 to p21Bax ratios and inhibits taxol-induced apoptosis of human breast cancer MCF-7 cells. Breast Cancer Res Treat. 1997;42:73–81. doi: 10.1023/a:1005777219997. [DOI] [PubMed] [Google Scholar]
- 4.Razandi M, Pedram A, Levin ER. Plasma membrane estrogen receptors signal to antiapoptosis in breast cancer. Mol Endocrinol. 2000;14:1434–47. doi: 10.1210/mend.14.9.0526. [DOI] [PubMed] [Google Scholar]
- 5.Swiatecka J, Dzieciol J, Anchim T, Dabrowska M, Pietruczuk M, Wolczynski S. Influence of estrogen, antiestrogen and UV-light on the balance between proliferation and apoptosis in MCF-7 breast adenocarcinoma cells culture. Neoplasma. 2000;47:15–24. [PubMed] [Google Scholar]
- 6.Teixeira C, Reed JC, Pratt MA. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl-2 proto-oncogene expression in human breast cancer cells. Cancer Res. 1995;55:3902–7. [PubMed] [Google Scholar]
- 7.Landis SH, Murray T, Bolden S, Wingo PA. Cancer statistics, 1998. CA Cancer J Clin. 1998;48:6–29. doi: 10.3322/canjclin.48.1.6. [DOI] [PubMed] [Google Scholar]
- 8.Toda M, Rabkin SD, Martuza RL. Treatment of human breast cancer in a brain metastatic model by G207, a replication-competent multimutated herpes simplex virus 1. Human Gene Therapy. 1998;9:2177–85. doi: 10.1089/hum.1998.9.15-2177. [DOI] [PubMed] [Google Scholar]
- 9.Walker JR, McGeagh KG, Sundaresan P, Jorgensen TJ, Rabkin SD, Martuza RL. Local and systemic therapy of human prostate adenocarcinoma with the conditionally replicating herpes simplex virus vector G207. Hum Gene Ther. 1999;10:2237–43. doi: 10.1089/10430349950017211. [DOI] [PubMed] [Google Scholar]
- 10.Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1:938–43. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 11.Ebright MI, Zager JS, Malhotra S, Delman KA, Weigel TL, Rusch VW, Fong Y. Replication-competent herpes virus NV1020 as direct treatment of pleural cancer in a rat model. Journal of Thoracic & Cardiovascular Surgery. 2002;124:123–9. doi: 10.1067/mtc.2002.122297. [DOI] [PubMed] [Google Scholar]
- 12.Kooby DA, Carew JF, Halterman MW, Mack JE, Bertino JR, Blumgart LH, Federoff HJ, Fong Y. Oncolytic viral therapy for human colorectal cancer and liver metastases using a multi-mutated herpes simplex virus type-1 (G207) FASEB Journal. 1999;13:1325–34. doi: 10.1096/fasebj.13.11.1325. [DOI] [PubMed] [Google Scholar]
- 13.Carew JF, Kooby DA, Halterman MW, Federoff HJ, Fong Y. Selective infection and cytolysis of human head and neck squamous cell carcinoma with sparing of normal mucosa by a cytotoxic herpes simplex virus type 1 (G207) Human Gene Therapy. 1999;10:1599–606. doi: 10.1089/10430349950017608. [DOI] [PubMed] [Google Scholar]
- 14.Galvan V, Roizman B. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type-dependent manner. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3931–6. doi: 10.1073/pnas.95.7.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roizman B. The function of herpes simplex virus genes: a primer for genetic engineering of novel vectors. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:11307–12. doi: 10.1073/pnas.93.21.11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galvan V, Brandimarti R, Munger J, Roizman B. Bcl-2 blocks a caspase-dependent pathway of apoptosis activated by herpes simplex virus 1 infection in HEp-2 cells. J Virol. 2000;74:1931–8. doi: 10.1128/jvi.74.4.1931-1938.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu A, Mazumder A, Martuza RL, Liu X, Thein M, Meehan KR, Rabkin SD. Biological purging of breast cancer cells using an attenuated replication-competent herpes simplex virus in human hematopoietic stem cell transplantation. Cancer Res. 2001;61:3009–15. [PubMed] [Google Scholar]
- 18.Wong RJ, Joe JK, Kim SH, Shah JP, Horsburgh B, Fong Y. Oncolytic herpesvirus effectively treats murine squamous cell carcinoma and spreads by natural lymphatics to treat sites of lymphatic metastases. Human Gene Therapy. 2002;13:1213–23. doi: 10.1089/104303402320138998. [DOI] [PubMed] [Google Scholar]
- 19.Leopardi R, Roizman B. The herpes simplex virus major regulatory protein ICP4 blocks apoptosis induced by the virus or by hyperthermia. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:9583–7. doi: 10.1073/pnas.93.18.9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He B, Gross M, Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:843–8. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Darzynkiewicz Z, Traganos F. Measurement of apoptosis. Adv Biochem Eng Biotechnol. 1998;62:33–73. doi: 10.1007/BFb0102305. [DOI] [PubMed] [Google Scholar]
- 22.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–6. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 23.Whitley RJ, Roizman B. Herpes simplex viruses: is a vaccine tenable? J Clin Invest. 2002;110:145–51. doi: 10.1172/JCI16126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, Palmer CA, Feigenbaum F, Tornatore C, Tufaro F, Martuza RL. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–74. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 25.Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, Petty R, Maclean A, Harland J, McKie E, Mabbs R, Brown M. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Therapy. 2000;7:859–66. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 26.Nakamura H, Kasuya H, Mullen JT, Yoon SS, Pawlik TM, Chandrasekhar S, Donahue JM, Chiocca EA, Chung RY, Tanabe KK. Regulation of herpes simplex virus gamma(1)34.5 expression and oncolysis of diffuse liver metastases by Myb34.5. Journal Of Clinical Investigation. 2002;109:871–82. doi: 10.1172/JCI10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennett JJ, Fong Y. Viral-based therapies for liver cancer. HPB. 2000;2:289–98. [Google Scholar]
- 28.Molino A, Micciolo R, Turazza M, Bonetti F, Piubello Q, Bonetti A, Nortilli R, Pelosi G, Cetto GL. Ki-67 immunostaining in 322 primary breast cancers: associations with clinical and pathological variables and prognosis. Int J Cancer. 1997;74:433–7. doi: 10.1002/(sici)1097-0215(19970822)74:4<433::aid-ijc12>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 29.Gerdes J, Pickartz H, Brotherton J, Hammerstein J, Weitzel H, Stein H. Growth fractions and estrogen receptors in human breast cancers as determined in situ with monoclonal antibodies. Am J Pathol. 1987;129:486–92. [PMC free article] [PubMed] [Google Scholar]
- 30.Somlo G, Simpson JF, Frankel P, Chow W, Leong L, Margolin K, Morgan R, Jr, Raschko J, Shibata S, Forman S, Kogut N, McNamara M, Molina A, Somlo E, Doroshow JH. Predictors of long-term outcome following high-dose chemotherapy in high-risk primary breast cancer. Br J Cancer. 2002;87:281–8. doi: 10.1038/sj.bjc.6600450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dickson RB, Stancel GM. The Bcl-2 protein family: arbiters of cell survival. J Natl Cancer Inst Monographs. 2000;27:135–45. doi: 10.1093/oxfordjournals.jncimonographs.a024237. [DOI] [PubMed] [Google Scholar]
- 32.Warri AM, Huovinen RL, Laine AM, Martikainen PM, Harkonen PL. Apoptosis in toremifene-induced growth inhibition of human breast cancer cells in vivo and in vitro. J Natl Cancer Inst. 1993;85:1412–8. doi: 10.1093/jnci/85.17.1412. [DOI] [PubMed] [Google Scholar]
- 33.Kyprianou N, English HF, Davidson NE, Isaacs JT. Programmed cell death during regression of the MCF-7 human breast cancer following estrogen ablation. Cancer Res. 1991;51:162–6. [PubMed] [Google Scholar]
- 34.Diel P, Smolnikar K, Michna H. The pure antiestrogen ICI 182780 is more effective in the induction of apoptosis and down regulation of BCL-2 than tamoxifen in MCF-7 cells. Breast Cancer Res Treat. 1999;58:87–97. doi: 10.1023/a:1006338123126. [DOI] [PubMed] [Google Scholar]
- 35.Wilson JW, Wakeling AE, Morris ID, Hickman JA, Dive C. MCF-7 human mammary adenocarcinoma cell death in vitro in response to hormone-withdrawal and DNA damage. Int J Cancer. 1995;61:502–8. doi: 10.1002/ijc.2910610413. [DOI] [PubMed] [Google Scholar]
- 36.Koyama AH, Miwa Y. Suppression of apoptotic DNA fragmentation in herpes simplex virus type 1-infected cells. J Virol. 1997;71:2567–71. doi: 10.1128/jvi.71.3.2567-2571.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zachos G, Koffa M, Preston CM, Clements JB, Conner J. Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication. J Virol. 2001;75:2710–28. doi: 10.1128/JVI.75.6.2710-2728.2001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Fernandez Y, Gu B, Martinez A, Torregrosa A, Sierra A. Inhibition of apoptosis in human breast cancer cells: role in tumor progression to the metastatic state. Int J Cancer. 2002;101:317–26. doi: 10.1002/ijc.10628. [DOI] [PubMed] [Google Scholar]
- 39.Gee JM, Robertson JF, Ellis IO, Willsher P, McClelland RA, Hoyle HB, Kyme SR, Finlay P, Blamey RW, Nicholson RI. Immunocytochemical localization of BCL-2 protein in human breast cancers and its relationship to a series of prognostic markers and response to endocrine therapy. Int J Cancer. 1994;59:619–28. doi: 10.1002/ijc.2910590508. [DOI] [PubMed] [Google Scholar]
- 40.Bonetti A, Zaninelli M, Leone R, Cetto GL, Pelosi G, Biolo S, Menghi A, Manfrin E, Bonetti F, Piubello Q. bcl-2 but not p53 expression is associated with resistance to chemotherapy in advanced breast cancer. Clin Cancer Res. 1998;4:2331–6. [PubMed] [Google Scholar]
- 41.Stanziale SF, Petrowsky H, Adusumilli PS, Ben Porat L, Gonen M, Fong Y. Infection with oncolytic herpes simplex virus-1 induces apoptosis in neighboring human cancer cells: a potential target to increase anticancer activity. Clinical Cancer Research. 2004;10:3225–32. doi: 10.1158/1078-0432.ccr-1083-3. [DOI] [PubMed] [Google Scholar]
- 42.Simoes-Wust AP, Schurpf T, Hall J, Stahel RA, Zangemeister-Wittke U. Bcl-2/bcl-xL bispecific antisense treatment sensitizes breast carcinoma cells to doxorubicin, paclitaxel and cyclophosphamide. Breast Cancer Res Treat. 2002;76:157–66. doi: 10.1023/a:1020543004400. [DOI] [PubMed] [Google Scholar]
- 43.Davis JM, Navolanic PM, Weinstein-Oppenheimer CR, Steelman LS, Hu W, Konopleva M, Blagosklonny MV, McCubrey JA. Raf-1 and bcl-2 induce distinct and common pathways that contribute to breast cancer drug resistance. Clin Cancer Res. 2003;9:1161–7. [PubMed] [Google Scholar]
- 44.Chi KC, Wallis AE, Lee CH, De Menezes DL, Sartor J, Dragowska WH, Mayer LD. Effects of Bcl-2 modulation with G3139 antisense oligonucleotide on human breast cancer cells are independent of inherent Bcl-2 protein expression. Breast Cancer Res Treat. 2000;63:199–212. doi: 10.1023/a:1017371013487. [DOI] [PubMed] [Google Scholar]
- 45.Johnston SR. Acquired tamoxifen resistance in human breast cancer--potential mechanisms and clinical implications. Anticancer Drugs. 1997;810:911–30. doi: 10.1097/00001813-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 46.Osborne CK, Yochmowitz MG, Knight WA, III, McGuire WL. The value of estrogen and progesterone receptors in the treatment of breast cancer. Cancer. 1980;(12 Suppl):2884–8. doi: 10.1002/1097-0142(19801215)46:12+<2884::aid-cncr2820461429>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 47.Johnston SR, Lu B, Dowsett M, Liang X, Kaufmann M, Scott GK, Osborne CK, Benz CC. Comparison of estrogen receptor DNA binding in untreated and acquired antiestrogen-resistant human breast tumors. Cancer Res. 1997;57:3723–7. [PubMed] [Google Scholar]
- 48.Bennett JJ, Adusumilli P, Petrowsky H, Burt BM, Roberts G, Delman KA, Zager JS, Chou TC, Fong Y. Up-regulation of GADD34 mediates the synergistic anticancer activity of mitomycin C and a gamma134.5 deleted oncolytic herpes virus (G207) FASEB J. 2004;18:1001–3. doi: 10.1096/fj.02-1080fje. [DOI] [PubMed] [Google Scholar]
- 49.Stanziale SF, Petrowsky H, Joe JK, Roberts GD, Zager JS, Gusani NJ, Ben Porat L, Gonen M, Fong YM. Ionizing radiation potentiates the antitumor efficacy of oncolytic herpes simplex virus G207 by upregulating ribonucleotide reductase. Surgery. 2002;132:353–9. doi: 10.1067/msy.2002.125715. [DOI] [PubMed] [Google Scholar]