

Abstract
Cytoskeletal alterations in endothelial cells have been linked to nitric oxide generation and cell-cell interactions. Transforming growth factor (TGF)-β has been described to affect cytoskeletal rearrangement in numerous cell types; however, the underlying pathway is unclear. In the present study, we found that human umbilical vein endothelial cells (HUVEC) have marked cytoskeletal alterations with short-term TGF-β treatment resulting in filipodia formation and F-actin assembly. The cytoskeletal alterations were blocked by the novel TGF-β type I receptor/ALK5 kinase inhibitor (SB-505124) but not by the p38 kinase inhibitor (SB-203580). TGF-β also induced marked stimulation of reactive oxygen species (ROS) within 5 min of TGF-β exposure. TGF-β stimulation of ROS was mediated by the NAPDH oxidase homolog Nox4 as DPI, an inhibitor of NADPH oxidase, and dominant-negative Nox4 adenovirus blocked ROS production. Finally, inhibition of ROS with ROS scavengers or dominant-negative Nox4 blocked the TGF-β effect on cytoskeleton changes in endothelial cells. In conclusion, our studies show for the first time that TGF-β-induced ROS production in human endothelial cells is via Nox4 and that TGF-β alteration of cytoskeleton in HUVEC is mediated via a Nox4-dependent pathway.
Keywords: human umbilical vein endothelial cells, cell-cell interactions, cytoskeletal rearrangement, transforming growth factor-β
endothelial function is closely linked to the cytoskeleton. Alterations in endothelial cytoskeleton have been linked to endothelial adhesion (10) and regulation of endothelial nitric oxide synthase activity (34). The regulation of endothelial cytoskeleton has been linked to reactive oxygen species (ROS) activity (10) and several growth factors (26). Endothelial dysfunction, characterized by deterioration of endothelial adhesion and vasodilator function, has been linked to numerous vascular diseases including diabetes and cardiovascular disease (16, 25, 33) and may be influenced by alterations in the cytoskeleton. Proposed mechanisms for endothelial dysfunction in diabetes involve generation of ROS (4). However, the role of transforming growth factor (TGF)-β, a growth factor closely linked to diabetic microvascular complications and a stimulator of ROS production (11, 31, 44), has not been previously examined in the regulation of human endothelial cytoskeleton and ROS production.
TGF-β is a multifunctional cytokine that can inhibit endothelial and epithelial cellular proliferation, stimulate matrix accumulation, and suppress inflammation (29). TGF-β is also a major factor in diabetic microvascular complications (31, 37). The major pathway involved in TGF-β signaling is via binding of active TGF-β to the type II receptor and subsequent binding of the type I receptor/ALK5 (18). It has recently been reported that endothelial cells also contain ALK1 as well as ALK5 and ALK1 may mediate certain effects of TGF-β (9). The activated ALK5 receptor then phosphorylates Smad2 or Smad3, which then translocates to the nucleus with the Co-Smad4 to regulate gene transcription. Although the role of Smads appears critical to TGF-β-induced gene regulation (19, 38), the role of Smads in mediating rapid effects of TGF-β has been questioned. In particular, cytoskeletal alterations by TGF-β occur in a biphasic manner in human prostate carcinoma cells and require Smad4 in the chronic phase (12-24 h) but are Smad4 independent and Rho GTPase dependent in the acute phase (5-30 min) (32). In murine mesangial cells, TGF-β induced rapid cytoskeletal changes via a calcium entry pathway (20), whereas in cat endothelial cells TGF-β induced F-actin assembly (26) via uncharacterized mechanisms.
A potential pathway of mediating cytoskeletal alterations by TGF-β is via ROS generation. ROS generation has been found to regulate the cytoskeleton in several cell types, including endothelial cells (10). Several studies identified NADPH oxidase as a major source of ROS production in endothelial cells (21, 43) and human umbilical vein endothelial cells (HUVEC) primarily express the NADPH oxidase isoform, Nox4 (1). Nox4 is a member of the family of gp91phox homologs that have recently been characterized in numerous cell types (15). In endothelial cells, hydrogen peroxide addition stimulates cytoskeletal changes and tyrosine phosphorylation of several proteins intimately involved in cytoskeletal regulation, including focal adhesion kinase (FAK), paxillin, and p130cas (10). Interestingly, TGF-β has been found to stimulate ROS production in a variety of cell types (13, 35), including endothelial cells (11), and also to stimulate tyrosine phosphorylation of cytoskeletal proteins (14). However, prior studies have not identified the enzyme responsible for ROS production by TGF-β in endothelial cells. In addition, the role of ROS in TGF-β-induced cytoskeletal alterations has not been addressed.
In the present study, we show that TGF-β can stimulate cytoskeletal alterations in HUVEC as characterized by filipodia formation and F-actin assembly. We found that TGF-β stimulation of ROS in endothelial cells is via Nox4 and that Nox4-induced ROS generation mediates TGF-β-induced cytoskeletal alterations.
MATERIALS AND METHODS
Reagents. Endothelial growth medium (EGM) culture medium was from Cambrex Bio Science Walkersville (Walkersville, MD). SB-505124 (ALK 5 inhibitor) was from Glaxo SmithKline (King of Prussia, PA, courtesy of N. Laping) and SB-203580 was from Biomol (Plymouth Meeting, PA). Type B Gelatin, EGTA, NAC, and DPI were purchased from Sigma (St. Louis, MO). SlowFade light antifade kit, Hoechst 33342, CM-H2DCFDA, rhodamine-phalloidin, and FITC-DNaseI were from Molecular Probes (Eugene, OR). TGF-β1 was from R&D Systems (Minneapolis, MN). Formaldehyde 37%, Triton X-100, and No. 1 coverslips were purchased from Fisher Scientific (Pittsburgh, PA). Black walled 96-well plates were purchased from Corning (Corning, NY).
Cell culture. HUVEC were isolated from human umbilical cord veins as described previously (23, 39) and cultured on 0.2% gelatin-coated dishes in EGM culture medium containing 10% FBS. The cells were routinely passaged with trypsin-EDTA and used for experiments between passages 2-6; 293 cells were obtained from American Type Culture Collection.
F- and G-actin staining by confocal microscopy. F-actin was stained with rhodamine phalloidin, and G-actin was stained with FITC DNase I. Cells were plated on coverslips coated with gelatin. Cells were kept quiescent in EGM culture media containing 0.5% BSA overnight before experimental treatment. Immediately after experimental protocol, cells were fixed with 3.7% formaldehyde and permeabilized with 0.1% Triton X-100. Coverslips were then incubated at room temperature with rhodamine-phalloidin (0.165 μM) alone or with FITC-DNaseI (0.3 μM) for 20 min. Cells were washed three times with PBS, and the coverslips were then mounted in SlowFade mounting media and sealed. Cells were directly visualized by fluorescence microscopy or confocal microscopy and representative photographs were obtained.
F- and G-actin quantitation with fluorescent plate reader. HUVEC were cultured to reach 80-90% confluence in 96-well black plates (Corning) coated with gelatin. After treatment, cells went through the same staining procedure as described above but FITC-phalloidin was used instead of rhodamine-phalloidin. Staining with FITC-phalloidin or FITC-DNase I was performed in separate wells. After being stained, the cells were washed three times with PBS and then 100 μl of PBS were left in each well and the plate was read with Millipore Cytofluor 2300 fluorescent plate reader using filter setting for excitation wavelength at 485 nm and emission wavelength at 530 nm. In selected experiments, cells were treated with kinase inhibitors, NAC or DPI, before TGF-β exposure or transduced with adenovirus for 72 h before TGF-β exposure.
ROS visualization and quantitation. HUVEC were plated on coverslips and made quiescent overnight in 0.5% BSA before stimulation with TGF-β. Cells were loaded for 10 min with 5 μM CM-H2DCFDA in phenol-red-free medium in the dark and then treated with TGF-β in the presence or absence of various inhibitors. In separate experiments, cells were transduced with adenoviral LacZ or dominant-negative ΔNADPH Nox4 vectors 72 h before experiments. Cells were then visualized by immunofluorescence microscopy after fixation and staining of the nuclei with DAPI or examined live using confocal microscopy. DCF fluorescence was visualized at an excitation wavelength of 488 nm and emission at 515 to 540 nm. To avoid photooxidation of the indicator dye, images were collected with a single rapid scan using identical parameters for all samples.
As a quantitative index of ROS generation, the Amplex Red reagent [10-acetyl-3,7-dihydroxyphenoxazine; Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit, Molecular Probes (A-22188)] was used. Amplex Red reacts with hydrogen peroxide in the presence of horseradish peroxidase (HRP) with a 1:1 stoichiometry to form resorufin. HUVEC were cultured on 96-well black plates coated with gelatin. The cells were rested overnight with 0.5% BSA in EGM medium and modulated with adenovirus or inhibitors. After the completion of pretreatment of HUVEC, Amplex Red and peroxidase in Kreb’s Ringer phosphate buffer (pH 7.4) were introduced into each well in the dark. The final reaction volume in each well was 100 μl. TGF-β at different concentrations was introduced just before reading the plate. The contents of the plate were shaken for 5 s before the commencement of the first cycle of measurement. Fluorescence intensity was kinetically recorded with excitation at 544 nm and emission of 590 nm at 37°C over a 20-cycle period with 10 flashes per well via fluorescent plate reader (POLARstar OPTIMA, BMG Labtechnologies). Measurements were made at 1-min intervals over a 20-min period. The data are reported as the mean value from each well over a 20-cycle period with n = 4-6 wells per experiment. Each experiment was repeated three times.
Immunoblot analysis of Nox4. Whole cell lysates from 293 cells and HUVEC were prepared with lysis buffer that contained 50 mM Tris·HCl (pH 7.2), 150 mM NaCl, 1% (wt/vol) Triton X-100, 1 mM EDTA, 1 mM PMSF, and 5 μg/ml aprotonin and leupeptin. Forty micrograms of cell lysate were resolved on a 10% reducing SDSPAGE and immunoblotted with antibody to Nox4 at 1:1,500 dilution. A secondary HRP-conjugated anti-rabbit antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used at 1:2,000 dilution. The antibody to Nox4 was raised against the COOH-terminal end of Nox4 and detected by ECL chemiluminescence as previously described (17). Two hundred ninety-three cells were transfected with a cDNA construct for human Nox4 as previously described (6, 17).
Immunostaining for Nox4 in HUVEC. HUVEC were plated on coverslips and made quiescent overnight in 0.5% BSA before stimulation with TGF-β. After stimulation, primary antibody at 1:100 dilution (rabbit polyclonal anti-Nox4 antibody) and secondary antibody (goat anti-rabbit IgG) conjugated to Alexa-Fluor-568 at 10 μg/ml final concentration (Molecular Probes) were used to detect Nox4. Bio-Rad MRC-600 confocal laser-scanning microscope mounted on a Zeiss Axiovert 100 fluorescent microscope equipped with a ×63 objective with rhodamine filter was used. Negative control was performed by omitting the primary antibody.
Preparation of NADPH Nox4 adenoviral construct and transduction of HUVEC. Nox4 deletion construct was generated as described previously (17). Briefly, cDNA for dominant-negative Nox4 lacking the NADPH binding domain was generated by removing the COOH-terminal sequence from wild-type Nox4 cDNA encoding 578 amino acids (GenBank accession number AF254621) for NADPH (amino acids 1 to 383). The construct was introduced into Ad-5 adenovirus using the Adeno-X expression system (BD Biosciences, Palo Alto, CA). A recombinant adenovirus encoding β-galactosidase was used as a control. Adenoviral constructs (600 infection forming units/cell) were preincubated for 100 min at room temperature with EGM media containing 0.5% BSA and 0.5 mg polylysine/ml. Transduction efficiency as measured by percent of cells expressing β-galactosidase was 90-95%, similar to prior studies with adenoviral transduction efficiency in HUVEC (22, 24). After preincubation, adenovirus transduction of HUVEC was performed by incubating cells in adenovirus-containing media overnight. The next day medium was changed to fresh EGM medium with 10% FBS. Seventy-two hours posttransduction, cells were used for experiments, after being made quiescent overnight in media containing 0.5% BSA.
Statistical analysis. In all experiments, data were evaluated for significance by one-way ANOVA using Student’s t-test. P < 0.05 was considered statistically significant.
RESULTS
TGF-β1 stimulates filipodia formation in HUVEC. To investigate whether TGF-β1 affects cytoskeletal alterations in endothelial cells, we treated HUVEC with TGF-β1 for different times and stained for F actin with rhodamine-phalloidin. Compared with control, TGF-β1 induced a rapid induction of lamelipodia between 5 and 15 min, which was then followed by development of long, thin membrane actin spikes, or filipodia, on the cell surface (Fig. 1). Using dual labeling of cells, we found that TGF-β increased F-actin staining coincident with reduced G-actin staining (Fig. 2A). The amount of F-actin and G-actin was quantified and the data are presented in Fig. 2B. After TGF-β treatment, F:G ratio progressively increased from 1.31 to 1.86 from 5 to 30 min (Fig. 2B).
Fig. 1.
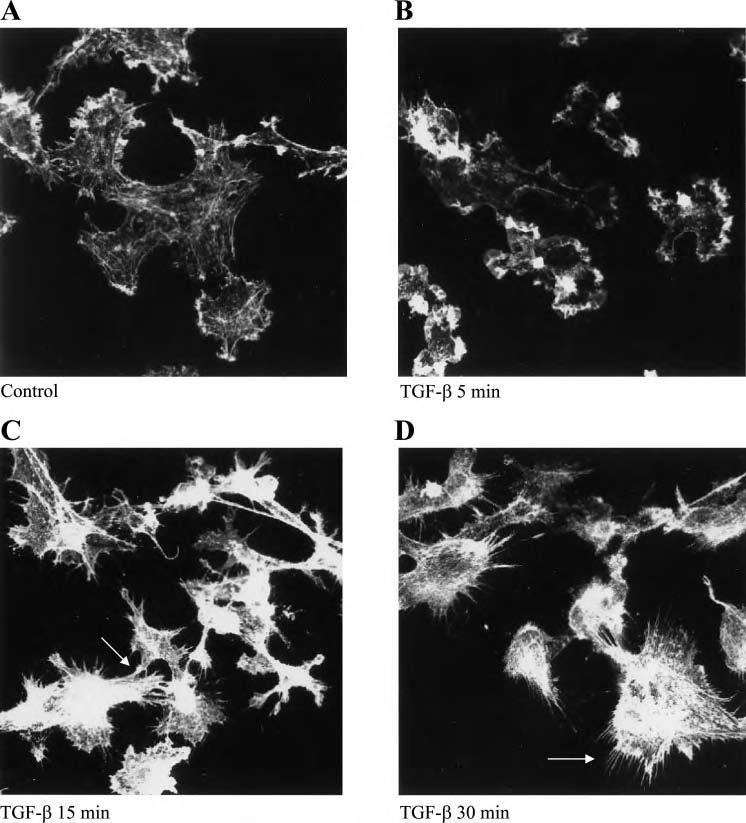
Induction of filipodia formation by transforming growth factor (TGF)-β in human umbilical vein endothelial cells (HUVEC). HUVEC were treated with vehicle or TGF-β1 (10 ng/ml) for various time points and cells were fixed and stained with rhodamine-phalloidin (A-D). Confocal microscopy was used to visualize cells and representative photographs were obtained. There is a dramatic increase in F-actin following TGF-β treatment at 15 and 30 min. Filipodia formation was noted in between cells as well (C) as away from neighboring cells (arrows; D).
Fig. 2.
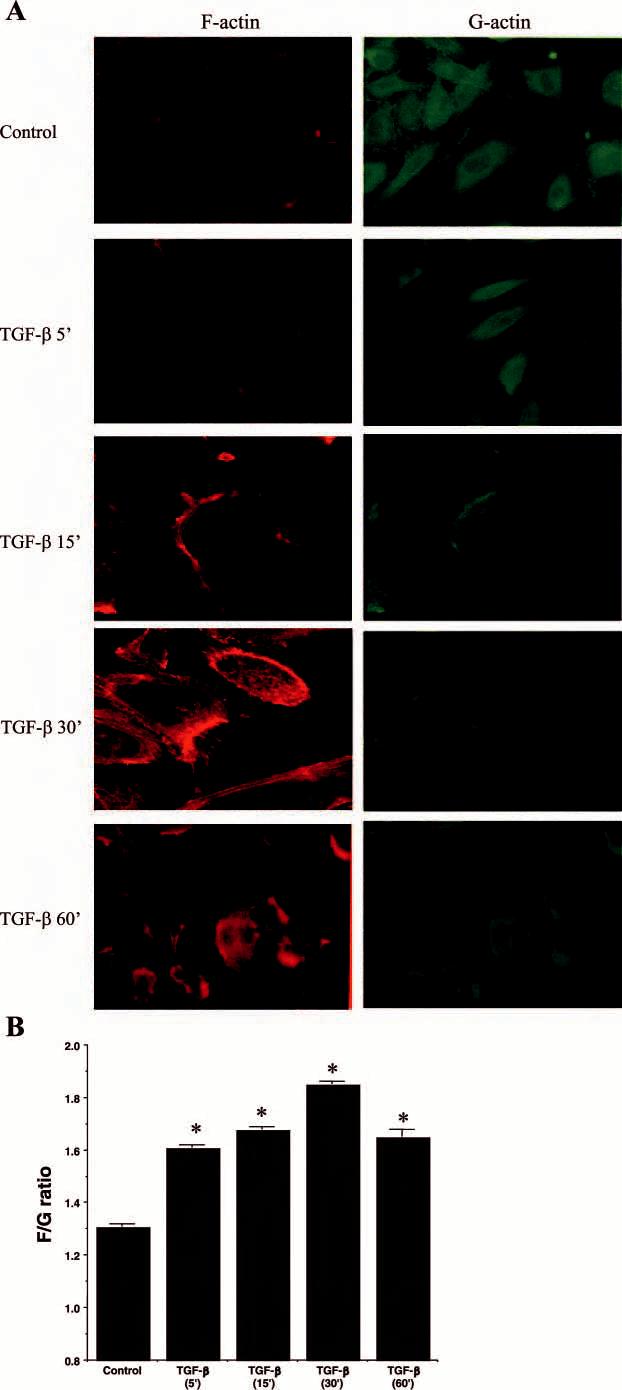
Induction of F-actin assembly by TGF-β in HUVEC. A: F-actin and G-actin were imaged on the same cells using rhodamine-phallodin and FITC-DNaseI, respectively, on HUVEC after TGF-β (10 ng/ml) treatment. There is a progressive increase in F-actin staining with a coincident decrease in G-actin staining. Also noted were prominent filipodial protrusions composed of F-actin. These results were most marked at 15 and 30 min after TGF-β treatment. B: F/G actin quantitation was performed in cells plated on a 96-well plate and stained with FITC-phalloidin or FITC-DNaseI. Fluorescence was measured using a fluorescent plate reader. There were 6 replicates per condition and experiments were repeated 3 times. Data are presented as the mean F/G actin ratio ± SE from a representative experiment. *P < 0.05 vs. control.
Role of TGF-β type I receptor/ALK5 and p38 on cytoskeletal changes in HUVEC. TGF-β primarily signals via TGF-β receptors located at the cell surface. Specific inhibition of the TGF-β type I receptor/ALK5 serine-threonine kinase with SB-505124 abolished the effects of TGF-β1 on F-actin assembly (Fig. 3, A and B). p38 Kinase activity has been implicated in regulating cytoskeletal assembly of endothelial cells (40, 42) as well as TGF-β-induced cytoskeletal changes in other cell types (32). However, pretreatment with SB-203580, an inhibitor of p38 MAP kinase, had no effect on F-actin assembly by TGF-β (Fig. 3, A and B).
Fig. 3.
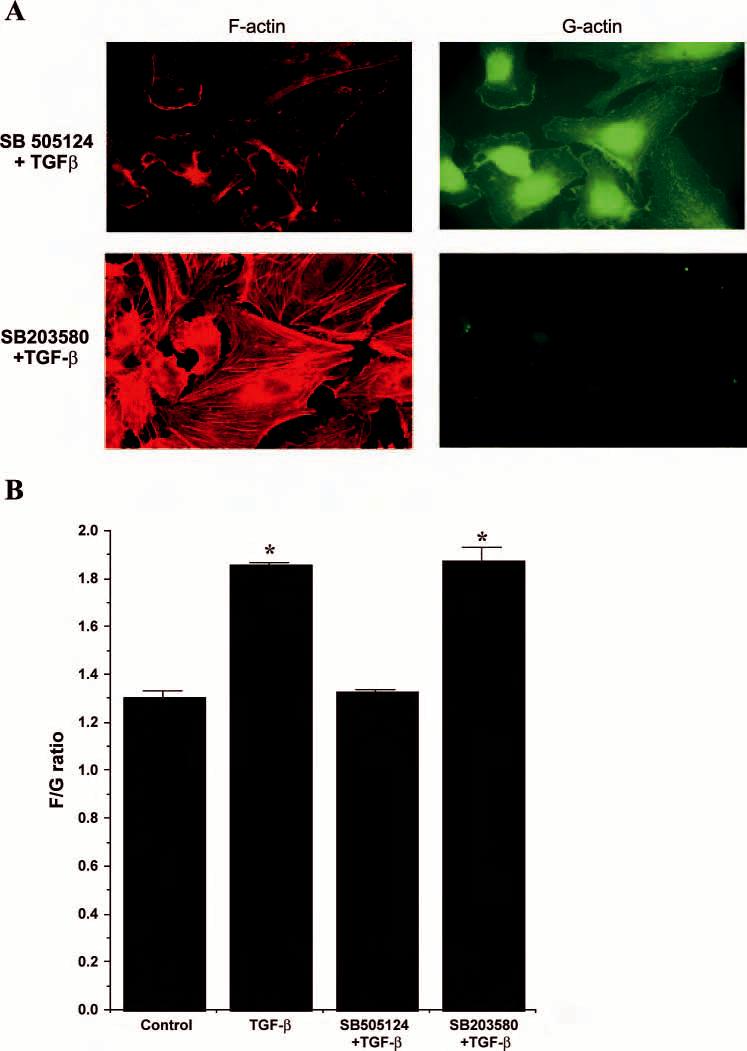
Inhibition of TGF-β type I receptor/ALK5 kinase, but not p38 kinase, blocks F-actin assembly by TGF-β. HUVEC were treated with TGF-β1 (10 ng/ml) for 30 min alone or preincubated with inhibitors for the TGF-β type I receptor kinase (SB-505124, 500 nM) or p38 kinase inhibitor (SB-203580, 5 μM) and imaged by confocal analysis (A). F/G actin quantitation was performed using a fluorescent plate reader (B). There were 6 replicates per condition and 3 separate experiments were performed. Data are presented as the mean F/G actin ratio ± SE from a representative experiment. *P < 0.05 vs. control.
TGF-β stimulates ROS production in HUVEC. As ROS production has been implicated in cytoskeletal changes, we examined whether TGF-β induced ROS. Using DCF fluorescence, we found that TGF-β induced a marked stimulation of ROS within 5 min of TGF-β exposure (Fig. 4). The increased ROS production was maintained for 60 min following TGF-β exposure. TGF-β-induced ROS requires the TGF-β type I receptor as the Alk5 inhibitor (SB-505124) prevented ROS production (Fig. 4F). ROS production was completely inhibited using the ROS scavenger NAC (Fig. 4G). The pathway involved in TGF-β-induced ROS production is likely via NADPH oxidase as inhibition of NADPH oxidase with DPI completely inhibited ROS production (Fig. 4H).
Fig. 4.
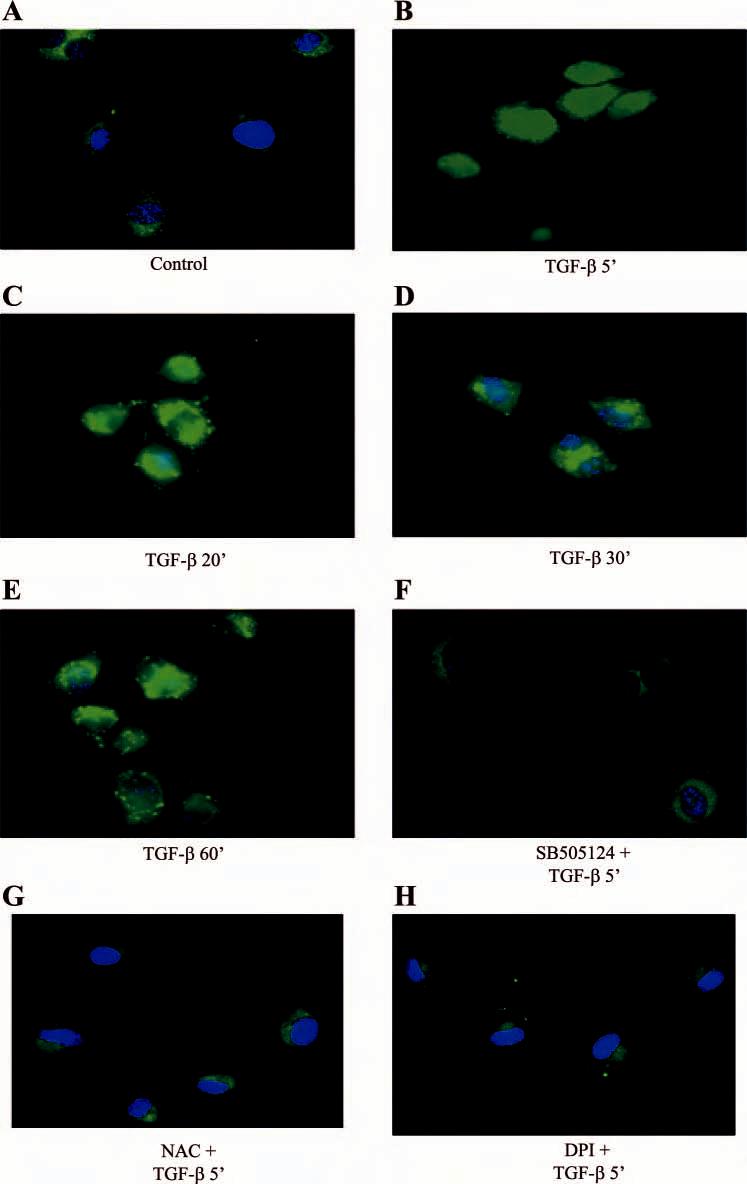
TGF-β stimulates reactive oxygen species (ROS) production. TGF-β1 (10 ng/ml)-induced ROS production was evaluated in HUVEC using DCF-loaded cells and costained with DAPI. A: control. B: TGF-β1 treatment 5 min. C: TGF-β1 treatment 20 min. D: TGF-β1 treatment 30 min. E: TGF-β1 treatment 60 min. F: ROS imaging after pretreatment with SB-505124 (500 nM, 4 h). G: 10 mM NAC (30 min). H: 10 μM DPI (30 min) and followed by TGF-β1 (10 ng/ml) treatment for 5 min. Experiments were repeated 3 times and representative photographs are shown.
TGF-β stimulation of ROS is via Nox4. Several isoforms of NADPH oxidase have been identified; however, Nox4 is the predominant isoform expressed in HUVEC by RT-PCR studies (1). We identified a distinct 65-kDa band for Nox4 in HUVEC as demonstrated by Western analysis (Fig. 5A) corresponding to the same sized band in 293 cells transfected with human Nox4, and as previously described in 3T3-L1 cells (17) and pancreatic cancer cells (41). Immunostaining of Nox4 revealed a diffuse pattern of Nox4 in HUVEC (Fig. 5B). Following short-term TGF-β treatment, there was increased localization of Nox4 to nucleus and the periphery of the cell (Fig. 5B).
Fig. 5.
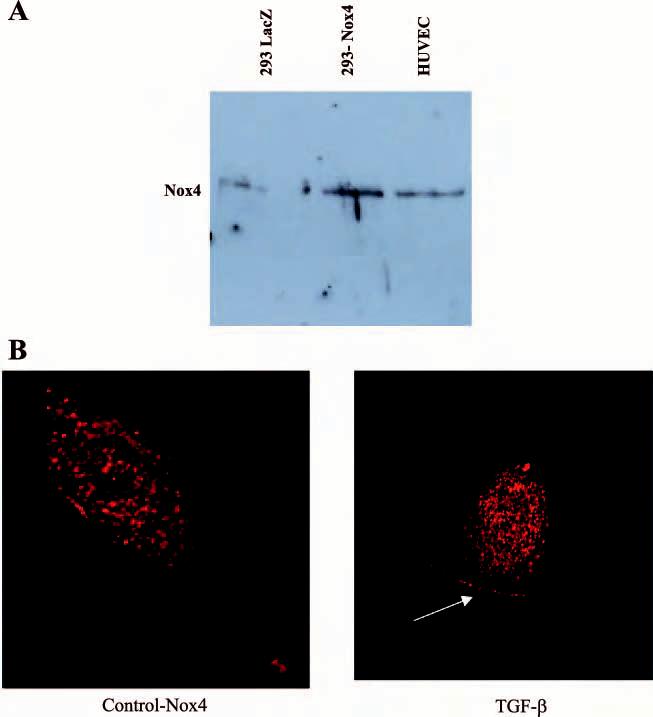
Nox 4 protein in HUVEC. A: cell lysates from 293 cells and HUVEC cells were analyzed for Nox4 protein by Western blot analysis. There is a distinct band at ∼65 kDa corresponding to Nox4 in HUVEC. A faint band is seen in lacZ-transfected 293 cells and is increased with transfection with wild-type Nox4. B: Nox4 protein was visualized by immunostaining of HUVEC with antibody to Nox4 and confocal microscopy. A Bio-Rad MRC-600 confocal laser-scanning microscope mounted on a Zeiss Axiovert 100 fluorescent microscope equipped with a ×63 objective with rhodamine filter was used. Under control conditions, Nox4 is present throughout the cell but concentrated in the nuclear region. With short-term TGF-β treatment, there is increased Nox4 in nuclear compartment and the periphery of the cell (see arrow).
To demonstrate the role of Nox4 in mediating TGF-β stimulation of ROS, we employed an adenoviral vector encoding Nox4 with a deletion of the NADPH binding site. This construct has been previously shown to have dominant-negative activity (17). Using adenovirus delivery, we found that dominant-negative NADPH-deficient Nox4 completely blocked TGF-β-induced ROS production (Fig. 6, Ac and Ad), whereas a control adenovirus vector encoding lacZ had no effect in inhibiting TGF-β-induced ROS production (Fig. 6Aa). Similar results were found with a quantitative assay using Amplex red (Fig. 6B).
Fig. 6.
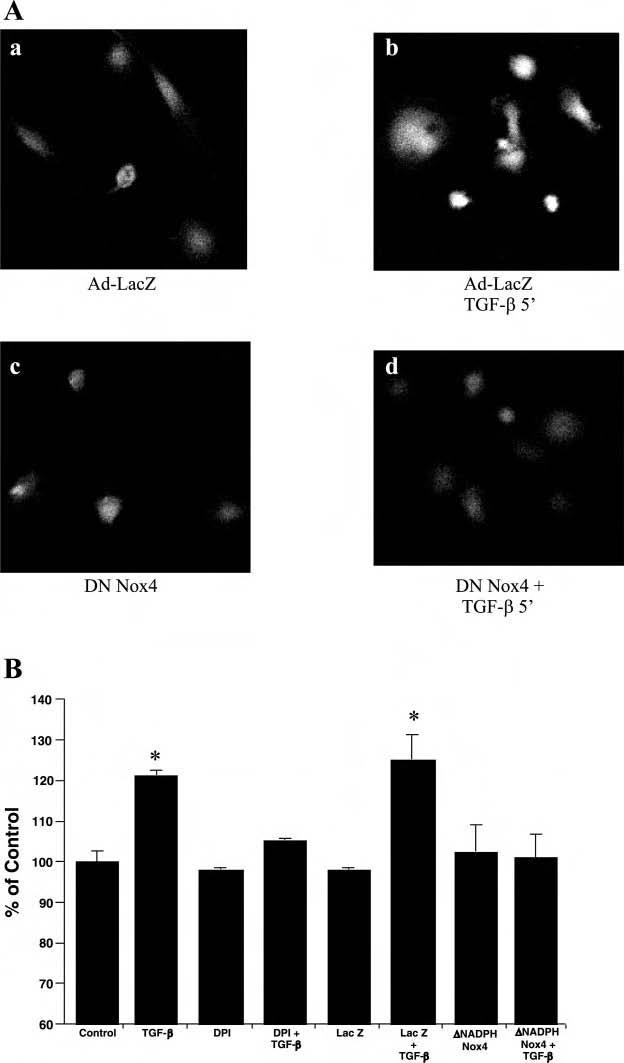
Nox 4 in HUVEC mediates TGF-β-induced ROS production. A: HUVEC were transduced with adenovirus encoding lacZ (a, b), or dominant-negative mutant Nox4 lacking NADPH binding site (c, d). Cells were loaded with DCF and treated with TGF-β (10 ng/ml, 5 min) and imaged using live confocal microscopy. Representative photographs were taken from 3 separate experiments. B: to quantitate ROS production, the Amplex Red assay was used to measure H2O2 production in HUVEC following TGF-β treatment alone and in the presence of DPI, LacZ adenovirus, and dominant-negative mutant Nox4. There were 6 replicates per condition and 3 separate experiments were performed. Data are presented as the mean percent control of H2O2 production ± SE from a representative experiment. *P < 0.05 vs. control.
TGF-β-induced cytoskeletal changes are mediated via ROS production. To determine the role of NADPH oxidase and ROS production by TGF-β in cytoskeletal alterations, we employed NAC, DPI, and the dominant-negative Nox4 adenovirus. Induction of filipodia by TGF-β was not affected in LacZ adenoviral transduced cells; however, filipodia formation was completely blocked by dominant-negative Nox4 (Fig. 7A). Similar findings were noted with NAC and DPI. NAC, DPI, and dominant-negative Nox4 blocked the TGF-β-induced increase in F/G-actin ratio (Fig. 7B).
Fig. 7.
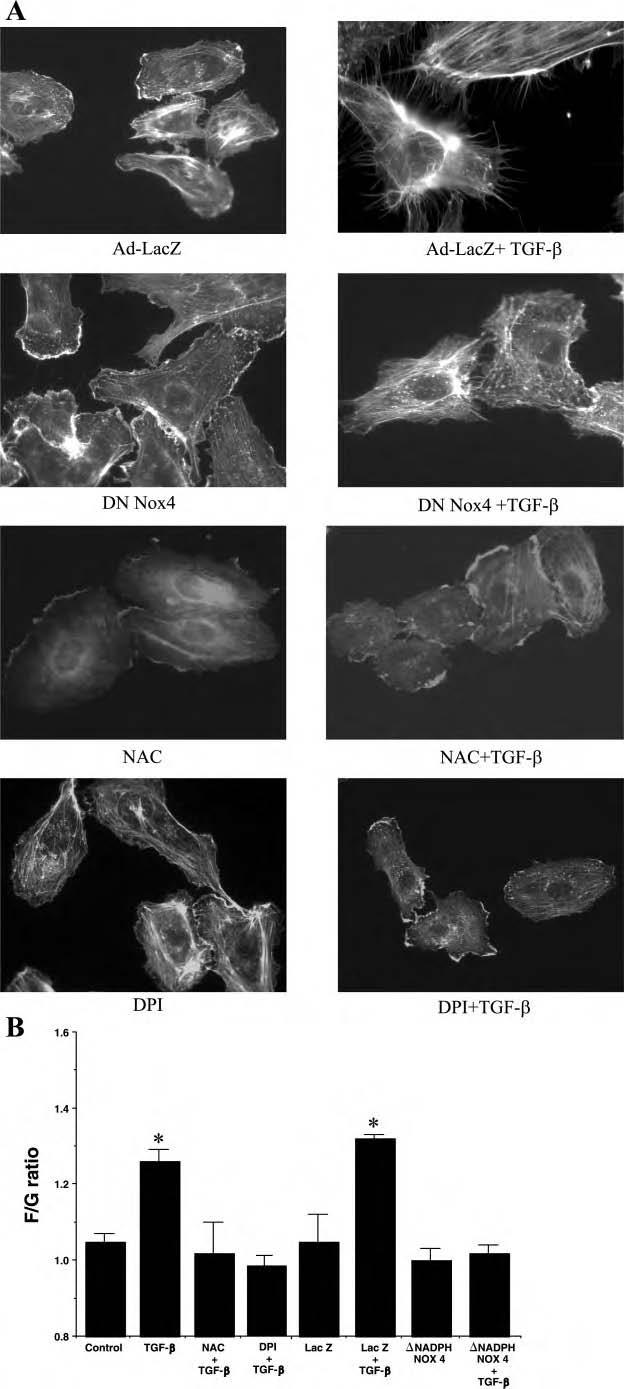
Inhibition of ROS and NADPH oxidase blocks TGF-β-induced cytoskeletal changes. A: TGF-β treatment induced F-actin assembly and filipodia formation in LacZ adenoviral-treated HUVEC but not in HUVEC transduced with adenovirus encoding dominant-negative Nox4. Experiments were repeated 3 times and representative photographs are shown. B: quantitation of F/G actin ratios was performed in wild-type HUVEC with and without TGF-β and in the presence of NAC (10 mM, 30 min) and DPI (10 μM, 30 min). LacZ adenoviral-transduced and NADPH-Nox4 adenoviral-transduced HUVEC were assessed under control conditions and following TGF-β1 treatment for 30 min. There were 6 replicates per condition and 3 separate experiments were performed. Data are presented as mean F/G actin ratio ± SE from a representative experiment. *P < 0.05 vs. control.
DISCUSSION
In the present study, we show that the actin cytoskeleton in HUVEC undergoes dramatic changes in response to TGF-β. TGF-β type I receptor/ALK5 kinase inhibition completely blocks TGF-β-induced cytoskeletal alterations, whereas p38 MAPkinase does not appear to be involved in mediating cytoskeletal changes by TGF-β in HUVEC. The intracellular signaling pathway involves generation of ROS by TGF-β as TGF-β stimulates ROS, and inhibitors of ROS block TGF-β-induced F/G-actin alterations. The major Nox isoform in HUVEC is Nox4, and inhibition of Nox4 activity blocks TGF-β-induced ROS production and cytoskeletal alterations.
F-actin assembly is associated with focal adhesion formation and generation of lamilipodia and filipodia structures. The formation of focal adhesion complexes may contribute to cell-cell contact. This appears to be the case with HUVEC as TGF-β-induced filipodia formation primarily occurred at sites adjacent to other cells. Other processes associated with F-actin assembly include regulation of cell growth, matrix adhesion, and apoptosis. Interestingly, TGF-β is well known to regulate endothelial cell differentiation, matrix production, and apoptosis (27, 28, 30). TGF-β-induced cytoskeletal changes are also likely related to chemotaxis of endothelial cells and effects on cell-cell communication and adhesion.
The signaling pathway by which TGF-β exerts cytoskeletal changes has been found to be both Smad independent and Smad dependent. Dominant-negative Smad4-transfected cells have been demonstrated to undergo rapid actin cytoskeletal alterations with the same kinetics as nontransfected epithelial cells (32) and in mesangial cells (unpublished observations, Sharma K). However, chronic exposure to TGF-β (24 h) induced cytoskeletal changes in epithelial cells in a Smad4-dependent manner (32). In our present studies, the rapid effects by TGF-β to regulate cytoskeletal changes suggest that the effect is Smad independent. Further studies are required to evaluate the chronic effects of TGF-β on endothelial cell actin cytoskeleton. The role of ROS stimulation to mediate TGF-β-induced cytoskeletal changes has not been previously described. ROS stimulation may mediate cytoskeletal changes via stimulation of pathways to regulate phosphorylation of the cytoskeletal proteins, paxillin, p130 cas, and FAK (10). Interactions of ROS with calcium, the RhoGTPase pathway, and cofilin and profilin are all likely involved in regulating the conversion of G-actin monomers to F-actin and development of filipodia (5, 8). Interestingly, TGF-β-induced cytoskeletal changes have been linked to calcium entry (20), RhoGTPase Cdc42 activation (32), and cofilin regulation (3). Furthermore, regulation of profilin was found to occur in the context of diabetic complications and regulated by ROS production (7).
The stimulation of ROS by TGF-β has been demonstrated in several cell types, including endothelial cells, smooth muscle cells, and fibroblasts (11, 35, 36). However, the timing of ROS stimulation in most studies has been described to occur in a time frame of 6 - 8 h, whereas we find stimulation of ROS within minutes of TGF-β exposure. In one study with human lung fibroblasts, TGF-β also induced ROS stimulation in a similar time frame (13); however, the source of ROS production was not identified. The stimulation of ROS by TGF-β in HUVEC appears to be via Nox4 as NADPH oxidase inhibition completely blocks ROS production and the use of dominant-negative Nox4 also prevents TGF-β-induced ROS production. It is conceivable that Nox1 may also contribute to ROS production as Nox1 has also been identified in HUVEC; however, the predominant isoform in HUVEC is Nox4 (1). The cellular distribution of Nox4 in HUVEC is similar to the reported distribution in vascular smooth muscle cells (12). It is interesting to note that Nox4 has been identified previously to colocalize with actin fibers suggesting an intimate relationship with the cytoskeleton (12). TGF-β treatment increased peripheral membrane localization of Nox4, suggesting possible association with NADPH oxidase subunits, such as phox22. Although phox22 may be integral to activation of Nox4 (2), the mechanism of stimulation of Nox4 by growth factors is unclear at present and is the subject of intense investigation.
In summary, we find that TGF-β stimulates F-actin assembly and filipodia formation in endothelial cells. The regulation of this effect is via the NADPH oxidase isoform Nox4. Further studies to understand the mechanisms of Nox4 activation by TGF-β and consequent effects on actin cytoskeleton and filipodia formation will likely reveal important insight into the relevance of these pathways in a variety of pathophysiological conditions, including diabetic vascular complications.
ACKNOWLEDGMENTS
We thank Dr. X. Ma who offered very helpful advice on the studies to evaluate ROS production and Dr. D. Lambeth who provided the original Nox4 cDNA clones.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
GRANTS
The studies were performed with partial support from National Institutes of Health Grants R01-DK-53867 (to K. Sharma) and R01-DK-43396 (to B. J. Goldstein).
REFERENCES
- Ago T, Kitazono T, Ooboshi H, Iyama T, Han Y, Takada J, Wakisaka M, Ibayashi S, Utsami H, Iida M. Nox4 as the major catalytic component of an endothelial NADP(H) oxidase. Circulation. 2004;109:227–233. doi: 10.1161/01.CIR.0000105680.92873.70. [DOI] [PubMed] [Google Scholar]
- Ambasta RK, Kumar P, Griendling KK, Schmidt HHHW, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem. 2004;279:45935–45941. doi: 10.1074/jbc.M406486200. [DOI] [PubMed] [Google Scholar]
- Bratt C, Lindberg C, Marko-Varga G. Restricted access chromatographic sample preparation of low mass proteins expressed in human fibro-blast cells for proteomics analysis. J Chromatogr A. 2001;909:279–288. doi: 10.1016/s0021-9673(00)01103-1. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Carlier MF, Valentin-Ranc C, Combeau C, Fievez S, Pantoloni D. Actin polymerization: regulation by divalent metal ion and nucleotide binding, ATP hydrolysis and binding of myosin. Adv Exp Med Biol. 1994;358:71–81. doi: 10.1007/978-1-4615-2578-3_7. [DOI] [PubMed] [Google Scholar]
- Cheng G, Cao Z, Xu X, Van Meir E, Lambeth D. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269:131–140. doi: 10.1016/s0378-1119(01)00449-8. [DOI] [PubMed] [Google Scholar]
- Clarkson M, Murphy M, Gupta S, Lambe T, Mackenzie HS, Godson C, Martin F, Brady HR. High glucose-altered gene expression in mesangial cells. Actin-regulatory protein gene expression is triggered by oxidative stress and cytoskeletal disassembly. J Biol Chem. 2002;277:9707–9712. doi: 10.1074/jbc.M109172200. [DOI] [PubMed] [Google Scholar]
- Didry D, Carlier M, Pantaloni D. Synergy between depolymerizing factor/cofilin and profilin in increasing actin filament turnover. J Biol Chem. 1998;273:25602–25611. doi: 10.1074/jbc.273.40.25602. [DOI] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mum-mery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediators of lateral TGFb/ALK5 signaling. Mol Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- Gozin A, Franzini E, Andrieu V, Da Costa L, Rollet-Labelle E, Pasquier C. Reactive oxygen species activate focal adhesion kinase, paxillin and P130CAS tyrosine phosphorylation in endothelial cells. Free Radic Biol Med. 1998;25:1021–1032. doi: 10.1016/s0891-5849(98)00134-8. [DOI] [PubMed] [Google Scholar]
- Hertig I, Hassoun P, Zuleta J, Thannickal VJ, Fanburg BL. Mechanism of basal and transforming growth factor β1 stimulated H2O2 release by endothelial cells. Trans Assoc Am Physicians. 1993;106:179–186. [PubMed] [Google Scholar]
- Hilenski L, Clempus R, Quinn M, Lambeth JD, Greindling K. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:1–8. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- Junn E, Lee KN, Ju HR, Han SH, Im JY, Kang HS, Lee TH, Bae SY, Lee ZW, Rhee SG. Requirement of hydrogen peroxide generation in TGF-β1 signal transduction in human lung fibroblast cells: involvement of hydrogen peroxide and Ca2+ in TGF-β1-induced IL-6 expression. J Immunol. 2000;165:2190–2197. doi: 10.4049/jimmunol.165.4.2190. [DOI] [PubMed] [Google Scholar]
- Kim JT, Joo CK. Involvement of cell-cell interaction in the rapid stimulation of cas tyrosine phosphorylation and src kinase activity by transforming growth factor β1. J Biol Chem. 2002;277:31938–31948. doi: 10.1074/jbc.M201178200. [DOI] [PubMed] [Google Scholar]
- Lambeth J, Cheng G, Arnold R, Edens W. Novel homologs of gp91phox. TIBS. 2000;25:459–461. doi: 10.1016/s0968-0004(00)01658-3. [DOI] [PubMed] [Google Scholar]
- Luscher TF, Tanner FC, Tschudi MR, Noll G. Endothelial dysfunction in coronary artery disease. Annu Rev Med. 1993;44:395–418. doi: 10.1146/annurev.me.44.020193.002143. [DOI] [PubMed] [Google Scholar]
- Mahadev K, Motoshima H, Wu X, Ruddy J, Arnold R, Cheng G, Lambeth J, Goldstein B. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol. 2004;24:1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J. How cells read TGF-β signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- Massague J, Wotton D. Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan T, Madesh M, Zhu Y, Wang L, Russo M, Deelman L, Henning R, Joseph SK, Hajnoczky G, Sharma K. TGF-β-induced Ca2+ influx involves the type III IP3 receptor and regulates actin cytoskeleton. Am J Physiol Renal Physiol. 2002;282:F910–F920. doi: 10.1152/ajprenal.00252.2001. [DOI] [PubMed] [Google Scholar]
- Mohazzab H, Kaminski P, Wolin M. NADH oxidoreductase is a major source or superoxide anion in bovine coronary artery endothelium. Am J Physiol Heart Circ Physiol. 1994;266:H2568–H2572. doi: 10.1152/ajpheart.1994.266.6.H2568. [DOI] [PubMed] [Google Scholar]
- Nakatsu M, Sainson R, Aoto J, Taylor K, Aitkenhead M, Perez-del-Pulgar S, Carpenter P, Hughes C. Angiogenic sprouting and capillary lumen formation modeled by human umbilical vein endothelial cells (HUVEC) in fibrin gels: the role of fibroblasts and angiopoietin-1. Microvasc Res. 2003;66:102–112. doi: 10.1016/s0026-2862(03)00045-1. [DOI] [PubMed] [Google Scholar]
- O’Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, Brass LF. Thrombin responses in human endothelial cells. Contributions from receptors other than Par1 include the transactivation of Par2 by thrombin-cleaved Par1. J Biol Chem. 2000;275:13502–13509. doi: 10.1074/jbc.275.18.13502. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, Funahashi T, Walsh K. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem. 2004;279:1304–1309. doi: 10.1074/jbc.M310389200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parving H, Nielsen F, Bang L, Smidt U, Svendsen T, Chen J, Gall M, Rossing P. Macro-microangiopathy and endothelial dysfunction in NIDDM patients with and without diabetic nephropathy. Diabetologia. 1996;39:1590–1597. doi: 10.1007/s001250050619. [DOI] [PubMed] [Google Scholar]
- Petroll WM, Jester JV, Barry-Lane PA, Cavanagh HD. Effects of basic FGF and TGF β1 on F-actin and ZO-1 organization during cat endothelial wound healing. Cornea. 1996;15:525–532. [PubMed] [Google Scholar]
- Pollman M, Naumovski L, Gibbons G. Vascular cell apoptosis: cell type-specific modulation by transforming growth factor-β1 in endothelial cells vs. smooth muscle cells. Circulation. 1999;99:2019–2026. doi: 10.1161/01.cir.99.15.2019. [DOI] [PubMed] [Google Scholar]
- RayChaudhury A, D’Amore P. Endothelial cell regulation by transforming growth factor-β. J Cell Biochem. 1991;47:224–229. doi: 10.1002/jcb.240470307. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB. The transforming growth factor-β. In: Sporn MB, Roberts AB, editors. Handbook of Experimental Pharmacology. Springer-Verlag; Heidelberg, Germany: 1990. pp. 419–472. [Google Scholar]
- Schulick A, Taylor A, Zuo W, Qiu C, Dong G, Woodward R, Agah R, Roberts A, Virmani R, Dichek D. Overexpression of transforming growth factor β1 in arterial endothelium causes hyperplasia, apoptosis, and cartilaginous metaplasia. Proc Natl Acad Sci USA. 1998;95:6983–6988. doi: 10.1073/pnas.95.12.6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Deelman L, Madesh M, Kurz B, Ciccone E, Siva S, Hu T, Zhu Y, Wang L, Henning R, Ma X, Hajnoczky G. Involvement of transforming growth factor-β in regulation of calcium transients in diabetic vascular smooth muscle cells. Am J Physiol Renal Physiol. 2003;285:F1258–F1270. doi: 10.1152/ajprenal.00145.2003. [DOI] [PubMed] [Google Scholar]
- Sofia E, Marene L, Heldin CH, Pontus S. Transforming growth factor-β induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell. 2002;13:902–914. doi: 10.1091/mbc.01-08-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano F, Virag L, Jagtap P, Szabo E, Mabley J, Liaudet L, Marton A, Hoyt D, Murthy K, Salzman A, Southan G, Szabo C. Diabetic enothelial dysfunction: the role of poly (ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- Su Y, Edwards-Bennett S, Bubb MR, Block ER. Regulation of endothelial nitric oxide synthase by the actin cytoskeleton. Am J Physiol Cell Physiol. 2003;284:C1542–C1549. doi: 10.1152/ajpcell.00248.2002. [DOI] [PubMed] [Google Scholar]
- Thannickal V, Day R, Klinz S, Bastien M, Larios J, Fanburg B. Ras-dependent and-independent regulation of reactive oxygen species by mitogenic growth factors and TGF-β1. FASEB J. 2000;14:1741–1748. doi: 10.1096/fj.99-0878com. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor β1. J Biol Chem. 1995;270:30334–30338. doi: 10.1074/jbc.270.51.30334. [DOI] [PubMed] [Google Scholar]
- Tsuchida KI, Cronin B, Sharma K. Novel aspects of transforming growth factor-β in diabetic kidney disease. Nephron. 2002;92:7–21. doi: 10.1159/000064486. [DOI] [PubMed] [Google Scholar]
- Tsuchida KI, Zhu Y, Siva S, Dunn SR, Sharma K. Role of Smad4 on TGF-β-induced extracellular matrix stimulation in mesangial cells. Kidney Int. 2003;63:2000–2009. doi: 10.1046/j.1523-1755.2003.00009.x. [DOI] [PubMed] [Google Scholar]
- Ukropec JA, Hollinger MK, Woolkalis MJ. Regulation of VE-cadherin linkage to the cytoskeleton in endothelial cells exposed to fluid shear stress. Exp Cell Res. 2002;273:240–247. doi: 10.1006/excr.2001.5453. [DOI] [PubMed] [Google Scholar]
- Van Nieuw Amerongen GP, Koolwijk P, Versteilen A, van Hinsbergh VWM. Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler Thromb Vasc Biol. 2003;23:211–217. doi: 10.1161/01.atv.0000054198.68894.88. [DOI] [PubMed] [Google Scholar]
- Vaquero EC, Edderkaoui M, Pandol SJ, Gukovsky I, Gukovskaya AS. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem. 2004;279:34643–34654. doi: 10.1074/jbc.M400078200. [DOI] [PubMed] [Google Scholar]
- Wang Q, Doerschuk C. The p38 mitogen-activated protein kinase mediates cytoskeletal remodeling in pulmonary microvascular endothelial cells upon intra-cellular adhesion molecule-1 ligation. J Immunol. 2001;166:6877–6884. doi: 10.4049/jimmunol.166.11.6877. [DOI] [PubMed] [Google Scholar]
- Zhang H, Schmeiber A, Garlichs C, Plotze K, Damme U, Mugge A, Daniel W. Angiotensin II-induced superoxide anion generation in human vascular enothelial cell: role of membrane-bound NADH/NADPH oxidases. Cardiovasc Res. 1999;44:215–222. doi: 10.1016/s0008-6363(99)00183-2. [DOI] [PubMed] [Google Scholar]
- Ziyadeh F, Hoffman B, Han D, Iglesias-de la Cruz C, Hong S, Isono M, Chen S, McGowan T, Sharma K. Long-term prevention of renal insufficiency excess matrix gene expression and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-β antibody in db/db diabetic mice. Proc Natl Acad Sci USA. 2000;97:8015–8020. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]