

Hydroboration has been an essential reaction in synthetic organic chemistry since Brown’s discovery that borane etherates are reactive at room temperature.1,2 Diverse hydroborating agents including THF borane, dimethyl sulfide borane, 9-BBN, and thexylborane are readily available and offer many options for selective hydroboration.1 However, each has limitations as well as advantages and all are air-sensitive. The far more stable pyridine borane (py·BH3) has also been considered as a hydroborating agent,3a,b but heating to 75-100 °C is required for dissociation to free borane, a prerequisite for π-complexation of the olefin and eventual hydroboration. Hindered amine boranes dissociate more readily and react at lower temperatures, but they are air-sensitive.3c The remaining challenge is to obtain high reactivity without compromising reagent stability and practicality.
We have explored the possibility of activating Py·BH3 by replacing one of the hydrides with a good leaving group (Scheme 1; Py·BH2X(1) with X ) I, Br, OTf, NTf2). If this approach is used, the strength of the B-N bond would no longer be problematic, provided that departure of the new leaving group X leads to hydroboration. This might occur by some process equivalent to SN2-like displacement of X to form the olefin π-complex 2 (path A) or an SN1-like heterolysis via 5 (path B), followed by 4-center addition of B-H(3) to give 4. A third possibility is dissociation of 1 to BH2X(6, path C), conventional hydroboration, and complexation with pyridine to afford 4. Prior studies show that intramolecular hydroborations using activated, unsaturated amine and phosphine boranes are consistent with internal versions of paths A or B.4 We now report that a similar hydroboration pathway is also viable as an intermolecular process.
Scheme 1.

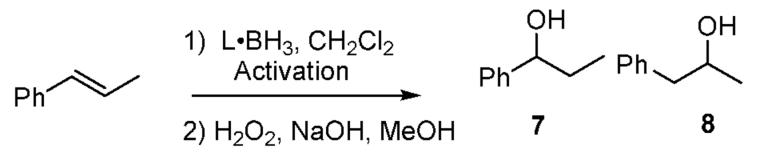
Several amine boranes and activation methods were compared to see if intermolecular hydroboration according to Scheme 1 is possible. The best results were achieved when commercially available pyridine borane (Py·BH3) was activated with 50 mol % of I2 in dichloromethane to generate Py·BH2I(1; rapid hydrogen evolution).5 Addition of β-methylstyrene followed by oxidative workup gave alcohol products (92%; 15:1 ratio, 7/8; entry 1, Table 1). This improved selectivity, compared to the 5:1 ratio using BH3·THF,6 suggests that activation produces a unique hydroborating agent and does not simply release BH3. Activation of Py·BH3 with bromine gave higher selectivity, but a much slower reaction (entry 2), while TfOH and HNTf2 (entries 3 and 4) induced faster but less selective hydroborations.
Table 1.
Hydroboration of β-methylstyrene with L·BH2X
 | |||||
|---|---|---|---|---|---|
| entry | L·BH3a | activation | time (h) | 7:8 | yield (%) |
| 1 | Py·BH3 | I2b | 2 | 15:1 | 92 |
| 2 | Py·BH3 | Br2b | 12 | >20:1 | 10d |
| 3 | Py·BH3 | TfOHc | 2 | 10:1 | 72 |
| 4 | Py·BH3 | HNTf2c | 2 | 10:1 | 90 |
| 5 | Lut·BH3 | I2b | 2 | 2.4:1 | 13d |
| 6 | Me2S·BH2Ie | 2 | 3.5:1 | 62 | |
1:1 ratio, L·BH3/alkene, room temperature.
50 mol %.
100 mol %.
Reaction quenched prior to completion.
Preformed (ref 7).
Next we compared the reagent 1 (X ) I) with Lut·BH2I (Table 1, entry 5; from lutidene borane + I2) and the known Me2S·BH2I (entry 6).7 Different hydroboration regioselectivity was found in each case, and unique 11B NMR signals were observed prior to the addition of alkene (Py·BH2I, δ -28.5 ppm; Lut·BH2I, δ -34.5 ppm; Me2S·BH2I, δ -20.5 ppm). The NMR data do not exclude the presence of BH2I in equilibrium with L·BH2I in one or more cases, but the regioselectivity results (entries 1, 5, and 6) prove that dissociation (as in path C) cannot be the only reaction pathway.
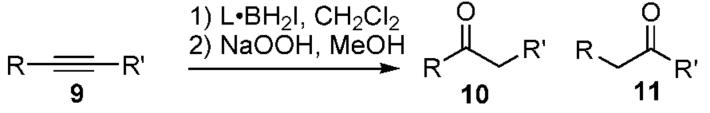
The hydroboration of 1-Ph-1-propyne (9) with BH3·THF is reported to give a 3:1 ratio of 10/11, while sia2BH, thexylBH2, catecholborane, and Br2BH·SMe2 afford mostly 11.8 In contrast, Py·BH2I produces a striking 15:1 selectivity favoring 10 (Table 2, entry 1), an effect that is amplified for p-CF3Ph-1-propyne and reversed for the p-MeOPh analogue (entries 2 and 3). Related trends are reported for styrene hydroboration.9 Lut·BH2I reacts nonselectively (entry 4), but Me2S·BH2I gives 10 with only traces of 11 (entry 5). Other alkynes (entries 8 and 9) are hydroborated with low regioselectivity, similar to the results with BH3·THF.8
Table 2.
Alkyne Hydroboration
 | ||||||
|---|---|---|---|---|---|---|
| entry | L | alkyne | R | R′ | 10:11 | yield (%) |
| 1 | Py | 9a | Ph | CH3 | 15:1 | 64 |
| 2 | Py | 9b | pCF3Ph | CH3 | >20:1 | NA |
| 3 | Py | 9c | pMeOPh | CH3 | 1:2 | NA |
| 4 | Lut | 9a | Ph | CH3 | 1.2:1 | 51 |
| 5 | Me2S | 9a | Ph | CH3 | 30:1 | 46 |
| 6 | Py | 9d | Ph | C2H5 | 10:1 | 66 |
| 7 | Py | 9e | C3H7 | C3H7 | 63 | |
| 8 | Py | 9f | CH3 | C5H11 | 1.5:1 | 61 |
| 9 | Py | 9g | CH3 | cC6H11 | 3:1 | 64 |
The simplest interpretation of the pyridine and lutidine borane results (Tables 1, 2) is that the ligand (L ) Py or Lut) remains attached to boron in the product-determining step for each reaction(Table 1, entries 1 and 5; Table 2, entries 1 and 4). However, the data require only that the Py·BH2I reagent follows a pathway different from path C (Scheme 1), assuming that the reaction of Me2S·BH2I involves dissociation to free BH2I.
Rate-determining dissociation of 1 (X ) I) to 5 (path B) is ruled out because the rate of methylstyrene hydroboration with Py·BH2I increases with alkene concentration (qualitatively, first order in alkene). The strong counterion dependence for hydroboration regiochemistry (Table 1) also argues against formal dissociation in an SN1-like mechanism, but neither the rate nor the regiochemistry data can rule out pathways where the conversion from 5 to 3 is rate-limiting if species analogous to tight ion pairs are involved. Path A (Scheme 1) is the simplest rationale that is consistent with facile hydroboration from Py·BH2I at room temperature. By way of analogy, Ryschkewitsch et al. have reported that Py·BH2I reacts readily with nitrogen nucleophiles, resulting in iodide displacement in an SN2-like process.5b Of course, the alkene is a much weaker nucleophile, and thus it would be premature to conclude that it can be sufficiently reactive to trigger the simplest version of path A. Furthermore, tight ion pair versions of path B cannot be ruled out, and other mechanistic variants remain to be evaluated.
Good functional group compatibility was observed with the Py·BH2I reagent (Table 3). Hydroboration of 12 followed by oxidative workup gave >95% primary alcohols 13 (NMR assay). Complete conversion of ester, amide, and amine substrates 12d-g required 2 equiv of Py·BH2I, but no reduction of these functional groups was observed within 2 h at room temperature. On the other hand, reduction of ketones and carboxylic acids (12,R = C(O)Me or CO2H) was fast compared to hydroboration of the alkene.
Table 3.
Functional Group Compatibility
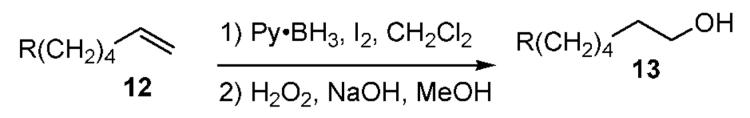 | |||
|---|---|---|---|
| entry | alkene | R | yield (%) |
| 1 | 12a | n-C6H13 | 98 |
| 2 | 12b | OBn | 83 |
| 3 | 12c | OTBS | 83 |
| 4 | 12d | OBz | 84a,b |
| 5 | 12e | NBn2 | 74a |
| 6 | 12f | NHBn | 80a |
| 7 | 12g | NHBz | 89a,b |
2:1:1 py·BH3/l2/alkene; 2 h at room temperature; NaOOH/MeOH.
Oxidative workup: NaBO3·H2O, THF/H2O.
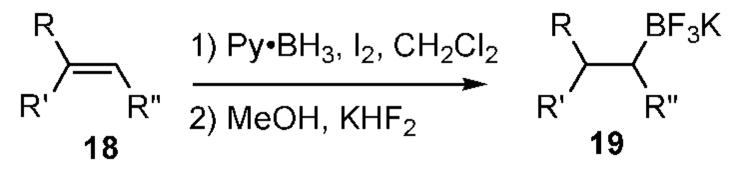
Monoalkyl boronic acid derivatives cannot be generated directly from unhindered alkenes using BH3·THF because the initially formed monoalkylborane is more reactive in hydroboration than is the parent BH3.10 However, the Py·BH2I method forms the 1:1 adducts considerably faster than 2:1 adducts, as might be expected according to path A (Scheme 1). Thus, hydroboration of 1-dodecene 12a was monitored after quenching in MeOH using positive ion detection ESMS. Strong signals for the 1:1 adducts 14 (Z ) MeO, Py) were observed, together with a weak signal for the 2:1 adduct 15 (Chart 1). Subsequent treatment with KHF211 allowed assay in the negative ion detection mode. A strong signal for 16 was observed, but 17 was not detected after precipitation from acetonitrile. Preparative experiments were performed from alkenes 18 to afford the corresponding potassium alkyltrifluoroborates 19 in 59-84% yield (Table 4). In all cases, ESMS with negative ion detection revealed the presence of 1:1 adducts, but not 2:1 adducts. On the other hand, use of excess alkene allowed the ESMS detection of a substantial signal for 17.
Table 4.
Preparation of Potassium Alkyltrifluoroborates 19
 | |||||
|---|---|---|---|---|---|
| entry | alkene | R | R′ | R″ | yield (%) |
| 1 | 18a | Ph | H | H | 84 |
| 2 | 18b | C4Hg | H | H | 76 |
| 3 | 18c | H | -C4H8- | 82 | |
| 4 | 18d | Ph | CH3 | H | 61 |
| 5 | 18e | Ph | -C4H8- | 59 | |
Molander has shown that alkyltrifluoroboratesalts are attractive reagents for Suzuki coupling applications,12but preparation of these salts required the use of catecholborane or BBr2H·SMe2. The Py·BH2I hydroboration is a simple alternative that cleanly affords the 1:1 adducts 19 and provides a high-yielding and convenient route to useful organoborane substrates.
In conclusion, we have presented evidence for an unusual hydroboration mechanism involving leaving group displacement from activated pyridine boranes 1. Hydroboration with Py·BH2Iis easily controlled to give the monoadducts and does not require handling sensitive trivalent boranes.
Supplementary Material
Chart 1.

Acknowledgments
This work was supported by NIH (CA17918; GM067146).
Footnotes
Supporting Information Available: Experimental procedures and characterization data (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Brown HC, Subba Rao BC. J. Am. Chem. Soc. 1959;81:6423. [Google Scholar]
- (2).Matteson DS. Stereodirected Synthesis with Organoboranes. Springer-Verlag; New York: 1995. Chapter 2. [Google Scholar]
- (3).(a) Hawthorne MF. J. Org. Chem. 1958;23:1788. [Google Scholar]; (b) Brown HC, Murray KJ, Murray LJ, Snover JA, Zweifel G. J. Am. Chem. Soc. 1960;82:4233. [Google Scholar]; (c) Brown HC, Kanth JVB, Dalvi PV, Zaidlewicz M. J. Org. Chem. 2000;65:4655. doi: 10.1021/jo0002745. and references therein. [DOI] [PubMed] [Google Scholar]
- (4).Scheideman M, Shapland P, Vedejs E. J. Am. Chem. Soc. 2003;125:10502. doi: 10.1021/ja034655m. [DOI] [PubMed] [Google Scholar]
- (5).(a) Ryschkewitsch GE. J. Am. Chem. Soc. 1966;88:3145. [Google Scholar]; (b) Ryschkewitsch GE, Garrett JM. J. Am. Chem. Soc. 1968;90:7234. [Google Scholar]
- (6).Brown HC, Zweifel G. J. Am. Chem. Soc. 1960;82:4708. [Google Scholar]
- (7).Cha JS, Min SJ, Kim JM, Kwon OO, Jeoung MK. Org. Prep. Proced. Int. 1993;25:466. [Google Scholar]
- (8).Brown HC, Scouten CG, Liotta R. J. Am. Chem. Soc. 1979;101:96. [Google Scholar]
- (9).Brown HC, Sharp RL. J. Am. Chem. Soc. 1966;88:5851. [Google Scholar]
- (10).Brown HC, Tsukamoto A, Bigley DB. J. Am. Chem. Soc. 1960;82:4703. [Google Scholar]
- (11).Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MR. J. Org. Chem. 1995;60:3020. [Google Scholar]
- (12).(a) Molander GA, Rivero M. Org. Lett. 2001;3:393. doi: 10.1021/ol006896u. [DOI] [PubMed] [Google Scholar]; (b) Molander GA, Yun C-S, Ribagorda M, Biolatto BJ. Org. Chem. 2003;68:5534. doi: 10.1021/jo0343331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.