

Abstract
We investigated the influence of the macrophage iron exporter ferroportin and its ligand hepcidin on intracellular Salmonella growth. Elevated ferroportin expression inhibited bacterial multiplication; hepcidin-induced ferroportin down-regulation enhanced it. Expression analysis of iron-responsive Salmonella genes indicated ferroportin-mediated iron deprivation. These results demonstrate a role for ferroportin in antimicrobial resistance.
Iron has a significant impact on infectious disease. Variations in dietary iron have been shown to influence the incidence and course of malaria, brucellosis, and tuberculosis (9, 21). Iron overload states such as sickle cell anemia and β-thalassemia increase the risk of infection with Salmonella and other pathogens (23, 28). Allelic variations in the macrophage metal transporter Nramp1 (Slc11a1) affect resistance to Salmonella, Leishmania, and mycobacteria, probably by influencing intraphagosomal iron concentrations (8). Some forms of hemochromatosis, an inherited disorder of iron metabolism, are also linked to susceptibility to intracellular pathogens, an association that has not been explained at a molecular level (11, 20). Since mutation or abnormal expression of the macrophage iron efflux protein ferroportin (FPN; Slc40a1) is a key feature of hemochromatosis (3, 10, 24), we examined the effect of FPN on the intracellular growth of Salmonella.
(Experiments carried out by S. Chlosta constituted part of a doctoral thesis submitted to the Charité Medical School of Berlin.)
We used the gentamicin protection assay to compare the growth of Salmonella (Salmonella enterica serovar Typhimurium strain SL1344) in stable, retrovirally transduced clones of J774, a murine macrophage line that carries the susceptible allele of Nramp1 (25). The J774-FPN1.RV2 clone, which expresses murine FPN and green fluorescent protein (GFP) from an integrated bicistronic vector, has two- to threefold-higher levels of FPN than the control J774-GFP.RV clone (16) (Fig. 1), a difference that is similar to the changes in FPN expression observed in both human hemochromatosis and mouse models of this disease (2, 15, 27, 29). As shown by the representative experiment whose data are presented in Fig. 1, the number of intracellular bacteria at 20 h postinfection was significantly lower in the J774-FPN1.RV2 cells. Similar data were obtained from four separate experiments, with the number of intracellular salmonellae in the J774-FPN1.RV2 cells being 2- to 25-fold less than in the J774-GFP.RV cells. Since the numbers of bacteria in the two cell lines were similar at 2 h postinfection, these results suggest that the intracellular multiplication of Salmonella was inhibited by the higher levels of FPN in the J774-FPN1.RV2 cells. FPN-dependent inhibition of Salmonella growth was also observed in transiently transfected HeLa cells, with the number of intracellular salmonellae at the 20-h time point being 5- to 18-fold lower in FPN-expressing cells in multiple experiments (Fig. 2). We found that the Q182H mutation of FPN, which is associated with hemochromatosis (14), also inhibited the growth of Salmonella in HeLa cells (Fig. 2), consistent with its wild-type iron export function (18). It is pertinent that the inhibitory effect of FPN on Salmonella growth is similar to that observed with Nramp1 (12, 17), which has an established in vivo role in regulating susceptibility to intracellular pathogens in mice and humans (8).
FIG. 1.

Effect of FPN on intracellular survival of Salmonella in J774 macrophages. J774-GFP.RV (G) or J774-FPN1.RV2 (F) cells were infected with Salmonella, and the number of intracellular bacteria surviving at 2 or 20 h postinfection was determined. Means (plus standard deviations; n = 3) from a single experiment are shown. The results are representative of four such experiments. The upper part of the figure shows FPN expression in the two J774 clones. The numbers below the Western blots show the relative intensities of the FPN bands based on densitometry (normalized to actin).
FIG. 2.

Effect of FPN expression on survival of Salmonella in HeLa cells. HeLa cells were transiently transfected with expression plasmids encoding GFP (G), wild-type FPN (F), or the Q182H mutant of FPN (Q). The results of Western blotting for FPN and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression are shown at the top. The lower part shows the number of intracellular Salmonella surviving at 2 or 20 h postinfection. Means (plus standard deviations; n = 3) from a single experiment are shown. The results are representative of two such experiments for the GFP expression construct and at least five such experiments for the FPN and Q182H mutant constructs.
Hepcidin is a hepatic peptide that is produced in response to inflammation and systemic iron overload (24). It binds FPN and induces its degradation, thus decreasing the export of iron from FPN-expressing cells such as macrophages and enterocytes (22). Abnormally low hepcidin levels are associated with at least two forms of hemochromatosis (24). To determine how hepcidin affects FPN bacteriostatic function, transiently transfected HeLa cells were treated with the peptide and infected with Salmonella. As shown previously (6, 22), hepcidin treatment reduced the level of wild-type FPN (Fig. 3). Correspondingly, FPN effects on intracellular Salmonella growth were reversed by hepcidin in both HeLa cells (Fig. 3) and the J774-FPN1.RV2 clone, although less dramatically in the latter (data not shown). Hepcidin did not affect Salmonella growth when added directly to bacterial cultures. The Q182H mutant was partially resistant to hepcidin-induced degradation, in agreement with other reports (18), and correspondingly, we found that unlike wild-type FPN, the mutant continued to inhibit bacterial growth in the presence of hepcidin (Fig. 3).
FIG. 3.

Effect of hepcidin on wild-type and Q182H FPN. HeLa cells were transfected to express either wild-type FPN or the Q182H mutant. Eighteen to 24 h later, hepcidin was added to the cells at 700 nM. For the Western blot, hepcidin was added for the indicated times after pretreatment of the cells for 60 min with 100 μM cycloheximide. The numbers below the blots indicate FPN band intensity, normalized to actin and expressed as a percentage of the control. For the gentamicin protection assay, the cells were infected with Salmonella about 18 h after the start of hepcidin treatment, and the number of intracellular bacteria surviving at 20 h postinfection was determined. Means (plus standard deviations; n = 3) from a single experiment are shown. The results are representative of three such experiments.
Two potential mechanisms could explain the FPN-mediated inhibition of Salmonella growth. FPN localizes to the plasma membrane and intracellular vesicles in primary macrophages (1, 4, 6) and J774 cells (F. Aydemir and M. Knutson, unpublished observations). Thus, it could inhibit bacterial growth by either iron deprivation or iron toxicity, depending on whether its iron export activity functions predominantly at the plasma membrane or the membrane of the Salmonella-containing vacuole. To distinguish between these mechanisms, we used reporter assays to examine the influence of FPN on expression of the Salmonella genes entF and iviXVII, which are induced by low and high iron concentrations, respectively (13). Salmonella strains containing β-galactosidase reporters controlled by either the entF or the iviXVII promoter were used to infect the J774-GFP.RV and J774-FPN1.RV2 cells. Four hours later, β-galactosidase expression was determined by a colorimetric assay (19). As shown in Fig. 4, entF-driven β-galactosidase expression (measured by absorbance at 420 nm and corrected for intracellular bacterial number) was reproducibly and significantly higher in the J774-FPN1.RV2 cells than in the J774-GFP.RV cells. The expression of the iviXVII reporter was slightly lower in the J774-FPN1.RV2 cells, and although this difference was not statistically significant, the trend was consistent with the behavior of the entF reporter. These results suggest that increased FPN expression inhibits Salmonella growth by decreasing the iron concentration in the intracellular microenvironment of the bacteria (although indirect effects on bacterial multiplication and reporter gene expression, possibly mediated by other iron transporters, could also contribute).
FIG. 4.
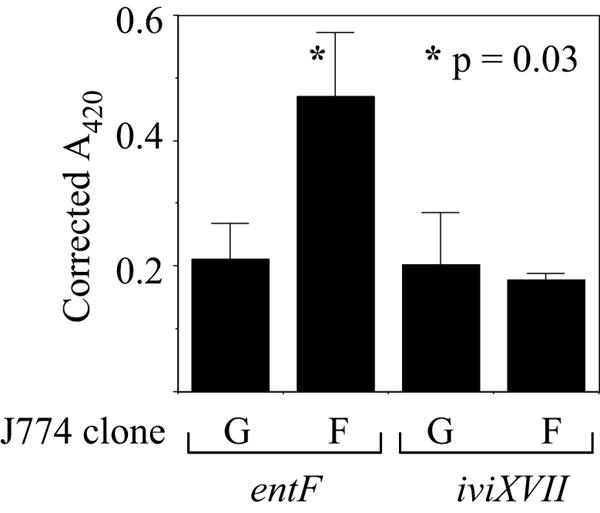
Effect of FPN on entF and iviXVII reporter gene expression. J774-GFP.RV (G) or J774-FPN1.RV2 (F) cells were infected with Salmonella strains carrying either the entF or iviXVII β-galactosidase reporters. β-Galactosidase expression was determined at 4 h postinfection and corrected for the number of intracellular bacteria. Means (plus standard deviations) from the results of three separate experiments are shown.
Our findings indicate an important role for FPN in controlling the growth of Salmonella inside cells and suggest that altered FPN expression and function in hemochromatosis, and possibly other iron overload states, may explain the susceptibility to intracellular pathogens in these conditions (11, 20, 23, 28). While up-regulation of hepcidin in response to infection is generally considered to be protective, our observations indicate that the opposite is true for intracellular pathogens. Hepcidin-resistant FPN mutations like Q182H may thus confer an advantage to the infected host by preserving resistance to intracellular pathogens even in the presence of elevated hepcidin. Several types of FPN mutations that differ in function and hepcidin sensitivity have been identified in patients with hemochromatosis (5, 7, 26). Determining how these mutations affect FPN′s ability to inhibit bacterial growth will help to identify individuals who are most susceptible to intracellular infection.
Acknowledgments
This work was supported by Public Health Service grants AI065619 (to B.J.C.) and DK05610 and DK064750 (to M.W.-R.).
We thank David Haile, South Texas Veterans Health Care System, for anti-FPN antibody and Michael Mahan, University of California, Santa Barbara, for entF and iviXVII Salmonella reporter strains. The technical assistance of Joan Lee is also gratefully acknowledged.
Editor: F. C. Fang
REFERENCES
- 1.Abboud, S., and D. J. Haile. 2000. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J. Biol. Chem. 275:19906-19912. [DOI] [PubMed] [Google Scholar]
- 2.Adams, P. C., Y. P. Barbin, Z. A. Khan, and S. Chakrabarti. 2003. Expression of ferroportin in hemochromatosis liver. Blood Cells Mol. Dis. 31:256-261. [DOI] [PubMed] [Google Scholar]
- 3.Beutler, E., J. C. Barton, V. J. Felitti, T. Gelbart, C. West, P. L. Lee, J. Waalen, and C. Vulpe. 2003. Ferroportin 1 (SLC40A1) variant associated with iron overload in African-Americans. Blood Cells Mol. Dis. 31:305-309. [DOI] [PubMed] [Google Scholar]
- 4.Canonne-Hergaux, F., A. Donovan, C. Delaby, H. Wang, and P. Gros. 2006. Comparative studies of duodenal and macrophage ferroportin proteins. Am. J. Physiol. Gastrointestinal Liver Physiol. 290:G156-G163. [DOI] [PubMed] [Google Scholar]
- 5.De Domenico, I., D. M. Ward, E. Nemeth, M. B. Vaughn, G. Musci, T. Ganz, and J. Kaplan. 2005. The molecular basis of ferroportin-linked hemochromatosis. Proc. Natl. Acad. Sci. USA 102:8955-8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delaby, C., N. Pilard, A. S. Goncalves, C. Beaumont, and F. Canonne-Hergaux. 2005. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 106:3979-3984. [DOI] [PubMed] [Google Scholar]
- 7.Drakesmith, H., L. M. Schimanski, E. Ormerod, A. T. Merryweather-Clarke, V. Viprakasit, J. P. Edwards, E. Sweetland, J. M. Bastin, D. Cowley, Y. Chinthammitr, K. J. Robson, and A. R. Townsend. 2005. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood 106:1092-1097. [DOI] [PubMed] [Google Scholar]
- 8.Forbes, J. R., and P. Gros. 2001. Divalent metal transport by Nramp proteins at the interface of host-pathogen interactions. Trends Microbiol. 9:397-403. [DOI] [PubMed] [Google Scholar]
- 9.Gangaidzo, I. T., V. M. Moyo, E. Mvundura, G. Aggrey, N. L. Murphree, H. Khumalo, T. Saungwenre, I. I. Kasvosve, Z. A. Gomo, T. Rouault, J. R. Boelaert, and V. R. Gordeuk. 2001. Association of pulmonary tuberculosis with increased dietary iron. J. Infect. Dis. 184:936-939. [DOI] [PubMed] [Google Scholar]
- 10.Gordeuk, V. R., A. Caleffi, E. Corradini, F. Ferrara, R. A. Jones, O. Castro, O. Onyekwere, R. Kittles, E. Pignatti, G. Montosi, C. Garuti, I. T. Gangaizdo, Z. A. Gomo, V. M. Moyo, T. Rouault, P. MacPhail, and A. Pietrangelo. 2003. Iron overload in Africans and African-Americans and a common mutation in the SLC40A1 (ferroportin 1) gene. Blood Cells Mol. Dis. 31:299-304. [DOI] [PubMed] [Google Scholar]
- 11.Gordeuk, V. R., C. E. McLaren, A. P. MacPhail, G. Deichsel, and T. H. Bothwell. 1996. Associations of iron overload in Africa with hepatocellular carcinoma and tuberculosis: Strachan's 1929 thesis revisited. Blood 87:3470-3476. [PubMed] [Google Scholar]
- 12.Govoni, G., F. Canonne-Hergaux, C. G. Pfeifer, S. L. Marcus, S. D. Mills, D. J. Hackam, S. Grinstein, D. Malo, B. B. Finlay, and P. Gros. 1999. Functional expression of Nramp1 in vitro in the murine macrophage line RAW264.7. Infect. Immun. 67:2225-2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heithoff, D. M., C. P. Conner, U. Hentschel, F. Govantes, P. C. Hanna, and M. J. Mahan. 1999. Coordinate intracellular expression of Salmonella genes induced during infection. J. Bacteriol. 181:799-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hetet, G., I. Devaux, N. Soufir, B. Grandchamp, and C. Beaumont. 2003. Molecular analyses of patients with hyperferritinemia and normal serum iron values reveal both L ferritin IRE and 3 new ferroportin (Slc11a3) mutations. Blood 102:1904-1910. [DOI] [PubMed] [Google Scholar]
- 15.Huang, F. W., J. L. Pinkus, G. S. Pinkus, M. D. Fleming, and N. C. Andrews. 2005. A mouse model of juvenile hemochromatosis. J. Clin. Investig. 115:2187-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knutson, M. D., M. Oukka, L. M. Koss, F. Aydemir, and M. Wessling-Resnick. 2005. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 over-expression and down-regulated by hepcidin. Proc. Natl. Acad. Sci. USA 102:1324-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lissner, C. R., R. N. Swanson, and A. D. O'Brien. 1983. Genetic control of the innate resistance of mice to Salmonella typhimurium: expression of the Ity gene in peritoneal and splenic macrophages isolated in vitro. J. Immunol. 131:3006-3013. [PubMed] [Google Scholar]
- 18.Liu, X. B., F. Yang, and D. J. Haile. 2005. Functional consequences of ferroportin 1 mutations. Blood Cells Mol. Dis. 35:33-46. [DOI] [PubMed] [Google Scholar]
- 19.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 20.Moyo, V. M., I. T. Gangaidzo, V. R. Gordeuk, C. F. Kiire, and A. P. Macphail. 1997. Tuberculosis and iron overload in Africa: a review. Central Afr. Med. J. 43:334-339. [PubMed] [Google Scholar]
- 21.Murray, M. J., A. B. Murray, M. B. Murray, and C. J. Murray. 1978. The adverse effect of iron repletion on the course of certain infections. Br. Med. J. 2:1113-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nemeth, E., M. S. Tuttle, J. Powelson, M. B. Vaughn, A. Donovan, D. M. Ward, T. Ganz, and J. Kaplan. 2004. Hepcidin regulates cellular iron efflux by binding ferroportin and inducing its degradation. Science 306:2090-2093. [DOI] [PubMed] [Google Scholar]
- 23.Onwubalili, J. K. 1983. Sickle cell disease and infection. J. Infect. 7:2-20. [DOI] [PubMed] [Google Scholar]
- 24.Pietrangelo, A. 2004. Hereditary hemochromatosis—a new look at an old disease. N. Engl. J. Med. 350:2383-2397. [DOI] [PubMed] [Google Scholar]
- 25.Ralph, P., J. Pritchard, and M. Cohn. 1975. Reticulum cell sarcoma: an effector cell in antibody-dependent cell-mediated immunity. J. Immunol. 114:898-905. [PubMed] [Google Scholar]
- 26.Schimanski, L. M., H. Drakesmith, A. T. Merryweather-Clarke, V. Viprakasit, J. P. Edwards, E. Sweetland, J. M. Bastin, D. Cowley, Y. Chinthammitr, K. J. Robson, and A. R. Townsend. 2005. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood 105:4096-4102. [DOI] [PubMed] [Google Scholar]
- 27.Viatte, L., J. C. Lesbordes-Brion, D. Q. Lou, M. Bennoun, G. Nicolas, A. Kahn, F. Canonne-Hergaux, and S. Vaulont. 2005. Deregulation of proteins involved in iron metabolism in hepcidin-deficient mice. Blood 105:4861-4864. [DOI] [PubMed] [Google Scholar]
- 28.Wanachiwanawin, W. 2000. Infections in E-β thalassemia. J. Pediatr. Hematol. Oncol. 22:581-587. [DOI] [PubMed] [Google Scholar]
- 29.Zoller, H., R. O. Koch, I. Theurl, P. Obrist, A. Pietrangelo, G. Montosi, D. J. Haile, and G. Weiss. 2001. Expression of the duodenal iron transporters divalent-metal transporter 1 and ferroportin 1 in iron deficiency and iron overload. Gastroenterology 120:1412-1419. [DOI] [PubMed] [Google Scholar]