

Abstract
Yeast glycoproteins are representative of low-complexity sequences, those sequences rich in a few types of amino acids. Low-complexity protein sequences comprise more than 10% of the proteome but are poorly aligned by existing methods. Under default conditions, BLAST and FASTA use the scoring matrix BLOSUM62, which is optimized for sequences with diverse amino acid compositions. Because low-complexity sequences are rich in a few amino acids, these tools tend to align the most common residues in nonhomologous positions, thereby generating anomalously high scores, deviations from the expected extreme value distribution, and small e values. This anomalous scoring prevents BLOSUM62-based BLAST and FASTA from identifying correct homologs for proteins with low-complexity sequences, including Saccharomyces cerevisiae wall proteins. We have devised and empirically tested scoring matrices that compensate for the overrepresentation of some amino acids in any query sequence in different ways. These matrices were tested for sensitivity in finding true homologs, discrimination against nonhomologous and random sequences, conformance to the extreme value distribution, and accuracy of e values. Of the tested matrices, the two best matrices (called E and gtQ) gave reliable alignments in BLAST and FASTA searches, identified a consistent set of paralogs of the yeast cell wall test set proteins, and improved the consistency of secondary structure predictions for cell wall proteins.
The ability to accurately align protein sequences is central to inferences about the evolutionary history of genes and therefore to the evolution of organelles and organisms as well. In addition, homology modeling and even functional inference through the annotation of similar sequences depends on alignment accuracy. For low-complexity sequences such as fungal cell wall proteins, errors caused by anomalous high scores for nonhomologous sequences will inevitably lead to erroneous inferences for evolution, structure, and function.
Low-complexity sequences in the proteome.
Proteins with low-complexity sequences are common and functionally important but are not well aligned by existing procedures. These proteins are rich in a few amino acids and thus have overall composition significantly different from the “average” compositions seen in the multiple alignments used to construct the BLOSUM alignment scoring matrices and for the BLAST statistical analyses (16). About 10% of known protein sequences have overall low complexity; eukaryotic genomes and some bacterial pathogens contain even higher percentages of low-complexity sequences (24, 32). The NCBI nonredundant database currently contains approximately 3.2 million sequences. Thus, there are about 320,000 low-complexity sequences that cannot be accurately compared or aligned and therefore cannot be compared on any large scale, either functionally or evolutionarily. In addition, there are low-complexity segments in half of all proteins (32). These segments also cannot be reliably aligned and so are currently “masked” by SEG or similar procedures and then ignored by the alignment tools (29, 34). In globular proteins, low-complexity sequences tend to occur as loops within and between globular domains (19, 21), regions often important for protein function. Recent papers have highlighted the need to solve this problem, and a logical solution is the modification of scoring matrices to compensate for the composition of the query sequence (6, 36, 37).
Fungal cell wall proteins are representative of low-complexity sequences; they average 35% Ser and Thr residues, with some 100-residue segments composed almost exclusively of these two amino acids (11, 20, 28). As a result, wall proteins are normally aligned only after SEG filtering to remove the low-complexity segments, so sequence comparisons cannot be made for the low-complexity regions. If there were rapid search and alignment protocols that could compare such compositionally biased segments, then both evolutionary and structural comparisons could be attempted.
The major alignment problem for low-complexity sequences is called low-complexity corruption (31). Intuitively, low-complexity corruption results from the alignment of high-frequency residues. In fungal cell wall proteins, the problem is most egregious for Ser, Thr, Pro, Ala, and Val. This phenomenon gives high alignment scores and low e values to nonhomologous pairs of protein segments (high-scoring pairs [HSPs]). For example, alignments of Ser with Ser and Thr with Thr in cell wall proteins give alignment scores of +4 and +5, respectively, in BLOSUM62, the standard scoring matrix. Because the residue alignment scores are summed over the segments being aligned, the many pairs of aligned Ser and Thr residues will give a high summed total alignment score, even if the frequently occurring amino acids are randomly distributed in the sequences. Indeed, in searches using low-complexity proteins as the query sequence, there are enough abnormally high-scoring pairs that the distribution of all scores is skewed by the overrepresentation of high scores (Fig. 1B). The skew means that the score distribution deviates from the expected extreme value distribution, and e values calculated from the scores are invalid because the underlying distribution is different. For low-complexity sequences, this combination of anomalous high scores and small e values appears with any search and alignment tool that uses BLOSUM matrices, including BLAST, FASTA, and the initial alignments in PSI-BLAST. Thus, if the alignment scores for frequently occurring amino acids were reduced appropriately, alignments of these residues would not artificially inflate the scores to generate HSPs from sequences with similar amino acid compositions but dissimilar sequences.
FIG. 1.

Alignments for best-scoring HSPs (e ≤ 10−3) for Aga1p, Muc1p, and Fig2p. The first 50 aligned residues in each alignment are shown, and identities are shown between the query sequence and the similar sequence. Residues S and T in boldface type are overrepresented in the query sequences. (A) Muc1p/Bsc1p alignment reported in all searches. e values for the alignments are shown on the left. (B to D) Other highest-scoring alignments and e values for BLAST searches with each listed matrix. There were no other HSPs with e values of <10−3 for the BLAST-E searches.
Matrices other than BLOSUM have been shown to be more appropriate for sequences of nonaverage composition. For example, to make discriminatory matrices and predict hydrophobic and transmembrane segments in proteins, the specialized matrices PHAT and SLIM use the background frequencies present in transmembrane alignments instead of standard amino acid frequencies (23, 25). Similarly, position-specific scoring matrices (PSSM) are used to predict coiled-coil structures and in all iterative searches after the first in PSI-BLAST (5, 7, 22). The effectiveness of these specialized matrices on their intended targets attests to the fact that adjustment of matrices to account for amino acid composition in the query and target sequences can be highly discriminating and sensitive.
Goals and evaluation criteria.
To improve the alignment of low-complexity sequences, we have developed and tested modifications to produce scoring matrices that are adjusted for the composition of each query. The goals are to prevent alignments of sequences that are compositionally similar but nonhomologous and to generate statistically significant, homology-driven alignments of low-complexity segments necessary for structural and evolutionary studies of the low-complexity portion of the proteome.
Each matrix modification method was evaluated based on the following criteria, as summarized in Table 1: sensitivity (the ability to find a high number of homologs) for both low-complexity and high-complexity query sequences, discrimination against randomized sequences and nonhomologous proteins with similar amino acid compositions, conformance with the expected extreme value distribution of alignment scores that should be generated during the search, accuracy of derived e values, and computational efficiency. The results demonstrated that two of the composition-based matrices are powerful adaptations for BLAST and FASTA searches and alignments for low-complexity Saccharomyces cerevisiae glycoprotein sequences.
TABLE 1.
Validation tests
| Test purpose | Methods and/or searches (matrices) | Data set
|
Results | ||
|---|---|---|---|---|---|
| Query | Database (positive control) | Database (negative control) | |||
| Sensitivity/discrimination (low complexity) | Sensitivity curves, BLAST and FASTA (11 modified and unmodified matrices) | 10 cell wall proteins | Yeast proteome | Locally randomized yeast proteome (pseudoprotein sequences) | Fig. 2 |
| Application in homology searching | Transitive closure, BLAST and FASTA (11 modified and unmodified matrices) | 10 cell wall proteins | GO and manually annotated cell wall proteins in yeast proteome | Non-cell wall proteins according to GO and manual annotations | Fig. 3, Table 4 |
| Sensitivity/discrimination (high complexity) | Sensitivity curves, BLAST and FASTA (11 modified and unmodified matrices) | Aravind (103 query sequences) | Aravind (true hits) | Aravind (false hits) | Table 5 |
| Conformance to extreme value distribution | Distribution of scores, BLAST and FASTA with B, PGP gtQ, and E | Flo1p fragment, random sequences of low and high complexity | None | 10,000 globally and locally random sequences with low and high complexity | Table S1; chi-square tests, κ and λ estimates |
| Distribution of scores (11 modified and unmodified matrices) | Aravind (103 query sequences) | Homologous sequences in yeast proteome | Nonhomologous sequences in yeast proteome | Fig. S2 | |
| Accuracy of false-hit e values | Mean and best e values | 10 cell wall proteins; Aravind | None | Nonhomologous sequences in yeast proteome | Table S2 |
MATERIALS AND METHODS
Summary of methods.
We searched for sequences similar to each of 10 yeast cell wall proteins. These proteins have low-complexity regions that constitute 40 to 100% of the open reading frame (ORF) length. For each query sequence, the amino acid frequencies were determined, and the scoring matrix was altered by the rules described below. The modified scoring matrices (Table 2) were rescaled to the same κ and λ statistical parameters as the standard scoring matrix (BLOSUM62) so that the reported e values were distributed similarly to those from BLOSUM62-based searches of high-complexity sequences (see Table S1 in the supplemental material). A similar rescaling strategy is used in PSI-BLAST (5). (Note that additional mathematical definitions and relationships are described in the supplemental material.) The query sequence was then used as the query in BLAST or FASTA. HSPs were ranked by e values.
TABLE 2.
Search methods and modified scoring matrices
| Search tool | Matrix | Description |
|---|---|---|
| BLAST | B | Standard BLOSUM62 (SEG filter not used) |
| BF | Standard BLOSUM62 with SEG filtering | |
| PGP | BLOSUM62 with 32-fold expanded scaling and 32-fold gap costs; score distributions adjusted to reflect the composition of the query | |
| E | Adjust scores to maintain expected score equal | |
| Q | Adjust scores to maintain target frequency equal | |
| gtE | BLOSUM62 with E modifications for overrepresented amino acids | |
| gtQ | BLOSUM62 with Q modifications for overrepresented amino acids | |
| FASTA | B | Standard BLOSUM62 (SEG filter not used) |
| BF | Standard BLOSUM62 with SEG filtering | |
| E | Adjust scores to maintain expected score equal | |
| Q | Adjust scores to maintain target frequency equal | |
| gtE | BLOSUM62 with E modifications for overrepresented amino acids | |
| gtQ | BLOSUM62 with Q modifications for overrepresented amino acids | |
| gtE32 | 32-fold gtE modifications and 32-fold gap costs | |
| gtQ32 | 32-fold gtQ modifications and 32-fold gap costs |
FASTA calculates e values for each search by comparison to scores generated by randomized query sequences. Therefore, the e values reported for FASTA searches are appropriate for each query sequence and scoring matrix.
Matrix modification Q.
One way to change scoring matrices is to adjust each scoring element, Sij, to compensate for the probability of a match at random. This approach keeps the target frequencies, Qij, equal to the standard target frequencies, in the hope that this will reduce random alignments of frequently appearing amino acids. Each new matrix element, S*ij, can be calculated as follows:
 |
(1) |
where Pi is the probability of the occurrence of an individual amino acid, i, and P*i is the probability of amino acid i in the query sequence, and the new score is calculated from Sij and Pj. Pj and Qij are taken to be unchanged, so one compensates for the low complexity in the query but not in the database sequence. λ predicts the width of the extreme value score distribution. In essence, each score, S*ij, is reduced or raised to compensate for the degree to which the frequency for i in the query sequence differs from the frequency for i in the standard ratios used in BLOSUM62 (16, 17). The new matrix will have the same target frequencies in the context of the amino acid composition of the query sequence that the original matrix had in the context of standard amino acid composition. Because target frequencies, Qij, are kept constant, equation 1 guarantees that the λ of the matrix in the context of the amino acid composition of the query sequence should not change. BLAST, however, requires that the matrix entries be integers, so λ does change after rounding of the score. For each search, λ* can be set to the λ of the original matrix by multiplying each score by the ratio of the λ* of the unscaled matrix to the λ of original matrix, as described previously (31). We call this matrix modification Q, for target frequency.
Matrix modification E.
The problem of complexity corruption can be thought of in another manner. The expected score, E, of a given matrix is as follows:
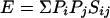 |
(2) |
The BLAST statistical model requires that value E in equation 2 be negative (18). If the probability of amino acid i in the query sequence is larger than the standard probability for i used in the database or score distribution simulation, the expected score for the ij pair will unduly contribute to the total score of alignments and will select for randomly aligned segments that have amino acid compositions similar to the query. Once again, we can adjust the score of the matrix to compensate for the fluctuation in the amino acid composition of a query from the standard amino acid composition and yet retain the intrinsic property (i.e., expected score, E) of the matrix in the context of the query's amino acid composition as follows:
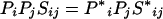 |
(3) |
We call this matrix modification E, for expected score. This modification significantly changes the value of λ, which is then reset according to equation 1.
Fig. S1 in the supplemental material shows the impact of matrix modifications on positive and negative scores relative to the ratio between the probability that amino acid i occurs in the query and the standard Robinson and Robinson probability for that amino acid (3). These matrix modifications decrease positive scores for frequent amino acid pairs but increase negative ones. Matrix modification E increases the negative scores for frequent pairs, but such a negative score never becomes positive nor does a positive score ever become negative. In contrast, Q modifications can convert a negative score into a positive one or vice versa. As a result, the two types of matrix modifications produced distinct total scores and alignments.
“gt” and “32” modifications.
A “greater-than” (gt) matrix modification was also implemented. Under this modification, scores are reduced only if a residue is more frequent in the query sequence than in “standard” frequencies calculated according to Robinson and Robinson frequencies (i.e., P*i/Pi > 1) (3). When applied to matrix modification E or to matrix modification Q, this produces scoring matrices gtE and gtQ, respectively.
PSI-BLAST uses BLAST-PGP with a 32-fold scale-up of BLOSUM62 to enhance sensitivity during the first round of comparisons. We have used the same scaling factor to augment the BLOSUM62 matrix before adjusting for amino acid composition deviation. This generates the gtE32 and gtQ32 matrices; gap costs are also scaled up (Table 2). The gtE32 and gtQ32 matrix modifications were implemented for FASTA only.
Implementation.
As a test of the effects of composition-based matrix modifications, we carried out searches on two sets of proteins. The first was a test to find homologs of low-complexity yeast cell wall proteins in a combined database of the yeast proteome and three complete sets of randomized yeast ORF pseudosequences. Randomizations of the sequences were global (the entire sequence randomized for each ORF) or local. For local randomizations, the sequence was randomized within contiguous windows of 12 residues. This window length corresponds to that of the SEG filter and maintains the local entropy of the sequences. For searches with cell wall proteins as queries, HSPs with authentic yeast ORFs were counted as “true” hits, and HSPs with randomized sequences were counted as “false.” This designation favors nondiscriminating matrices such as BLOSUM62, because some nonhomologous sequences were counted as “true” hits for the tests shown in Fig. 2. Inspection of alignments (Fig. 1) and comparison of annotations (see Table 4) showed that these nonhomologous “true” hits were not reported in searches with gtQ and E matrices. The other search set was the Aravind data set, which contains 103 domain-specific query sequences and a total of 1,005 true positives in the yeast proteome, curated as described previously by Schaffer et al. (31). We used those definitions of “true” and “false” hits.
FIG. 2.

Sensitivity curves for BLAST (B) and FASTA (F) with different scoring matrices (Mat). Matrix B is the traditional BLOSUM62 matrix; E, Q, gtE, gtQ, gtE32, and gtQ32 are described in the text. (A) S. cerevisiae cell wall proteins searched against the yeast proteome and three locally randomized copies of the yeast proteome. Search output was binned into groups of hits by e value (10−130, 10−8, 5 × 10−8, 10−7, 10−5, 5 × 10−2, 10−1, 5 × 10−1, 1, 5, and 10). True positives (yeast ORFs) and false positives (locally randomized yeast pseudoprotein sequences) were counted and plotted for each group of hits. “Gap” is the gap cost (cost to open and cost to extend). For PGP, gtE32, and gtQ32 modifications, the listed gap values were multiplied by 32 before alignments were evaluated. The designated gap penalties gave maximal discrimination for each tested matrix.
TABLE 4.
Comparison of transitive closure sets
| Search | Matrix | No. of rounds to close | Hits
|
GO cell wall
|
No. of curated proteins
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Type | No. | No. | % of hits | Cell wall | Wall biogenesis | Not cell wall | Unknown or ambiguous | |||
| BLAST | B | >5 | Total | 863 | 66 | 8 | NDa | ND | ND | ND |
| BF | >8 | Total | 784 | 60 | 8 | ND | ND | ND | ND | |
| PGP | 7 | Total | 192 | 28 | 15 | 41 | 6 | 122 | 23 | |
| Uniqueb | 135 | 4 | 3 | 0 | 5 | 122 | 8 | |||
| E | 3 | Total | 48 | 18 | 38 | 35 | 0 | 0 | 13 | |
| Unique | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |||
| gtQ | 4 | Total | 64 | 26 | 41 | 46 | 2 | 2 | 14 | |
| Unique | 13 | 5 | 38 | 8 | 2 | 2 | 1 | |||
| FASTA | B | 13 | Total | 397 | 51 | 13 | ND | ND | ND | ND |
| BF | 10 | Total | 158 | 43 | 27 | 61 | 6 | 60 | 31 | |
| Unique | 16 | 2 | 12 | 2 | 0 | 14 | 0 | |||
| E | 3 | Total | 48 | 18 | 38 | 35 | 0 | 0 | 13 | |
| Unique | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |||
| gtQ | 3 | Total | 51 | 21 | 41 | 38 | 0 | 0 | 13 | |
| Unique | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |||
| gtQ32 | 3 | Total | 51 | 21 | 41 | 38 | 0 | 0 | 13 | |
| Unique | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |||
ND, not determined.
Hits were classified as unique if they were identified only in the specified search. For example, transitive closure with BLAST-PGP identified 192 homologous ORFs, 135 of which were identified only in the BLAST-PGP search, and 57 were also identified in at least one other search.
To test the sensitivity and selectivity of pairwise search algorithms for high-complexity sequences with the modified scoring matrices, stand-alone versions of BLAST (version 2.2.2) and FASTA (version 3) were used. As recommended previously (2, 16, 27), gap costs of 9 to 13 were used with BLAST, lower costs, 5 to 9, were used for FASTA searches, and the gap extension cost was set at 1. Each search used one of the scoring matrices (described above) based upon the amino acid frequencies in the individual query sequence. The notations that describe each type of search with each type of matrix are summarized in Table 2.
All BLAST searches were implemented using the command-line executable “blastall” with the BLAST-x matrices or the command-line executable “blastpgp” with the BLAST-PGP matrix and the composition-based statistics flag on “(-t T).” Both command-line executables produce gapped pairwise alignments, but BLASTPGP uses composition-based statistics to assess significance and can be used to generate PSSM from first-round hits. The PSSM is used to score the second round of searches in PSI-BLAST. To preserve comparability, blastpgp searches were relegated to one round “(-j 1).” The FASTA searches were conducted with the command line-executable “fasta34.” Command line options were default options unless specified otherwise. All matrix modification searches were PERL and BASH shell scripts executed on a Sun Microsystems SunBlade100 workstation running Debian GNU Linux. Searches were performed without SEG filtering unless specifically designated and were repeated for several different gap values.
Transitive closure tests.
We tested whether the similarity sets were closed for yeast cell wall proteins. These tests compared output from BLAST-PGP, FASTA-B, and the four matrix modifications that are sufficiently sensitive and discriminating to support searches with low-complexity sequences: E, gtQ, gtQ32, and gtE32. These searches used the 10 yeast cell wall proteins as query sequences to search the yeast protein database (retrieved from the NCBI). Searches were done with the gap costs shown in Fig. 2. HSPs with e values less than the specified cutoff for distinct new proteins in each round became the query set for the next round, still against the same database. This process continued until no new, distinct proteins with e values below the specified cutoff were obtained (14, 35).
Comparisons of the transitive closure sets were performed using a Java web application and other Java codes. The WAR file for the web application is available from the authors. The glycosylphosphatidylinositol (GPI) protein set was taken from data described previously (8, 11). Using the Gene Ontology (GO) database terms “cell wall (sensu fungi)” and “cell wall organization and biogenesis,” the Gene Ontology sets were obtained from the Saccharomyces Genome Database website (http://www.yeastgenome.org/). We curated the “cell wall protein,” “non-cell wall protein,” “wall biogenesis,” and “unknown or ambiguous” classifications shown in Table 4.
Availability.
The PERL and shell scripts, customized databases, and supplemental sensitivity curves described in this paper can be obtained from the authors.
RESULTS
General approach.
Two types of score adjustments, with several variations of each, were designed and evaluated in tests using a variety of query sequences and databases summarized in Table 1. The score adjustments and queries are described below.
Score changes for frequently occurring amino acids.
The E and Q matrix modification methods reduce the alignment score, Sij, for aligned residues i and j for amino acids occurring at a high frequency in a query sequence but preserve the net negative value for the matrix that is required for accurate statistical analyses of the alignments (3). Each modification method yields a different scoring matrix for each query sequence. Each modification method and its variants compensate in different ways for the deviation from the standard Robinson and Robinson frequencies used to derive the gapped BLAST statistical parameters for BLOSUM62, as summarized in Materials and Methods (3, 18). The E method keeps the expected score of the matrix constant, while the Q method keeps the target frequencies, Qij, constant, where Qij is the expected frequency that a residue, i, in one sequence is replaced by j in randomly aligned sequences (18). These frequencies are determined in a set of standard alignments using BLOSUM62.
All matrix modifications are summarized in Table 2 and are described in detail in Materials and Methods. Throughout, we append suffixes to indicate which modifications were applied to a search method (Table 2). For example, A “BLAST-BF” search indicates that unmodified BLOSUM62 (“B”) was used with SEG filtering (“F”). FASTA-gtE32 indicates that we carried out a FASTA search with three modifications to the BLOSUM62 matrix: E, gt, and 32. We adopt the BLAST filtering criterion as a working definition for a low-complexity sequence, that is, one with Shannon entropy less than 2.2 over a window of at least 12 amino acid residues (5, 34).
Query sets.
The cell wall query set for most searches with low-complexity queries was a group of 10 cell wall GPI class mannoproteins (8, 11, 20): Cwp2p, Sag1p, Ssr1p, Tip1p, Sed1p, Tir1p, Flo11p, Aga1p, Flo1p, and Fig2p, with lengths of 92, 650, 238, 210, 338, 254, 1,367, 725, 1,537, and 1,609 residues, respectively (8, 11). These sequences are representative of GPI-anchored fungal cell wall proteins and include six unique genes, two members of the FLO gene family, and two members of the TIR/TIP family. These and other cell wall proteins are mosaics of high-complexity and low-complexity segments (8, 10, 11, 20).
Tests with high-complexity queries used a standard data set of 103 yeast signal transduction proteins as queries in searches of the S. cerevisiae proteome and three copies of the proteome with the ORF sequences randomized (31).
Effects of matrix modifications on searches with low-complexity query sequences.
The problem of low-complexity corruption is illustrated in Fig. 1. BLOSUM62-based BLAST or FASTA searches with yeast cell wall proteins as queries identified homologs with highly similar sequences (Fig. 1A) but also returned HSPs with randomized sequences and nonhomologous proteins, even when score statistics were adjusted by PGP or when low-complexity regions were masked with SEG (Fig. 1B to D). These alignments were based on high frequencies of matched Ser and Thr residues and therefore identified many nonhomologous sequences as highly similar, a known consequence of low-complexity corruption (31, 34). In the BLAST-B search, the highest-scoring match to Muc1p was a random pseudoprotein segment derived from Dan4p. Similarly, the three highest-scoring matches to Fig2p (e < 10−62) were randomized versions of Muc1p. Like the BLAST- BLOSUM62 searches, BLAST-BF and BLAST-PGP, which uses composition-based statistical analyses with BLOSUM62, gave matches in which >80% of the identities were Ser or Thr (Fig. 1C and D). Other residues were seldom aligned. PGP also identified a large number of best hits with similar compositions but unlikely homology: among the highest-scoring matches for Aga1p was Snt1p, a histone deacetylase subunit, and for Muc1p, the third highest-scoring match was to the Sec31p subunit of the endoplasmic reticulum protein translocation pore. These proteins are unlikely to be homologous on the basis of functional analogy, cellular localization, or alignment of conserved sequence motifs. In addition, BLAST-B, PGP, and BF searches identified many randomized sequences as HSPs with an e value of <10−3.
Alignments were greatly improved after matrix scores were adjusted to reflect the composition of the query sequences. Of the matrix variants listed in Table 2, the E and gtQ variations with BLAST or FASTA, as well as gtQ32 with FASTA, gave more specific alignments. (Our website, http://diverge.hunter.cuny.edu:8080/modmat, has automated, composition-based matrix modifications and search capability for any query sequence.) E matrices were highly specific; they required regions of extensive identity to achieve HSPs with significant e values. The Muc1p/Bsc1p homology (Fig. 1A) was the only significant hit for any of the three query proteins illustrated in Fig. 1. gtQ matrices showed more high-quality HSPs, a result of acquisition of significant scores over even relatively short but highly similar segments (Fig. 1E). All of the significant HSPs were to proteins that are also localized to cell walls. Note that with gtQ, the best match for Aga1p was in a segment that was aligned with a randomized Muc1p pseudoprotein in the best match of the BLOSUM62-based search (Fig. 1B).
Thus, the alignments showed that searches with BLOSUM62 matrices were subject to low-complexity corruption, even with PGP statistics or SEG filtering. These findings were confirmed in the structural comparisons and the sensitivity and transitive closure tests described below. In contrast, gtQ matrices were highly sensitive, reaching significant e values in relatively short segments of both low-complexity and high-complexity compositions. The E matrices were highly discriminatory and identified only long HSPs with a high likelihood of homology.
Structural correlations and matrix modification.
Alignments are especially important in structural searches. There are few structures known for low-complexity proteins, and indeed, structures for low-complexity sequences are severely underrepresented in the Protein Databank (21). Therefore, apparent matches to nonhomologous sequences may be used mistakenly as the basis for alignment and modeling. Use of gtQ and E matrices can assure better alignments and more accurate structural predictions.
If aligned regions are homologous, they should have similar secondary structures (15). We tested the composition-modified matrices as predictors of concordant secondary structure predictions for pairs of HSPs with e values of ≤10−3. The cell wall query proteins were used to search the S. cerevisiae genome database. Each aligned sequence segment was used as the input for GOR IV, a secondary structure predictor that does not depend on BLOSUM62-based alignment to homologous sequences (13). The GOR IV secondary structure predictions of α-helix or β-sheet were compared (Table 3). The gtQ matrices gave the highest degree of concordance, over 80%, followed by E and B matrices. However, the concordance values with PGP had high variance due to the inclusion of nonhomologous HSPs (Fig. 1). We repeated the test for the subset of HSPs with 10−5 ≥ e ≥ 10−30, values for the alignments most likely to be relevant for such predictions. For these HSPs, E and gtQ matrices outperformed BLOSUM62-based matrices. Again, PGP searches had poor concordance and the greatest standard deviation (not shown), indicating variation in the quality of the matches, as expected in situations where HSPs include nonhomologous matches. Thus, the use of modified matrices significantly improved the reliability of secondary structure predictions.
TABLE 3.
Concordance of GOR IV secondary structure predictions for cell wall-related aligned sequencesa
| Matrix | 10−3 ≥ e
|
10−5 ≥ e ≥ 10−30
|
||
|---|---|---|---|---|
| No. of aligned residuesb | Concordance H + E (%)c | No. of aligned residuesb | Concordance H + E (%)c | |
| E | 2,845 | 80 | 2,763 | 81 |
| gtQ | 3,010 | 82 | 650 | 65 |
| B | 3,937 | 74 | 1,812 | 51 |
| PGP | 6,245 | 63 | 243 | 58 |
The cell wall query set was used for BLAST searches of the S. cerevisiae genome. GOR IV was used to predict the conformation of all sequences in all HSPs within the designated range of e values. Each residue predicted to be in α-helix (H) or β-sheet (E) conformation was compared to its aligned partner and scored as concordant if the conformation predictions were identical.
Number of aligned residues predicted to be in α-helix (H) or β-sheet (E) conformation in all HSPs with the designated e values.
Percentage of instances where both members of an aligned pair of residues are predicted to be in the same α-helical or β-sheet conformation.
Sensitivity and discrimination.
Sensitivity curves are a standard way to illustrate the effectiveness of search strategies (31). These graphs (Fig. 2) illustrate sensitivity (number of homologs identified as HSPs) as horizontal displacement and discrimination (number of false hits identified as HSPs) as vertical displacement. Thus, good performance is indicated by a curve that has a long horizontal component with minimal verticality apparent only at the right-hand end of the curve. Previous work has defined false hits either as randomized sequences of composition similar to that of the true hits (3, 31) or as proteins known to be nonhomologous (31). To test the composition-modified matrices for discrimination against nonhomologous, low-complexity sequences similar to the query sequences, we searched the cell wall protein query set against the S. cerevisiae genome combined with the locally randomized pseudoprotein sequences described in Materials and Methods.
Figure 2 shows sensitivity plots for the cell wall protein query set against the S. cerevisiae proteome and three locally randomized copies. All tested matrix modification methods performed better than BLAST with B or BF and FASTA-B, which were unable to discriminate between authentic and randomized sequences. BLAST-PGP, which uses composition-based statistics with BLOSUM62, found 25 true hits (including the 10 query sequences themselves) at e values below that of the first false hit. Among the modified matrix searches, BLAST-E was highly discriminatory (it found very few false hits even with large e values). The gtQ matrices showed by far the best sensitivity (105 true hits with lower e values than the best-scoring false hit). Thus, FASTA-gtQ32 identified the 10 query sequences and 95 paralogs of the query proteins at e values that excluded false hits, whereas BLAST-PGP identified only 15 paralogs.
Transitive closure tests.
We used transitive closure as an empirical test of the usefulness of the composition-based matrix modifications. The 10 cell wall proteins were used as query sequences in BLAST and FASTA searches. Each query was used with different matrices derived from its own composition. The ORFs corresponding to all hits with e values of <10−3 were used as the query sequences in the next round of searches, again with scoring matrices derived from each specific composition. This procedure was repeated until no new HSPs were identified. If a search method discriminates between similar and nonsimilar sequences, transitive closure should terminate after a relatively small set of sequences is identified. On the other hand, low-complexity corruption or other artifacts will result in frequent identification of nonhomologous proteins with low e values. The consequences will include a larger number of search rounds to achieve closure, and the significant “hits” will potentially include much of the proteome.
As expected, BLAST-B failed to achieve closure on the low-complexity query sequences, even with a cutoff e value of ≤10−9. With a standard cutoff e value of ≤10−3, there were many new hits in each round, with a total of 863 sequences after five rounds (15% of the yeast proteome) (Fig. 3 and Table 4). BLAST-BF also failed to close. The other methods achieved closure in 3 to 10 rounds (Table 4). There were 192 different ORFs identified in one or more of the searches with composition-modified matrices. Of these, 47 ORFs were identified in all searches, with 1 more ORF identified by five of the six modified matrix methods. Thus, there was a core of 48 hits that were most similar to the query sequences.
FIG. 3.

Transitive closure trial of BLAST (B)-PGP, BLAST-E, BLAST-gtQ, FASTA (F)-B, FASTA-E, FASTA-gtQ, FASTA-gtE32, and FASTA-gtQ32. Iterative searches of the yeast cell wall protein query set (10 proteins) against the Saccharomyces Genome Database were run as described in the text. The cutoff e value was 10−3 for all searches. The hits marked “query” are the identities to the query sequences, which have the smallest e values in the first-round searches. The NCBI gi accession numbers for the identified ORFs are shown in Table S3 in the supplemental material.
BLAST-PGP was the most sensitive method that closed, but it did not discriminate against nonhomologous sequences. The BLAST-PGP test identified 135 hits not found in any other search. Most of these extra hits were due to low-complexity corruption, similar to that seen in Fig. 1 and 2. The alignments were rich in pairings of nonhomologous Ser and Thr, and there were multiple different alignments in the same segments of the protein pairs with the same score. Such multiple equivalent HSPs are typical of low-complexity corruption. Furthermore, the vast majority of these hits were for proteins that are unlikely to be related to cell wall proteins (Table 4).
We reasoned that the most likely homologs of the query sequences would be other cell wall and cell surface proteins, since their composition and domain structures are similar to each other and substantially different from those of globular proteins (20). Therefore, we functionally classified the hits identified in the transitive closure tests. The 343 ORFs identified in any modified matrix search or BLAST-PGP or FASTA-BF were labeled cell wall or not cell wall, either in accordance with the GO database or as curated by the authors. BLAST-PGP and FASTA-BF searches included many non-cell wall proteins among the significant hits (12). In contrast, searches with E and gtQ composition-modified matrices identified a highly similar set of ORFs, almost all of which were classified as cell wall proteins in either BLAST or FASTA. A complete list of hits for BLAST-PGP and composition-modified matrix searches is shown in Table S4 in the supplemental material.
Effects of matrix modifications on searches with high-complexity query sequences.
To assess the effects of composition-based matrix modification on searches with high-complexity sequences, we also tested our methods in searches with globular (high-complexity) proteins as queries. The Aravind data set is a set of curated signal transduction proteins within the S. cerevisiae proteome (31). A total of 103 of these proteins were used as queries in BLAST and FASTA searches, counting the number of alignments with curated “true” and “false” homologs within the previously established criterion that the e value was ≤10−2 (Table 5) (31). As previously reported, BLAST with BLOSUM62 was the most sensitive method, returning 46% of the known homologs at this e value (31). Among the composition-modified matrices, searches with gtQ performed well, with 82 to 86% of BLOSUM62's sensitivity in BLAST and 75% sensitivity in FASTA searches. B and gtQ had similar levels of discrimination against false hits. Again, the E matrices were highly discriminatory and gave no false hits, but the searches were less sensitive. Thus, composition-modified matrices provided moderately lower sensitivity but similar (gtQ) or increased (E) discrimination in searches with sequences whose composition is near the Robinson and Robinson average.
TABLE 5.
Results of modified-matrix searches on the Aravind data set
| Method | No. of true homologs (e ≤ 10−2) | No. of false hits (e ≤ 10−2) | % of BLAST-B sensitivity |
|---|---|---|---|
| BLAST-Ba | 460 | 3 | 100 |
| BLAST-PGP | 434 | 2 | 94 |
| BLAST-BF | 436 | 2 | 95 |
| BLAST-E | 231 | 0 | 50 |
| BLAST-Q | 348 | 1 | 76 |
| BLAST-gtE | 401 | 1 | 87 |
| BLAST-gtQ | 390 | 3 | 85 |
| FASTA-B | 388 | 0 | 84 |
| FASTA-BF | 398 | 0 | 86 |
| FASTA-E | 242 | 0 | 53 |
| FASTA-Q | 223 | 80 | 48 |
| FASTA-gtE | 339 | 1 | 74 |
| FASTA-gtE32 | 318 | 0 | 69 |
| FASTA-gtQ | 319 | 0 | 69 |
| FASTA-gtQ32 | 345 | 1 | 75 |
BLAST-B identified 45.8% of the total “true” homologs.
Score distributions.
The reliability of e values depends on the statistical distribution of the alignment scores, which must conform to the Gumbel extreme value distribution (18). We tested this conformance for BLOSUM62 and the gtQ and E modifications. Each test used a 1,000-residue segment from Flo1p, a randomized sequence with the same composition as the yeast cell wall query data set, and a random sequence of the same composition as the Robinson and Robinson high-complexity data set as queries. As in previous tests of searches and matrices (3), each query was tested for Smith-Waterman alignments (27) against four databases, each with a size of 104: high-complexity sequences randomized globally, high-complexity sequences locally randomized, low-complexity sequences randomized globally, and low-complexity sequences randomized locally. For each search, the 104 alignment scores were binned and compared to expected scores in the extreme value distribution with Pearson's χ2 test. The distributions of alignment scores generated by the composition-modified matrices, as they should be, were similar to the extreme value distribution with a P value of <0.005. However, in BLOSUM62-based searches for low-complexity sequences in both low-complexity databases, the P value was >0.03 to 0.07. Thus, BLOSUM62 conformed less well than the modified matrices to an extreme value distribution. The detailed data appear in Table S1 in the supplemental material.
The score distributions were used to estimate the statistical parameters κ and λ of the distributions as well (3). For FASTA searches, assuming conformance with the extreme value distribution, κ and λ are calculated and e values are derived from the distribution for each search (26). In contrast, standard BLAST assumes values for these parameters that were derived from empirical estimates in gapped searches of high-complexity sequences. It is noteworthy that for the BLOSUM62-based searches of cell wall queries against the randomized cell wall pseudosequences, the value of κ was as much as 106 times greater than the standard value of 0.0243. This difference is probably the major source of the inaccuracy of e values and subsequent low-complexity corruption in low-complexity searches using BLOSUM62. In contrast, the composition-modified matrices generated score distributions with κ values that differed from the standard by less than fourfold. The λ values were all close to the BLAST-assumed value of 0.24, again with the exceptions of the BLOSUM62-based cell wall searches against the low-complexity and low-complexity pseudosequence databases (see Table S1 in the supplemental material).
Another test for conformance is probability plots of the inverse Poisson distribution P values for alignment scores. Although such plots are often used to compare scores for two samplings of a population, they can also be used to illustrate the number of scores at each probability in two distributions (9). The plots in Fig. S2 show the cumulative fraction of scores above given index scores for comparisons of the E and gtQ matrices compared to the distribution in the BLAST-PGP search of the high-complexity query and database (31). The plots are linear, as expected for comparable score distributions.
e values for false hits.
In an extreme value distribution, the mean best e value of false hits should be 1 (26). We therefore calculated this quantity for each matrix modification in both BLAST and FASTA searches using high- and low-complexity queries. In searches with high-complexity queries, all matrices had mean first false hit scores between 0.41 and 11.7 (see Table S2 in the supplemental material). Again, E matrices were the most discriminatory and had the largest e values for false hits. In contrast, in searches with low-complexity queries, the composition-modified matrices far outperformed BLOSUM62. For BLOSUM62, even with SEG filtering or composition-modified statistics, the mean e values for the first false hits were between 10−3 and 10−46. Furthermore, the best-scoring false hit in a BLOSUM62-based search had an e value of 10−110. In contrast, the modified matrices generated mean e values of between 10−2 and 102. Thus, in high-complexity searches, the E and gtQ modifications produced e values close to 1 for the first false hit, as expected. For low-complexity sequences, the E and gtQ modifications produced e values much closer to the expected value of 1 than in searches with BLOSUM62.
Computational efficiency.
In BLAST, the major computational burden is the time needed to extend the two- to four-letter words from the query sequence that find similarity to sequences in the database (4, 5). We therefore measured the computation times in BLAST and FASTA. BLAST-E and BLAST-gtQ ran faster than BLAST-B and BLAST-PGP for low-complexity sequences for both the S. cerevisiae genome database and the database that consisted of the genome with randomized sequences (Table 6). The maximum difference was about a 25-fold speed-up for the BLAST-E search with low-complexity queries. For high-complexity sequences, E matrices were slightly more efficient and gtQ matrices were 40% slower than standard BLAST methods. In contrast, composition-based matrix modifications had little effect on the scan times for searches by FASTA (data not shown).
TABLE 6.
Efficiency of modified-matrix BLAST searches
| Query set | Matrix | Computation time (s)
|
|
|---|---|---|---|
| S. cerevisiae genome | S. cerevisiae genome with random sequences | ||
| Cell wall proteins | E | 3 | 9 |
| gtQ | 8 | 26 | |
| BLOSUM-PGP | 60 | 230 | |
| BLOSUM62 | 55 | 213 | |
| Aravind | E | 15 | 43 |
| gtQ | 24 | 80 | |
| BLOSUM-PGP | 18 | 56 | |
| BLOSUM62 | 17 | 55 | |
DISCUSSION
There is an acute need for bioinformatic tools that align and compare low-complexity sequences. Most available programs merely identify or mask such segments (1, 19, 33, 34). We have shown that strategies that base alignment scores on the frequency of specific amino acids in the query sequence greatly improve the reliability and usefulness of BLAST and FASTA searches for low-complexity query sequences. These E and gtQ matrix modification methods decreased the scores for common residues and were highly discriminatory against nonhomologous sequences. The searches using these matrices identified a consistent set of paralogs of known yeast wall proteins (Table 4). These proteins share homologous sequence regions and motifs that have not been identified in BLOSUM62-based searches (J. Coronado et al., unpublished data). Searches with composition-modified matrices also improved structural concordance in aligned sequences (Table 3).
The modified matrices yielded alignment scores in BLAST and FASTA that conformed to the extreme value distribution (see Table S1 and Fig. S2 in the supplemental material) and generated e values more accurately than BLOSUM62-based searches (see Table S2 in the supplemental material) for low-complexity sequences. In searches with high-complexity queries, the distributions also conformed to the expected extreme value distribution, but the increased discriminatory power of the modified matrices decreased sensitivity somewhat (Table 5). This finding is consistent with a previous report that BLOSUM62 is the most sensitive matrix for searches with high-complexity sequences (17).
Transitive closure with modified-matrix searches identified a consistent set of yeast proteins.
The transitive closure tests demonstrated that searches with E or gtQ modified matrices reliably identified apparent homologs of cell wall query sequences (Table 4, GO annotation and manually curated sets). In contrast, BLOSUM62-based searches with standard statistics did not close and hit a large fraction of the yeast proteome. The transitive closure test closed with BLAST-PGP, but the majority of the hits with e values of <10−3 were not cell wall-related proteins (Table 4). Indeed, inspection revealed that most of them were low-complexity sequences in mobile elements or RNA-processing enzymes.
The BLAST and FASTA transitive closure tests with the three best-performing composition-based matrices (BLAST with E or gtQ and FASTA with E, gtQ, or gtQ32) identified 61 apparent homologs of the yeast cell wall proteins with alignment e values smaller than 10−3. Of those apparent homologs, 48 were retrieved by all five of these modified-matrix searches; FASTA-E retrieved only these 48 ORFs. One additional ORF, Ylr110c, was retrieved by the four other modified-matrix searches. Nine more ORFs were identified by BLAST-gtQ, FASTA-gtQ, and FASTA-gtQ32. Based on inspection of the significant alignments and resistance to low-complexity corruption, the E and gtQ modifications used in BLAST, or used with high gap costs in FASTA, define a consistent set of potentially homologous low-complexity proteins efficiently and accurately (Table 4; see Table S4 in the supplemental material).
Other matrix modifications.
Matrices modified for composition of both query and target sequences might further increase sensitivity but at the cost of calculating a new matrix for each HSP. An analysis of reciprocal hits in the transitive closure test shows that query-based modifications were sufficient to find all known paralogous pairs (see the supplemental material).
In a different approach, Yu and colleagues (6, 36, 37) previously proposed composition-based modifications of BLOSUM scoring matrices to do alignments of low-complexity sequences without SEG filtering. The scoring matrices described previously (37) are corrected by keeping the total entropy of each matrix constant, a strategy to maximize sensitivity for queries of unusual composition. Thus, these modifications would apply to a different aspect of the low-complexity search and alignment problem. The consequences of such matrices on a large scale have not yet been published.
Structural consequences.
Disordered regions of proteins often include low-complexity sequences. DISORDER, a scoring matrix specific for disordered regions of structurally well-characterized proteins, improves scores for homologous protein pairs with 40 to 50% identity (30). The discrimination ability is similar to that of BLOSUM62, and the increase in sensitivity appears to be twofold. In contrast, the E and gtQ matrices increased discrimination for any query sequence, and gtQ showed a greater sensitivity. The result was better agreement in predicted secondary structures of the aligned segments.
Summary.
We have presented several ways to normalize the alignment scores and statistical parameters for individual query sequences (Table 2). Of these, the E and gtQ modifications support sensitive, discriminating, and accurate search and scoring statistics for proteins or segments whose amino acid composition is far outside the Robinson and Robinson amino acid frequencies originally used to estimate the statistical parameters of λ and κ.
The scoring matrix modifications E and gtQ rendered SEG filtering unnecessary and generated alignment scores that conformed to the extreme value distribution, which BLOSUM62-based searches could not do for these sequences of unusual composition. The composition-based matrix modifications also generated score distributions with statistical parameters much closer to those assumed in gapped BLAST statistics, so the resultant e values were more accurate than those from BLOSUM62 and at least as accurate as composition-based statistics in BLAST-PGP. Therefore, BLAST or FASTA with the E or gtQ modified matrices showed great resistance to low-complexity corruption and reliably identified apparent homologs of these important, low-complexity sequences without masking out the low-complexity segments. Furthermore, for these sequences, the efficiency of BLAST was improved, and the efficiency of FASTA was not significantly changed. For query sequences containing low-complexity regions, BLAST-gtQ and FASTA-gtQ32 were the most sensitive search methods and had good discrimination against nonhomologous sequences with similar amino acid compositions. Matrix modification E with either BLAST or FASTA searches had maximal discrimination against nonhomologous sequences but was somewhat less sensitive. The results presented here demonstrate that composition-based matrix modifications discriminate against nonhomologous alignments and therefore make accurate comparative studies of low-complexity sequences possible. This accuracy is necessary for phylogenetics and for structural comparisons.
Another benefit of these matrices will be an analogous improvement in the accuracy of genomic annotations, which are often based on functional analogies for homologous sequences. For instance, transitive closure identified a set of 48 sequences in S. cerevisiae that are similar to the cell wall protein queries. Searches through fungal genomes have revealed that apparent homologs of these proteins are present in other ascomycetes and basidiomycetes (Coronado et al., unpublished). These homologies in turn imply commonalities in cell wall structure and function for fungi whose walls are not as well characterized as those of S. cerevisiae.
Supplementary Material
Acknowledgments
This work was funded by the NIH/NCRR/RCMI program (grant RR 03037), the NIH/MBRS-SCORE program (grant S06 GM 60654), and the Howard Hughes Medical Institute (grant 52002679). J.C. is a Fellow of the NSF-MAGNET and RCMI programs.
Footnotes
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1.Alba, M. M., R. A. Laskowski, and J. M. Hancock. 2002. Detecting cryptically simple protein sequences using the SIMPLE algorithm. Bioinformatics 18:672-678. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F. 1998. Generalized affine gap costs for protein sequence alignment. Proteins 32:88-96. [PubMed] [Google Scholar]
- 3.Altschul, S. F., and W. Gish. 1996. Local alignment statistics. Methods Enzymol. 266:460-480. [DOI] [PubMed] [Google Scholar]
- 4.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 5.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altschul, S. F., J. C. Wootton, E. M. Gertz, R. Agarwala, A. Morgulis, A. A. Schaffer, and Y. K. Yu. 2005. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 272:5101-5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berger, B., D. B. Wilson, E. Wolf, T. Tonchev, M. Milla, and P. S. Kim. 1995. Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. USA 92:8259-8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caro, L. H., H. Tettelin, J. H. Vossen, A. F. Ram, H. van den Ende, and F. M. Klis. 1997. In silico identification of glycosyl-phosphatidylinositol-anchored plasma-membrane and cell wall proteins of Saccharomyces cerevisiae. Yeast 13:1477-1489. [DOI] [PubMed] [Google Scholar]
- 9.Chambers, J., W. Cleveland, B. Kleiner, and P. Tukey. 1983. Graphical methods of data analysis. Duxbury, Pacific Grove, Cailf.
- 10.Colussi, P. A., and P. Orlean. 1997. The essential Schizosaccharomyces pombe gpil+ gene complements a bakers' yeast GPI anchoring mutant and is required for efficient cell separation. Yeast 13:139-150. [DOI] [PubMed] [Google Scholar]
- 11.De Groot, P. W., K. J. Hellingwerf, and F. M. Klis. 2003. Genome-wide identification of fungal GPI proteins. Yeast 20:781-796. [DOI] [PubMed] [Google Scholar]
- 12.Dwight, S. S., M. A. Harris, K. Dolinski, C. A. Ball, G. Binkley, K. R. Christie, D. G. Fisk, L. Issel-Tarver, M. Schroeder, G. Sherlock, A. Sethuraman, S. Weng, D. Botstein, and J. M. Cherry. 2002. Saccharomyces Genome Database (SGD) provides secondary gene annotation using the Gene Ontology (GO). Nucleic Acids Res. 30:69-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garnier, J., J. F. Gibrat, and B. Robson. 1996. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 266:540-553. [DOI] [PubMed] [Google Scholar]
- 14.Gerstein, M. 1998. Measurement of the effectiveness of transitive sequence comparison, through a third ‘intermediate’ sequence. Bioinformatics 14:707-714. [DOI] [PubMed] [Google Scholar]
- 15.Grigorescu, A., M. H. Chen, H. Zhao, P. C. Kahn, and P. N. Lipke. 2000. A CD2-based model of yeast alpha-agglutinin elucidates solution properties and binding characteristics. IUBMB Life 50:105-113. [DOI] [PubMed] [Google Scholar]
- 16.Henikoff, S., and J. G. Henikoff. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 89:10915-10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henikoff, S., and J. G. Henikoff. 1993. Performance evaluation of amino acid substitution matrices. Proteins 17:49-61. [DOI] [PubMed] [Google Scholar]
- 18.Karlin, S., and S. F. Altschul. 1990. Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc. Natl. Acad. Sci. USA 87:2264-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li, X., P. Romero, M. Rani, A. K. Dunker, and Z. Obradovic. 1999. Predicting protein disorder for N-, C-, and internal regions. Genome Inform. Ser. Workshop Genome Inform. 10:30-40. [PubMed] [Google Scholar]
- 20.Lipke, P. N., and J. Kurjan. 1992. Sexual agglutination in budding yeasts: structure, function, and regulation of adhesion glycoproteins. Microbiol. Rev. 56:180-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, J., H. Tan, and B. Rost. 2002. Loopy proteins appear conserved in evolution. J. Mol. Biol. 322:53-64. [DOI] [PubMed] [Google Scholar]
- 22.Lupas, A. 1996. Prediction and analysis of coiled-coil structures. Methods Enzymol. 266:513-525. [DOI] [PubMed] [Google Scholar]
- 23.Muller, T., S. Rahmann, and M. Rehmsmeier. 2001. Non-symmetric score matrices and the detection of homologous transmembrane proteins. Bioinformatics 17(Suppl. 1):S182-S189. [DOI] [PubMed] [Google Scholar]
- 24.Nandi, T., D. Dash, R. Ghai, C. B-Rao, K. Kannan, S. K. Brahmachari, C. Ramakrishnan, and S. Ramachandran. 2003. A novel complexity measure for comparative analysis of protein sequences from complete genomes. J. Biomol. Struct. Dyn. 20:657-668. [DOI] [PubMed] [Google Scholar]
- 25.Ng, P. C., J. G. Henikoff, and S. Henikoff. 2000. PHAT: a transmembrane-specific substitution matrix. Predicted hydrophobic and transmembrane. Bioinformatics 16:760-766. [DOI] [PubMed] [Google Scholar]
- 26.Pearson, W. R. 1998. Empirical statistical estimates for sequence similarity searches. J. Mol. Biol. 276:71-84. [DOI] [PubMed] [Google Scholar]
- 27.Pearson, W. R. 2000. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol. Biol. 132:185-219. [DOI] [PubMed] [Google Scholar]
- 28.Popolo, L., and M. Vai. 1999. The Gas1 glycoprotein, a putative wall polymer cross-linker. Biochim. Biophys. Acta 1426:385-400. [DOI] [PubMed] [Google Scholar]
- 29.Promponas, V. J., A. J. Enright, S. Tsoka, D. P. Kreil, C. Leroy, S. Hamodrakas, C. Sander, and C. A. Ouzounis. 2000. CAST: an iterative algorithm for the complexity analysis of sequence tracts. Complexity analysis of sequence tracts. Bioinformatics 16:915-922. [DOI] [PubMed] [Google Scholar]
- 30.Radivojac, P., Z. Obradovic, C. J. Brown, and A. K. Dunker. 2002. Improving sequence alignments for intrinsically disordered proteins. Pac. Symp. Biocomput. 2002:589-600. [PubMed] [Google Scholar]
- 31.Schaffer, A. A., L. Aravind, T. L. Madden, S. Shavirin, J. L. Spouge, Y. I. Wolf, E. V. Koonin, and S. F. Altschul. 2001. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 29:2994-3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sim, K. L., and T. P. Creamer. 2002. Abundance and distributions of eukaryote protein simple sequences. Mol. Cell. Proteomics 1:983-995. [DOI] [PubMed] [Google Scholar]
- 33.Wise, M. J. 2001. 0j.py: a software tool for low complexity proteins and protein domains. Bioinformatics 17(Suppl. 1):S288-S295. [DOI] [PubMed] [Google Scholar]
- 34.Wootton, J. C., and S. Federhen. 1996. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 266:554-571. [DOI] [PubMed] [Google Scholar]
- 35.Yona, G., N. Linial, and M. Linial. 2000. ProtoMap: automatic classification of protein sequences and hierarchy of protein families. Nucleic Acids Res. 28:49-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu, Y. K., and S. F. Altschul. 2005. The construction of amino acid substitution matrices for the comparison of proteins with non-standard compositions. Bioinformatics 21:902-911. [DOI] [PubMed] [Google Scholar]
- 37.Yu, Y. K., J. C. Wootton, and S. F. Altschul. 2003. The compositional adjustment of amino acid substitution matrices. Proc. Natl. Acad. Sci. USA 100:15688-15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.