

Abstract
Suppressor of cytokine signaling (Socs) 3 is a cytokine-inducible inhibitor with critical but selective cell-specific effects. We show that deficiency of Socs3 in T cells had minimal effects on differentiation of T cells to the T helper (Th) 1 or Th2 subsets; accordingly, Socs3 had no effect on IL-12-dependent signal transducer and activator of transcription (Stat) 4 phosphorylation or IL-4-dependent Stat6 phosphorylation. By contrast, Socs3 was found to be a major regulator of IL-23-mediated Stat3 phosphorylation and Th17 generation, and Stat3 directly binds to the IL-17A and IL-17F promoters. We conclude that Socs3 is an essential negative regulator of IL-23 signaling, inhibition of which constrains the generation of Th17 differentiation.
Keywords: signal transducer and activator of transcription 3, T lymphocytes
Suppressor of cytokine signaling (Socs) proteins are rapidly induced by cytokines and thus represent classic feedback inhibitors (1, 2). As such, they are critical regulators of cytokine signaling (3) and the Janus kinase (Jak)/signal transducer and activator of transcription (Stat) pathway (4). The eight members of the Socs protein family include Socs1–7 and cytokine inducible Src homology domain 2 (SH2) domain-containing protein (CIS) (5). Each has a central SH2 domain that targets the protein to phosphorylated tyrosine residues on activated cytokine receptors or the associated Jak and thus interferes with signaling (6). Socs1 and Socs3 are additionally able to inhibit cytokine signaling by virtue of an N-terminal kinase inhibitory region that acts as a pseudosubstrate for Jaks (7). Socs proteins also have a C-terminal Socs box that acts as an E3 ubiquitin ligase, which is thought to promote degradation of the receptor/Jak complex and thus serves as yet another mechanism to modify cytokine signaling (8, 9).
The essential in vivo functions of Socs proteins are best illustrated in gene-targeted mice lacking these factors (3). These studies have documented remarkably specific functions of Socs proteins and have also illustrated the limitations of overexpression studies, which have tended to exaggerate Socs proteins' physiological roles (10, 11). For instance, lack of Socs1 leads to death within the first few weeks of life from autoimmune disease, which is reversed by interrupting IFNγ signaling (12). Thus, despite the numerous ligands reported to induce Socs1, its in vivo role is fairly limited. Similarly, Socs2 has a very specific role in constraining growth hormone, as illustrated by gigantism in the corresponding gene knockout mice (13, 14).
Understanding the function of Socs3 has been hampered by the fact that Socs3 knockout mice die in utero with placental defects because of exaggerated leukemia inhibitor factor signaling (15, 16). However, conditional knockouts of Socs3 have nonetheless documented specific roles for this factor in various tissues, including the brain (17), bone marrow (18, 19), and the liver (20, 21). Mice deficient for Socs3 within the central nervous system show increased levels of phospho-Stat3 within the hypothalamus in response to stimulation by leptin and are resistant to obesity when fed a high-fat diet (17). Similarly, macrophages and liver cells lacking Socs3 have enhanced IL-6-dependent Stat3 phosphorylation, whereas IL-10-dependent Stat3 phosphorylation is unaffected (20, 22, 23), which is consistent with the idea that the inhibitory actions of individual Socs proteins are selective for a given cytokine.
By contrast with the innate immune system, the role of Socs3 in the adaptive immune system has been less well studied. Several groups have explored the role of Socs3 within the immune system using Socs3 knockout fetal bone marrow reconstitution of irradiated Rag2−/− mice (22), one of which reported that development of both T and B cell lineages was unaffected (24).
After thymic development, naïve T CD4+ T cells have classically been described as having two possible cell fates: T helper (Th) 1 or Th2. Th1 cells produce IFNγ, which promotes cell-mediated immune responses that eliminate intracellular pathogens. Th2 differentiated cells produce IL-4, IL-5, and IL-13, which promote humoral immunity and are critical in eradicating helminth infestations (25). The polarization of naïve Th cells into Th1 or Th2 cells is regulated by IL-12 and IL-4, which activate Stat4 (26–28) and Stat6 (29, 30), respectively. By using overexpression models, Socs3 has been implicated as a regulator of Th1/2 polarization via its ability to inhibit Stat4 (31, 32).
Recently, a third lineage of Th cells that selectively produce IL-17 (Th17) has been identified, and they are thought to be key regulators of inflammation (33–36). IL-17 consists of a family of related cytokines (IL-17A–F), IL-17 itself being synonymous with IL-17A. A related cytokine, IL-17F, shares a similar structure, chromosomal location, and receptor usage as IL-17A (37, 38). IL-17A and IL-17F secretion is regulated by a cytokine related to IL-12, namely IL-23 (39). IL-12 and IL-23 share a subunit, p40, and bind to a common receptor subunit, IL-12Rβ1. However, each cytokine has a ligand-specific subunit and distinct in vivo functions (40).
To assess directly the role of Socs3 in Th development we used a conditional knockout approach. In this study we found that Socs3 has little effect on Th1/2 polarization but has a significant role in constraining the generation of Th17 cells. We show that IL-23-induced Stat3 phosphorylation is enhanced in the absence of Socs3 and that Stat3 is able to bind to the promoter regions of both IL-17A and IL-17F. Thus, our data point to an important role of Socs3 in regulation of Th17 differentiation.
Results
Socs3 Is Not Essential for Normal T Cell Development.
Previous studies have shown that Cre expressed under the control of the mouse mammary tumor virus (MMTV) promoter results in deletion within the lymphoid compartment (41, 42). To confirm loss of Socs3 in T cells, spleens, thymi, and peripheral lymph nodes of CreMMTV Socs3fl/fl mice and wild-type littermate controls were analyzed for the Socs3 expression by using real-time quantitative PCR (q-PCR). The results showed loss of >99% of Socs3 mRNA in thymocytes, lymphocytes, and activated CD4+ T cells (Fig. 1a and b). To assess whether T cell development was altered by absence of Socs3, thymocyte, lymphocyte, and splenocyte numbers from CreMMTV Socs3fl/fl and littermate controls were counted. CreMMTV Socs3fl/fl mice were found to have a small but significant increase in total thymocyte numbers (Fig. 1c; P = 0.015), with no significant difference in numbers of lymph node cells and splenocytes. Despite this finding, CreMMTV Socs3fl/fl mice demonstrated normal proportions of the major thymic populations as assessed by expression of CD4 and CD8 or the constituents of the double-negative compartment (Fig. 1 d and e). Analysis of CreMMTV Socs3fl/fl peripheral T cells demonstrated no differences in the proportions of CD4+ and CD8+ T cells or the expression of activation markers in these cells compared with littermate controls (Fig. 5, which is published as supporting information on the PNAS web site).
Fig. 1.

Socs3 is not essential for T cell development. Loss of Socs3 mRNA expression in thymocytes, splenocytes, isolated splenic CD4+ T cells (a), and activated CD4+ T cells (b) is shown. In these latter experiments, CD4+ T cells were isolated from thymi and spleen of Socs3fl/fl or CreMMTV Socs3fl/fl mice and activated by plate-bound anti-CD3 and anti-CD28 for 2 days. Total RNA extraction, cDNA synthesis, and real-time q-PCR were performed as described in Materials and Methods. Relative gene expression was normalized against the gene expression level of thymocytes from Socs3fl/fl mice. (c) Thymocytes, lymph node cells, and splenocytes were harvested and counted from 8- to 10-week-old CreMMTV Socs3fl/fl mice and their littermate controls. Data shown are the average from four mice. Student's t test was used to compare the data from CreMMTV Socs3fl/fl mice and their littermate controls. (d and e) Thymocytes from CreMMTV Socs3fl/fl mice and their littermate controls were stained with CD4-allophycocyanin and CD8-FITC. Thy1.2-positive, lineage-negative, CD4/8-double-negative thymocytes were stained for expression for CD25-FITC and CD44-allophycocyanin.
Socs3 Deficiency Is Not a Critical Regulator of Th1/2 Polarization.
Previous studies employing transgenic expression of Socs3 have suggested a major role of Socs3 in the regulation of Th1/Th2 polarization (31). We therefore next investigated whether T cells lacking Socs3 would have significant alterations in Th cell differentiation. To this end, naïve CD4+ T cells were isolated and stimulated to mature into Th1 and Th2 cells by using the appropriate cytokines. The degree of polarization was assessed both by cytokine secretion using ELISA (Fig. 2c) and by intracellular staining using flow cytometry (Fig. 2 a and b); no consistent differences between Socs3-deficient T cells and littermate controls were observed with either method.
Fig. 2.

Socs3 deficiency has no significant effect on Th1 and Th2 differentiation. CD4+ T cells isolated from splenocytes of CreMMTV Socs3fl/fl and littermate control mice were cultured under Th1 or Th2 polarizing conditions for 7 days. (a and b) IFNγ- and IL-4-producing cells were detected by intracellular cytokine staining. Histograms are representative of four separate experiments. (c) IFNγ or IL-4 secreted by Th1 and Th2 cells from CreMMTV Socs3fl/fl and littermate control mice were measured from the cell culture supernatants by ELISA.
Socs3 Deficiency Leads to Increased Phosphorylation of Stat3 in Response to IL-23.
By using a transgenic system, it was reported that Socs3 regulates IL-12-dependent Stat4 phosphorylation (31). We therefore next analyzed cytokine-dependent Stat activation in Socs3-deficient T cells. As shown in Fig. 3, IL-12 and IL-4 induced comparable levels of Stat4 (Fig. 3a) and Stat6 (Fig. 3b) tyrosine phosphorylation in Socs3-deficient T cells versus littermate cells. Because IL-2 signaling has also been reported to influence Th polarization, via phosphorylation of Stat5 (43), we next analyzed Stat5 phosphorylation in T cells lacking Socs3 but again found no alteration (Fig. 3c).
Fig. 3.

Socs3 selectively regulates Stat3 phosphorylation and binding to IL-17 and IL-17F promoters. Splenic CD4+ T cells isolated from CreMMTV Socs3fl/fl and littermate mice were activated with plate-bound anti-CD3 and anti-CD28 for 2 days, expanded in IL-2-containing media, rested, and restimulated with IL-2, IL-4, IL-12, or IL-23 for 15 min to 6 h. Cell lysates were separated by SDS/PAGE, transferred to nitrocellulose membranes, and immunoblotted for phospho-Stat6, phospho-Stat3, and phospho-Stat4. Protein loading was assessed by blotting with anti-β-actin and reprobing the membranes with pan-Stat antibodies. Data in a–d are representative of two to three independent experiments. Lysates were from wild-type and CreMMTV Socs3fl/fl CD4+ lymphoblasts stimulated with IL-23 for 1 h. Stat3 binding to the IL-17A and IL-17F promoter regions was assessed by chromatin immunoprecipitation. The DNA eluted from Stat3 precipitated samples was quantified by q-PCR. (e) Values were normalized to input value and are expressed as fold enrichment relative to normal rabbit serum (NRS) for each experiment.
Recently, a new lineage of Th cells was identified (33–35). This distinct lineage is regulated positively by IL-23 (39) and negatively by IFNγ and IL-4. Polarized cells of this lineage selectively secrete IL-17 (33). IL-23 and IL-12 share a subunit, and both cytokines use IL-12Rβ1 as a subunit of their receptor, but each cytokine also utilizes a distinct receptor subunit and activates different Stat proteins. Specifically, IL-23 activates Stat3 (40). Because Socs3 has been shown to be a critical regulator of cytokine-dependent Stat3 phosphorylation in other systems (18, 20, 44, 45), we next explored whether this regulation occurred in Th cells stimulated with IL-23. As shown in Fig. 3d, IL-23-induced tyrosine phosphorylation of Stat3 was enhanced in the absence of Socs3. To confirm that increased Stat3 phosphorylation was not due to increased IL-23 receptor expression, we measured IL-23 receptor mRNA by q-PCR and found that expression was not enhanced (Fig. 6, which is published as supporting information on the PNAS web site).
The role of Stat3 as a regulator of IL-17A and IL-17F expression has yet to be defined. To this end, we next explored whether Stat3 was able to bind to the promoters of the IL-17A and IL-17F genes. Analysis of the IL-17A and IL-17F promoter regions revealed several potential Stat-binding sites, so we used chromatin immunoprecipitation and q-PCR to analyze whether Stat3 was able to bind to these promoters. Using CD4+ T cells from CreMMTV Socs3fl/fl mice and wild-type littermate controls, we found that Stat3 bound to both IL-17A and IL17F promoters (Fig. 3e). No constitutive enhancement of Stat3 binding to either promoter was evident in the absence of cytokine stimulation, but IL-23 stimulation resulted in significantly enhanced binding (Fig. 7, which is published as supporting information on the PNAS web site).
Loss of Socs3 Results in Enhanced Th17 Generation.
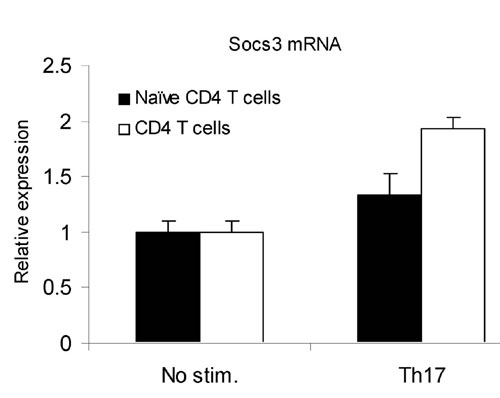
Given that Socs3 appeared to negatively regulate IL-23-dependent Stat3 phosphorylation and that Stat3 directly bound the IL-17A and IL-17F promoters, we next determined whether Socs3 had an effect on Th17 generation. Naïve CD4+ T cells were cultured under conditions that would be expected to promote Th1, Th2, and Th17 cell differentiation. First we confirmed that Socs3 mRNA was expressed in cells polarized with IL-23, anti-IFNγ, and anti-IL-4 (Fig. 8, which is published as supporting information on the PNAS web site). Next we explored Th17 maturation in CreMMTV Socs3fl/fl mice. As shown in Fig. 4a, increased production of IL-17A mRNA was noted in Socs3-null cells under all conditions. Not only were such cells noted to be enhanced in the presence of IL-23, anti-IFNγ, and anti-IL-4, but, strikingly, IL-17-producing cells were present under conditions that would be expected to promote Th1 or Th2 differentiation and block Th17 generation (33). We found that, like IL-17A, IL-17F mRNA showed a dramatic increase in expression in the absence of Socs3 (Fig. 4b); in fact, the magnitude of difference was even greater for this cytokine than for IL-17A itself. The overproduction of IL-17 mRNA was confirmed by investigation of IL-17 protein expression. IL-17 secreted into the cell supernatant was measured by ELISA (Fig. 4c), and cell expression of IL-17 was identified by intracellular staining (Fig. 4d). Once again, increased expression was seen under all conditions (Fig. 4e). Because there are no commercial antibodies for IL-17F we were unable to confirm whether increased IL-17F mRNA translates into an increased production of this cytokine in the absence of Socs3.
Fig. 4.

Socs3 negatively regulates IL-17 and IL-17F expression in T cells. Naïve splenic CD4+ T cells were enriched from CreMMTV Socs3fl/fl and littermate control mice and activated with plate-bound anti-CD3 and anti-CD28. (a and b) Cells were cultured in media containing Il-2 (40 international units/ml) under the following polarizing conditions: Th0 (no additional cytokines), Th1 [IL-12 (10 ng/ml) and anti-IL-4 (10 μg/ml)]; Th2 [IL-4 (10 ng;ml) and anti-IFNγ (10 μg/ml)]; and Il-23 (10 ng/ml) in the absence or presence of anti-IFNγ (10 μg/ml) or Th17 [IL-23 (10 ng/ml), anti-IFNγ (10 μg/ml), and anti-IL-4 (10 μg/ml)]. (c) IL-17A or IL-17F mRNA expression was detected by real-time q-PCR. (d and e) IL-17 protein production was measured in cell culture supernatants by ELISA and by intracellular cytokine staining using flow cytometry. Data shown are representative of two to four independent experiments.
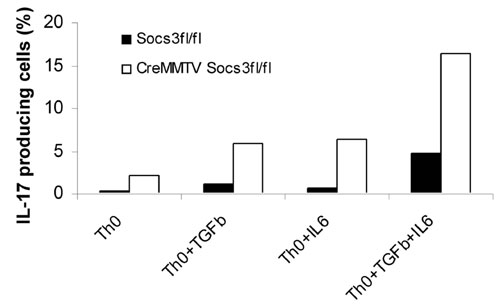
Recent work by Veldhoen et al. (46) has shown that TGF-β and IL-6 are potent inducers of Th17 polarization, so we next asked whether the absence of Socs3 would affect polarization in the presence of these cytokines. In keeping with recent findings, TGF-β was a potent inducer of IL-17 secretion, which was enhanced with the addition of IL-6. The effect of these cytokines was markedly enhanced in the absence of Socs3 (Fig. 9, which is published as supporting information on the PNAS web site). Thus, Socs3 appears to be a critical regulator of Th17 generation regardless of whether IL-6, TGF-β, or IL-23 are used and evidently serves to attenuate Th17 generation even under Th1 conditions.
Discussion
Using a conditional knockout approach, we have shown that Socs3 has a minimal role in thymocyte development and, in contrast to previous reports (31, 32, 43), no significant role on Th1 and Th2 polarization and Stat4 or Stat6 phosphorylation. By contrast, IL-23-dependent Stat3 phosphorylation was enhanced in the absence of Socs3, and Stat3 was found to bind both IL-17A and IL-17F promoters. Functionally, this enhanced signaling was associated with an increase in the proportion of naïve T cells polarizing into Th17 cells, under both optimal and suboptimal conditions for Th17 generation.
The notion that Th cells can be subdivided into two discrete populations on the basis of cytokine expression was first proposed by Mosmann et al. (47). Recently this dichotomous view was challenged by the discovery of a third lineage of mature Th cells that secrete IL-17, a cytokine that indirectly recruits neutrophils and is implicated in combating extracellular bacterial infections (33–36). Th17 cells have been generated in vitro by T cell receptor activation in the presence of IL-23 and antibodies blocking both IL-4 and IFNγ (33). Although IL-23 is known to activate Stat1, Stat3, and Stat4, the requirement for Stat signaling in the differentiation of Th17 cells is unknown (40). Our data would indicate that IL-23-activated Stat3 directly binds the IL-17A and IL-17F promoters. Adding weight to the importance of Stat3 in the generation of Th17 cells is the recent discovery that both TGF-β and IL-6 are potent inducers of IL-17 secretion (46). IL-6 is a known activator of Stat3 that is enhanced in the absence of Socs3 (20, 22, 23). Our data indicate that, regardless of whether Th17 cells are generated by IL-6 or IL-23, Socs3 deficiency resulted in marked enhancement. Indeed, even under optimal Th1 conditions, Socs3 evidently has an important role in constraining IL-17 production. Further work is required to delineate precisely how IL-6 and IL-23 regulate the transcription of IL-17A and IL-17F and how Stat3 contributes to this regulation. With regard to previous studies in which Socs3 was reported to affect Th1/Th2 differentiation, it must be considered that overexpression studies have often been misleading with respect to physiologic functions (48). Alternatively, it is important to bear in mind that our studies used stimulation with anti-CD3 antibody. In the future, it may be useful to revisit this issue of Th1/Th2 regulation using more physiological models.
The role of Socs3 in T cell biology has been difficult to assess, because Socs3-null mice die in utero with defects of placental development. This development defect is overcome in mice lacking both Socs3 and leukemia inhibitory factor; nonetheless, the mice ultimately succumb to a fatal inflammatory disease characterized by infiltration of peripheral tissues with neutrophils and monocytes (16). The disease mimics that seen in mice with a conditional bone marrow deletion of Socs3 and irradiated animals that have been rescued with Socs3−/− bone marrow (18). A contributor to this pathology is that Socs3 serves as an inhibitor of granulocyte/colony-stimulating factor (G-CSF) signaling. However, an enhanced response to G-CSF does not fully explain why adult Socs3-null mice develop a fatal inflammatory syndrome. Mice lacking Socs3 within the bone marrow compartment have normal-sized spleens and healthy full blood counts at 8 weeks of age, the inflammatory infiltrate appearing only after 17 weeks of age, suggesting a second trigger. We believe that our present findings suggest an additional explanation for the pathology associated with Socs3 deficiency.
IL-17 has been implicated in a number of autoimmune diseases both in humans and in mouse models (49). Increased levels of IL-17A have been found in samples from patients with extrinsic allergic alveolitis (50), arthritis (51), and Crohn's disease (52). Although IL-17 has no direct chemotactic activity, it is a potent inducer of IL-6, G-CSF, and chemokines (53, 54). These cytokines in turn recruit and activate circulating neutrophils (55). Transgenic mice overexpressing IL-17A (56, 57) or the p19 subunit of IL-23 (58) develop a systemic inflammatory pathology associated with increased expression of G-CSF. We have observed that CreMMTV Socs3fl/fl mice with advanced age also developed systemic inflammatory disease. Like IL-17 and IL-23 p19 transgenic mice, CreMMTV Socs3fl/fl mice have elevated levels of G-CSF in tissues (Fig. 10, which is published as supporting information on the PNAS web site), albeit at a delayed onset at 3–6 months of age. The extent to which the Socs3 deficiency phenotype is similar to these transgenic models warrants further inquiry.
To summarize, our data indicate that Socs3 has an important role in Th cell differentiation, limiting the development of Th17 cell polarization. It does so by attenuating the phosphorylation of Stat3, a transcription factor that is likely to be a direct regulator of IL-17 transcription. Defining the mechanisms underlying disease in Socs3-deficient mice and the relative contributions of Stat3 and IL-17 will be important areas of future investigation.
Materials and Methods
Generation of Socs3-Deficient T Cells.
Mice bearing loxP-flanked conditional (Socs3fl) alleles of Socs3 on a C57BL/6J inbred background were described in ref. 23. loxP sites flank the Socs3 exon 2, and expression of Cre deletes the intervening DNA. Socs3fl/fl mice were bred with mice expressing Cre under the control of the MMTV-LTR (CreMMTV), which allows expression in mammary gland, salivary gland, seminal vesicle, skin, and B and T cells (42, 59). Socs3fl/fl mice lacking transgenic Cre were used as controls.
Cells and Culture Conditions.
Thymocytes and peripheral lymphocytes were obtained by disrupting organs of healthy 8- to 10-week-old CreMMTV Socs3fl/fl mice together with littermate controls. Cell cultures were performed in RPMI medium 1640 supplemented with 10% FCS, 2 mM glutamine, 100 units/ml penicillin, 0.1 mg/ml streptomycin (BioSource International, Camarillo, CA), and 2 mM 2-mercaptoethanol. Th cells were enriched by positive selection by using Macs beads (Miltenyi Biotec, Bergisch-Gladbach, Germany). Cells were activated by plate-bound anti-CD3 and soluble anti-CD28 (BD Pharmingen, San Diego) and cultured under Th1 (IL-12, anti-IL4) or Th2 (IL-4, anti-IFNγ) polarizing conditions or in the presence of IL-23, IL-6 (10 ng/ml; R & D Systems), TGFβ (5 ng/ml; R & D Systems), or combinations thereof with anti-IFNγ and anti-IL-4.
Flow Cytometry and Cytokine Measurement.
Thymocytes were stained for surface expression of the following markers: CD4, CD8, CD25, CD44, T cell receptor γδ, B220, Thy1.2, pan NK DX5, Mac1, and Gr-1. Peripheral lymphocytes were stained for surface expression of the following markers: CD4, CD8, CD25, CD69, CD44 (all from BD Biosciences, San Diego). Detection of IFNγ-, IL-4-, and IL-17-producing cells was determined by intracellular cytokine staining with anti-IFNγ-allophycocyanin, anti-IL-17-phycoerythrin, or anti-IL-4-phycoerythrin (BD Biosciences). Briefly, cells were stimulated for 4 h with PMA and ionomycin, with GolgiStop (BD Biosciences) added after 2 h. Cell stimulation was terminated by fixing in 4% formyl saline. Fixed cells were stained with fluorescent antibodies in 0.1% saponin permeabilization buffer and analyzed on a FACSCalibur (BD Biosciences). Events were collected and analyzed by using flowjo software (Tree Star, Ashland, OR).
Total RNA was extracted by RNeasy kit (Qiagen, Valencia, CA) and assayed by real-time q-PCR. cDNA was synthesized with TaqMan Reverse Transcription kit (Applied Biosystems) by using random hexamers as primers according to the manufacturer's instruction. 18sRNA was used as endogenous control. TaqMan primers and probes for murine Socs3, IFNγ, IL-4, IL-17, IL-17F, and 18sRNA were purchased from Applied Biosystems, and samples were analyzed by using the ABI PRISM 7700 Sequence Detection System (Applied Biosystems).
Cytokine production in cell culture supernatants were analyzed by ELISA by using mouse IL-17, IL-4, and IFNγ Quantikine assay kits (R & D Systems) according to the manufacturer's instructions.
Immunoblotting.
T lymphoblasts in RPMI medium 1640 supplemented with 10% FCS were stimulated with 10 ng/ml IL-23, 10 ng/ml IL-12, or 20 ng/ml IL-4 for 0 min to 6 h. Stimulation was terminated by washing in ice-cold PBS and lysing in detergent buffer containing complete Protease Inhibitor Mixture (Roche Applied Science, Indianapolis) and sodium orthovanadate. Cell lysates were electrophoresed, transferred onto a nitrocellulose filter, and immunoblotted with antibodies against phospho-Stat6, phospho-Stat3, phospho-Stat5 (Cell Signaling Technology, Danvers, MA), and phospho-Stat4 (Invitrogen). Total Stat levels were assessed by using anti-Stat6, anti-Stat3, anti-Stat5 (Cell Signaling Technology), and anti-Stat4 (Santa Cruz Biotechnology).
Chromatin Immunoprecipitation.
Chromatin immunoprecipitation was performed as previously described (60). To summarize, CD4+ T cells polarized under Th17 conditions and expanded in IL-2 were stimulated with IL-23 for 1 h. DNA-bound transcription factors were subsequently crosslinked in vivo by using complete medium containing 1% formaldehyde for 10 min followed by sonication of the cell lysate. After preclearing with protein A agarose beads (Upstate Biotechnology, Charlottesville, VA), cell lysates were immunoprecipitated with anti-Stat3 antibody (Santa Cruz Biotechnology) or normal rabbit serum overnight at 4°C. After washing and elution, crosslinks were reversed at 65°C for 4 h. The eluted DNA was purified, and samples were analyzed by q-PCR by using IL-17A promoter site-specific primers.
Supplementary Material
Abbreviations
- Socs
suppressor of cytokine signaling
- Stat
signal transducer and activator of transcription
- Th
T helper
- G-CSF
granulocyte/colony-stimulating factor
- q-PCR
quantitative PCR.
Footnotes
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Brender C., Nielsen M., Kaltoft K., Mikkelsen G., Zhang Q., Wasik M., Billestrup N., Odum N. Blood. 2001;97:1056–1062. doi: 10.1182/blood.v97.4.1056. [DOI] [PubMed] [Google Scholar]
- 2.Davey H. W., McLachlan M. J., Wilkins R. J., Hilton D. J., Adams T. E. Mol. Cell. Endocrinol. 1999;158:111–116. doi: 10.1016/s0303-7207(99)00175-6. [DOI] [PubMed] [Google Scholar]
- 3.Alexander W. S., Hilton D. J. Annu. Rev. Immunol. 2004;22:503–529. doi: 10.1146/annurev.immunol.22.091003.090312. [DOI] [PubMed] [Google Scholar]
- 4.O'Shea J. J., Gadina M., Schreiber R. D. Cell. 2002;109(Suppl):S121–S131. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- 5.Yoshimura A., Ohkubo T., Kiguchi T., Jenkins N. A., Gilbert D. J., Copeland N. G., Hara T., Miyajima A. EMBO J. 1995;14:2816–2826. doi: 10.1002/j.1460-2075.1995.tb07281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Endo T. A., Masuhara M., Yokouchi M., Suzuki R., Sakamoto H., Mitsui K., Matsumoto A., Tanimura S., Ohtsubo M., Misawa H., et al. Nature. 1997;387:921–924. doi: 10.1038/43213. [DOI] [PubMed] [Google Scholar]
- 7.Nicholson S. E., Willson T. A., Farley A., Starr R., Zhang J. G., Baca M., Alexander W. S., Metcalf D., Hilton D. J., Nicola N. A. EMBO J. 1999;18:375–385. doi: 10.1093/emboj/18.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J. G., Farley A., Nicholson S. E., Willson T. A., Zugaro L. M., Simpson R. J., Moritz R. L., Cary D., Richardson R., Hausmann G., et al. Proc. Natl. Acad. Sci. USA. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamura T., Sato S., Haque D., Liu L., Kaelin W. G., Jr, Conaway R. C., Conaway J. W. Genes Dev. 1998;12:3872–3881. doi: 10.1101/gad.12.24.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krebs D. L., Hilton D. J. Stem Cells. 2001;19:378–387. doi: 10.1634/stemcells.19-5-378. [DOI] [PubMed] [Google Scholar]
- 11.Alexander W. S. Nat. Rev. Immunol. 2002;2:410–416. doi: 10.1038/nri818. [DOI] [PubMed] [Google Scholar]
- 12.Alexander W. S., Starr R., Fenner J. E., Scott C. L., Handman E., Sprigg N. S., Corbin J. E., Cornish A. L., Darwiche R., Owczarek C. M., et al. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 13.Metcalf D., Greenhalgh C. J., Viney E., Willson T. A., Starr R., Nicola N. A., Hilton D. J., Alexander W. S. Nature. 2000;405:1069–1073. doi: 10.1038/35016611. [DOI] [PubMed] [Google Scholar]
- 14.Greenhalgh C. J., Metcalf D., Thaus A. L., Corbin J. E., Uren R., Morgan P. O., Fabri L. J., Zhang J. G., Martin H. M., Willson T. A., et al. J. Biol. Chem. 2002;277:40181–40184. doi: 10.1074/jbc.C200450200. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi Y., Carpino N., Cross J. C., Torres M., Parganas E., Ihle J. N. EMBO J. 2003;22:372–384. doi: 10.1093/emboj/cdg057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robb L., Boyle K., Rakar S., Hartley L., Lochland J., Roberts A. W., Alexander W. S., Metcalf D. Proc. Natl. Acad. Sci. USA. 2005;102:16333–16338. doi: 10.1073/pnas.0508023102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mori H., Hanada R., Hanada T., Aki D., Mashima R., Nishinakamura H., Torisu T., Chien K. R., Yasukawa H., Yoshimura A. Nat. Med. 2004;10:739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 18.Croker B. A., Metcalf D., Robb L., Wei W., Mifsud S., DiRago L., Cluse L. A., Sutherland K. D., Hartley L., Williams E., et al. Immunity. 2004;20:153–165. doi: 10.1016/s1074-7613(04)00022-6. [DOI] [PubMed] [Google Scholar]
- 19.Kimura A., Kinjyo I., Matsumura Y., Mori H., Mashima R., Harada M., Chien K. R., Yasukawa H., Yoshimura A. J. Biol. Chem. 2004;279:6905–6910. doi: 10.1074/jbc.C300496200. [DOI] [PubMed] [Google Scholar]
- 20.Croker B. A., Krebs D. L., Zhang J. G., Wormald S., Willson T. A., Stanley E. G., Robb L., Greenhalgh C. J., Forster I., Clausen B. E., et al. Nat. Immunol. 2003;4:540–545. doi: 10.1038/ni931. [DOI] [PubMed] [Google Scholar]
- 21.Sun R., Jaruga B., Kulkarni S., Sun H., Gao B. Biochem. Biophys. Res. Commun. 2005;338:1943–1949. doi: 10.1016/j.bbrc.2005.10.171. [DOI] [PubMed] [Google Scholar]
- 22.Lang R., Pauleau A. L., Parganas E., Takahashi Y., Mages J., Ihle J. N., Rutschman R., Murray P. J. Nat. Immunol. 2003;4:546–550. doi: 10.1038/ni932. [DOI] [PubMed] [Google Scholar]
- 23.Yasukawa H., Ohishi M., Mori H., Murakami M., Chinen T., Aki D., Hanada T., Takeda K., Akira S., Hoshijima M., et al. Nat. Immunol. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- 24.Marine J. C., McKay C., Wang D., Topham D. J., Parganas E., Nakajima H., Pendeville H., Yasukawa H., Sasaki A., Yoshimura A., Ihle J. N. Cell. 1999;98:617–627. doi: 10.1016/s0092-8674(00)80049-5. [DOI] [PubMed] [Google Scholar]
- 25.Murphy K. M., Reiner S. L. Nat. Rev. Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 26.Jacobson N. G., Szabo S. J., Guler M. L., Gorham J. D., Murphy K. M. Adv. Exp. Med. Biol. 1996;409:61–73. doi: 10.1007/978-1-4615-5855-2_9. [DOI] [PubMed] [Google Scholar]
- 27.Jacobson N. G., Szabo S. J., Weber-Nordt R. M., Zhong Z., Schreiber R. D., Darnell J. E., Jr, Murphy K. M. J. Exp. Med. 1995;181:1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bacon C. M., Petricoin E. F., III, Ortaldo J. R., Rees R. C., Larner A. C., Johnston J. A., O'Shea J. J. Proc. Natl. Acad. Sci. USA. 1995;92:7307–7311. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimoda K., van Deursen J., Sangster M. Y., Sarawar S. R., Carson R. T., Tripp R. A., Chu C., Quelle F. W., Nosaka T., Vignali D. A., et al. Nature. 1996;380:630–633. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 30.Takeda K., Tanaka T., Shi W., Matsumoto M., Minami M., Kashiwamura S., Nakanishi K., Yoshida N., Kishimoto T., Akira S. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 31.Seki Y., Inoue H., Nagata N., Hayashi K., Fukuyama S., Matsumoto K., Komine O., Hamano S., Himeno K., Inagaki-Ohara K., et al. Nat. Med. 2003;9:1047–1054. doi: 10.1038/nm896. [DOI] [PubMed] [Google Scholar]
- 32.Takatori H., Nakajima H., Kagami S., Hirose K., Suto A., Suzuki K., Kubo M., Yoshimura A., Saito Y., Iwamoto I. J. Immunol. 2005;174:4105–4112. doi: 10.4049/jimmunol.174.7.4105. [DOI] [PubMed] [Google Scholar]
- 33.Harrington L. E., Hatton R. D., Mangan P. R., Turner H., Murphy T. L., Murphy K. M., Weaver C. T. Nat. Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 34.Park H., Li Z., Yang X. O., Chang S. H., Nurieva R., Wang Y. H., Wang Y., Hood L., Zhu Z., Tian Q., Dong C. Nat. Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sato S., Sanjo H., Takeda K., Ninomiya-Tsuji J., Yamamoto M., Kawai T., Matsumoto K., Takeuchi O., Akira S. Nat. Immunol. 2005;6:1087–1095. doi: 10.1038/ni1255. [DOI] [PubMed] [Google Scholar]
- 36.Happel K. I., Dubin P. J., Zheng M., Ghilardi N., Lockhart C., Quinton L. J., Odden A. R., Shellito J. E., Bagby G. J., Nelson S., Kolls J. K. J. Exp. Med. 2005;202:761–769. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolls J. K., Linden A. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 38.Moseley T. A., Haudenschild D. R., Rose L., Reddi A. H. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 39.Aggarwal S., Ghilardi N., Xie M. H., de Sauvage F. J., Gurney A. L. J. Biol. Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 40.Parham C., Chirica M., Timans J., Vaisberg E., Travis M., Cheung J., Pflanz S., Zhang R., Singh K. P., Vega F., et al. J. Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 41.Hennighausen L., Wall R. J., Tillmann U., Li M., Furth P. A. J. Cell Biochem. 1995;59:463–472. doi: 10.1002/jcb.240590407. [DOI] [PubMed] [Google Scholar]
- 42.Wagner K. U., McAllister K., Ward T., Davis B., Wiseman R., Hennighausen L. Transgenic Res. 2001;10:545–553. doi: 10.1023/a:1013063514007. [DOI] [PubMed] [Google Scholar]
- 43.Cote-Sierra J., Foucras G., Guo L., Chiodetti L., Young H. A., Hu-Li J., Zhu J., Paul W. E. Proc. Natl. Acad. Sci. USA. 2004;101:3880–3885. doi: 10.1073/pnas.0400339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forrai A., Boyle K., Hart A., Hartley L., Rakar S., Willson T. A., Simpson K. M., Roberts A. W., Alexander W. S., Voss A. K., Robb L. Stem Cells. 2005;24:604–614. doi: 10.1634/stemcells.2005-0323. [DOI] [PubMed] [Google Scholar]
- 45.Le Provost F., Miyoshi K., Vilotte J. L., Bierie B., Robinson G. W., Hennighausen L. Biochem. Biophys. Res. Commun. 2005;338:1696–1701. doi: 10.1016/j.bbrc.2005.10.138. [DOI] [PubMed] [Google Scholar]
- 46.Veldhoen M., Hocking R. J., Atkins C. J., Locksley R. M., Stockinger B. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 47.Mosmann T. R., Cherwinski H., Bond M. W., Giedlin M. A., Coffman R. L. J. Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 48.Berlato C., Cassatella M. A., Kinjyo I., Gatto L., Yoshimura A., Bazzoni F. J. Immunol. 2002;168:6404–6411. doi: 10.4049/jimmunol.168.12.6404. [DOI] [PubMed] [Google Scholar]
- 49.Katz Y., Nadiv O. Isr. Med. Assoc. J. 2000;2(Suppl):21–22. [PubMed] [Google Scholar]
- 50.Laan M., Palmberg L., Larsson K., Linden A. Eur. Respir. J. 2002;19:534–537. doi: 10.1183/09031936.02.00280902. [DOI] [PubMed] [Google Scholar]
- 51.Chabaud M., Garnero P., Dayer J. M., Guerne P. A., Fossiez F., Miossec P. Cytokine. 2000;12:1092–1099. doi: 10.1006/cyto.2000.0681. [DOI] [PubMed] [Google Scholar]
- 52.Fujino S., Andoh A., Bamba S., Ogawa A., Hata K., Araki Y., Bamba T., Fujiyama Y. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fossiez F., Djossou O., Chomarat P., Flores-Romo L., Ait-Yahia S., Maat C., Pin J. J., Garrone P., Garcia E., Saeland S., et al. J. Exp. Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wuyts W. A., Vanaudenaerde B. M., Dupont L. J., Van Raemdonck D. E., Demedts M. G., Verleden G. M. J. Heart Lung Transplant. 2005;24:875–881. doi: 10.1016/j.healun.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 55.Laan M., Cui Z. H., Hoshino H., Lotvall J., Sjostrand M., Gruenert D. C., Skoogh B. E., Linden A. J. Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- 56.Schwarzenberger P., La Russa V., Miller A., Ye P., Huang W., Zieske A., Nelson S., Bagby G. J., Stoltz D., Mynatt R. L., et al. J. Immunol. 1998;161:6383–6389. [PubMed] [Google Scholar]
- 57.Schwarzenberger P., Huang W., Ye P., Oliver P., Manuel M., Zhang Z., Bagby G., Nelson S., Kolls J. K. J. Immunol. 2000;164:4783–4789. doi: 10.4049/jimmunol.164.9.4783. [DOI] [PubMed] [Google Scholar]
- 58.Wiekowski M. T., Leach M. W., Evans E. W., Sullivan L., Chen S. C., Vassileva G., Bazan J. F., Gorman D. M., Kastelein R. A., Narula S., Lira S. A. J. Immunol. 2001;166:7563–7570. doi: 10.4049/jimmunol.166.12.7563. [DOI] [PubMed] [Google Scholar]
- 59.Wagner K. U., Wall R. J., St-Onge L., Gruss P., Wynshaw-Boris A., Garrett L., Li M., Furth P. A., Hennighausen L. Nucleic Acids Res. 1997;25:4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morinobu A., Kanno Y., O'Shea J. J. J. Biol. Chem. 2004;279:40640–40646. doi: 10.1074/jbc.M407576200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}