

Abstract
Relatedness estimators are widely used in genetic studies, but effects of population structure on performance of estimators, criteria to evaluate estimators, and benefits of using such estimators in conservation programs have to date received little attention. In this article we present new estimators, based on the relationship between coancestry and molecular similarity between individuals, and compare them with existing estimators using Monte Carlo simulation of populations, either panmictic or structured. Estimators were evaluated using statistical criteria and a diversity criterion that minimized relatedness. Results show that ranking of estimators depends on the population structure. An existing estimator based on two-gene and four-gene coefficients of identity performs best in panmictic populations, whereas a new estimator based on coancestry performs best in structured populations. The number of marker alleles and loci did not affect ranking of estimators. Statistical criteria were insufficient to evaluate estimators for their use in conservation programs. The regression coefficient of pedigree relatedness on estimated relatedness (β2) was substantially lower than unity for all estimators, causing overestimation of the diversity conserved. A simple correction to achieve β2 = 1 improves both existing and new estimators. Using relatedness estimates with correction considerably increased diversity in structured populations, but did not do so or even decreased diversity in panmictic populations.
ADDITIVE genetic relatedness between individuals plays an important role in many fields of genetics. In genetic analyses, knowledge of relatedness is used to estimate genetic parameters such as heritabilities and genetic correlations (Falconer and Mackay 1996). In artificial selection, estimation of breeding values relies on knowledge of relatedness of individuals (Henderson 1984; Lynch and Walsh 1998), and relatedness between individuals affects optimum designs of selection programs (e.g., Nicholas and Smith 1983). In evolutionary biology, knowledge of relatedness between interacting individuals is required to predict evolutionary consequences of social interaction (Hamilton 1964). In conservation genetics, knowledge of relatedness is required to optimize conservation strategies. In this article we focus on estimating relatedness for use in conservation strategies, but results are equally relevant for other fields in genetics. Throughout, we consider the traditional population-genetic definition of relatedness for diploid individuals, which equals twice the coefficient of coancestry (Malécot 1948; Lynch and Walsh 1998).
When pedigrees of populations are known, additive genetic relatedness between individuals can be calculated from the pedigree (Emik and Terrill 1949) and can be used to estimate additive genetic variance. Pedigree data are, however, often lacking or incomplete, especially between subpopulations of a species. In those cases, estimates of relatedness rely on molecular markers. Methods to estimate relatedness from molecular marker data described in the literature can be divided into two groups (Blouin 2003): (1) methods that estimate relatedness on a continuous scale (e.g., Lynch and Ritland 1999; Wang 2002), and (2) methods that categorize individuals into a limited number of discrete classes of relatives, such as full-sib, half-sib, or parent–offspring relationships.
Toro et al. (2002) compared estimators expressing relatedness on a continuous scale in a pedigreed population of pigs divided into two related strains, using actual and simulated markers. Molecular coancestry (Malécot 1948), the estimator of Lynch and Ritland (1999), and a maximum-likelihood estimator showed the highest correlation between pedigree and estimated relatedness. When both strains were analyzed together, molecular coancestry performed substantially better than more sophisticated estimators, indicating that quality of estimators depends on the population structure and that current estimators are not optimal in general. More recently, novel estimators have been proposed and compared by Wang (2002) and Milligan (2003) for their statistical performance in an “outbred” population structure, having only four degrees of relatedness, parent–offspring, full-sibs, first cousins, and unrelated individuals.
A number of issues remain unsolved, relating in particular to the population structure (Milligan 2003), the utility of estimated relatedness in conservation programs, and the criterion to judge the quality of an estimator. Estimators of Lynch and Ritland (1999) and Wang (2002) assume no inbreeding. Those estimators have been evaluated using simulated populations without pedigree, no inbreeding, and simple classes of relatives of full-sibs, half-sibs, parent–offspring, or unrelated individuals. Complex pedigree structures and high levels of relatedness and inbreeding, however, are typical for populations in need of conservation. There is a need for relatedness estimators that can be applied to fragmented populations, where interest is in both within and between subpopulation relatedness. Development of such estimators is not merely a statistical issue, but needs a connection with population genetic concepts such as drift. Furthermore, the utility of using estimators in conservation programs, with the aim to maximize the amount of additive genetic variance conserved, has not been investigated to our knowledge. Hence, more knowledge is needed of the usefulness of relatedness estimators to support conservation strategies, such as determining which individuals are genetically important.
In this article we introduce estimators that are based on the relationship between coancestry and relatedness, which holds irrespective of inbreeding. In total, we compare eight estimators expressing relatedness on a continuous scale, with a focus on supporting conservation strategies. Monte Carlo simulations produced populations with both pedigree and marker data. Behavior of the estimators is studied for alternative populations, differing in (a) the number of alleles per locus in the base generation, (b) the number of loci used, (c) the average relatedness compared to the base population, (d) the population structure (either panmictic or structured), and (e) the size of a subset of individuals selected to maximize the amount of genetic variation conserved. Relatedness was estimated using simulated marker data and analyzed against pedigree relatedness, using both statistical and diversity criteria.
METHODS
Here we describe (1) the relatedness estimators considered, (2) the simulated population structures in which estimators will be tested, and (3) the criteria used to assess quality of the estimators.
Relatedness estimators:
Eight relatedness estimators are compared, which we divide into three categories (Table 1). The first category is based on the relationship between additive genetic relatedness (r), population genetic coancestry (f, also known as “kinship”; Falconer and Mackay 1996), and molecular coancestry (fM) (Jacquard 1983; Lynch 1988; Toro et al. 2002) and consists of both existing and new estimators. The second category is based on the relationship between additive genetic relatedness and two-gene and four-gene coefficients of identity in “noninbred” populations and consists of the estimators of Lynch and Ritland (1999) and Wang (2002). The third category consists of the estimator of Queller and Goodnight (1989). All estimators express relatedness on a continuous scale.
TABLE 1.
Estimators used
| Abbreviation | Full name/reference | Equation | Category |
|---|---|---|---|
| fM | Molecular coancestry | 4 | 1 |
| UCS | Unweighted corrected similarity | 5 | 1 |
| WCS | Weighted corrected similarity | 6, 7 | 1 |
| WEDS | Weighted equal drift similarity | 6, 7, 8 | 1 |
| L&R | Lynch and Ritland (1999) | 13, 14 | 2 |
| Wang | Wang (2002)a | — | 2 |
| Q&G | Queller and Goodnight (1989) | 15, 16 | 3 |
See http://www.zoo.cam.ac.uk/ioz/software.htm for software.
Milligan (2003) presented a maximum-likelihood estimator for noninbred populations. In “inbred” populations, however, finding the maximum-likelihood value is computationally demanding because many modes of identity by descent (IBD) occur (see Table 1 in Milligan 2003). We did, therefore, not investigate maximum-likelihood estimators.
Category 1:
Estimators based on coancestry:
By definition, additive genetic relatedness (r) between diploid individuals equals twice the coefficient of coancestry (f, also known as kinship; r = 2f) (Malécot 1948; Falconer and Mackay 1996). Thus, conservation strategies based on coancestry are equivalent to strategies based on relatedness. Coancestry of two individuals is the probability that two alleles drawn randomly, one from each individual, are IBD, indicating that they descend from a common ancestor (Falconer and Mackay 1996). Coancestry and relatedness are expressed relative to a so-called base population, in which all alleles are defined as being not IBD, so that coancestry in the base population is zero by definition (Falconer and Mackay 1996; Lynch and Walsh 1998). Alleles that are molecularly identical in the base population are referred to as alike in state (AIS). Thus, in any generation, the proportion of alleles AIS is equal to expected homozygosity in the base population. When pedigrees are known, the founder generation is commonly used as the base population, so that relatedness among founders is zero by definition. In principle, base populations serve merely as a reference point, and the choice of the base population is arbitrary. However, not all choices are genetically meaningful and theoretically correct, particularly in structured populations (see discussion).
The new estimators presented in this article are based on the approach of Eding and Meuwissen (2003). Eding and Meuwissen (2003) developed estimators of between-population coancestry, using observations on molecular similarity between and within populations, in which case the definition of the base population is more obvious. We modify estimators of Eding and Meuwissen (2001, 2003) to estimate coancestries between individuals instead of between populations.
First we describe the theoretical background of estimators based on coancestry. Estimators based on coancestry make use of the molecular similarity index (Sxy,l), which refers to a single locus l in a pair of individuals xy and is defined as the probability that two marker alleles drawn from two individuals are molecularly identical (Jacquard 1983; Caballero and Toro 2000; Toro et al. 2002). In the following, Sxy,l is referred to as “similarity.” For locus l, similarity between individual x having alleles a and b and individual y having alleles c and d is defined as
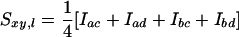 |
(1) |
(Li and Horvitz 1953), where indicator Iac is one when allele a of individual x is identical to allele c of individual y, and zero otherwise, etc. Similarity takes values of 0, 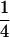 ,
, 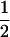 , or 1. Values of
, or 1. Values of 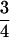 do not occur, because the fourth indicator must be equal to 1 when the previous three indicators are equal to 1. Similarities of
do not occur, because the fourth indicator must be equal to 1 when the previous three indicators are equal to 1. Similarities of 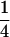 require at least three distinct alleles and therefore do not occur at biallelic loci.
require at least three distinct alleles and therefore do not occur at biallelic loci.
Similarity will vary between pairs of individuals and will be partly due to alleles that are IBD but also due to alleles AIS. When sl denotes the probability that two alleles at locus l are AIS, then expected similarity between individuals x and y at locus l is
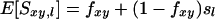 |
(2a) |
(Lynch 1988), where sl is the average similarity at locus l in the base population, and fxy is the coancestry between individuals x and y expressed relative to this base population. Equation 2a may be interpreted as the probability that alleles are IBD (fxy) plus the probability that they are not IBD but AIS [(1 − fxy)sl]. Equation 2a holds irrespective of inbreeding or random mating. Rearrangement of Equation 2a gives a convenient form resembling Wright's F-statistics (Wright 1978):
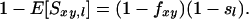 |
(2b) |
A so-called “method of moments estimator” of coancestry is obtained by rearranging Equation 2a, substituting expected similarity by observed similarity, and averaging over L loci, which gives
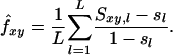 |
(3) |
Multiplying Equation 3 by a factor of 2 yields a relatedness estimator (see Ritland 1996).
Equation 3 shows that a value for sl is needed for each locus. Because allele frequencies in the base population are usually unknown, sl needs to be estimated, which involves two problems. First, when the average level of AIS is estimated incorrectly, the average estimated relatedness of the current population will be biased. The observed average similarity and the estimated probability of alleles AIS together implicitly define the base population. The lower the estimated AIS, the further back in time this base population is set, and the higher the average estimated relatedness of the current population. Vice versa, an overestimation of AIS will result in underestimating relatedness (Toro et al. 2003). For example, when the base population is set equal to the current population, which is done implicitly when sl is calculated from current allele frequencies assuming random mating, IBD between all pairs of individuals will be −1/2N on average, resulting in negative estimates of relatedness for many pairs of individuals. Negative estimates are difficult to interpret because relatedness is defined as twice the probability that alleles are IBD. The second problem is that, although probabilities of alleles AIS differ per locus, expected coancestry for a pair of individuals is equal at all neutral loci by definition. Ideally, this should be taken into account when estimating sl for each locus. In the following we describe estimators based on Equations 2 and 3, in order of increasing complexity.
Molecular coancestry (fM):
Toro et al. (2002; 2003) used fM as an estimator of coancestry. Molecular coancestry ignores alleles AIS by setting sl = 0 for all loci, so that estimated relatedness equals the average similarity over loci multiplied by a factor of two:
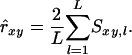 |
(4) |
When founder alleles are unique, 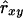 would be an unbiased estimator of relatedness.
would be an unbiased estimator of relatedness.
Unweighted corrected similarity:
For the unweighted corrected similarity (UCS) estimator, sl is estimated assuming that all distinct alleles in the current population had equal frequencies (pl) in the base population, pl = 1/nl, where nl is the number of distinct alleles at locus l observed in the current population, which is often referred to as allelic diversity (ADl) (Fernandez et al. 2005). Consequently, the probability that alleles are AIS equals 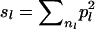 = 1/nl. Estimates for UCS were obtained by substituting sl = 1/nl into Equation 3 and multiplying by a factor of 2, giving
= 1/nl. Estimates for UCS were obtained by substituting sl = 1/nl into Equation 3 and multiplying by a factor of 2, giving
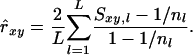 |
(5) |
The assumption that sl = 1/nl ignores differences in allele frequencies among loci and consequently does not necessarily satisfy the condition that expected coancestry of a pair of individuals is equal at all loci. However, it is simple to apply and may turn out to be robust.
Weighted corrected similarity:
Allele frequencies vary among loci. Consequently, different loci contribute differently to the estimated relatedness, and the variance of observed similarity around its expectation (Equation 2a) varies among loci. The weighted corrected similarity (WCS) estimator uses weights (wl) to optimize the impact of loci on estimated relatedness,
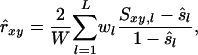 |
(6) |
where W is the sum of weights wl over all loci and 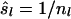 . When variance of an estimator varies among observations, using reciprocals of the variance as weights minimizes the mean squared error of the estimate (Lynch and Ritland 1999; Eding and Meuwissen 2001). The variance of estimated coancestry is proportional to
. When variance of an estimator varies among observations, using reciprocals of the variance as weights minimizes the mean squared error of the estimate (Lynch and Ritland 1999; Eding and Meuwissen 2001). The variance of estimated coancestry is proportional to 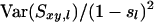 (Equation 3). An exact expression for Var(Sxy,l) follows from the probabilities of occurrence of each similarity value and is given in the appendix. A simple approximation for Var(Sxy,l) is obtained by assuming that Iac through Ibd in Equation 1 are mutually independent, in which case Var(Sxy,l) is proportional to Var(I..) and we can use Var(I) to obtain weights. Since I. is binomial, the reciprocal of weight wl for locus l having nl alleles equals
(Equation 3). An exact expression for Var(Sxy,l) follows from the probabilities of occurrence of each similarity value and is given in the appendix. A simple approximation for Var(Sxy,l) is obtained by assuming that Iac through Ibd in Equation 1 are mutually independent, in which case Var(Sxy,l) is proportional to Var(I..) and we can use Var(I) to obtain weights. Since I. is binomial, the reciprocal of weight wl for locus l having nl alleles equals
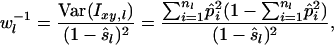 |
(7) |
where 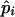 is the estimated allele frequency of allele i at locus l in the current population. Preliminary results showed that differences between exact or approximate weights were negligible. Values presented in results, therefore, are obtained using approximate weights (Equation 7), which are much simpler than exact weights.
is the estimated allele frequency of allele i at locus l in the current population. Preliminary results showed that differences between exact or approximate weights were negligible. Values presented in results, therefore, are obtained using approximate weights (Equation 7), which are much simpler than exact weights.
Weighted equal drift similarity:
The UCS and WCS estimators use the number of distinct alleles to estimate sl for each locus, which does not fully guarantee that coancestry between a pair of individuals is equal at all loci. The weighted equal drift similarity (WEDS) estimator solves this problem by calculating sl so that the increase in coancestry since the base population is equal at all loci. The WEDS estimator starts by setting sl = 0 for the locus having the lowest expected similarity (Smin) given its allele frequencies, 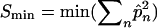 , where n is the number of alleles. This defines the base population such that estimated sl will be nonnegative for all loci. The next step is to calculate sl at other loci as the expected similarity at those loci, corrected with the same amount Smin of coancestry. It follows from Equation 2b, that for all loci
, where n is the number of alleles. This defines the base population such that estimated sl will be nonnegative for all loci. The next step is to calculate sl at other loci as the expected similarity at those loci, corrected with the same amount Smin of coancestry. It follows from Equation 2b, that for all loci
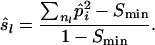 |
(8) |
Finally, coancestries are estimated using Equations 6 and 7.
Weighted log-linear model:
Eding and Meuwissen (2003) estimated average coancestries within and between populations, by using the logarithm of Equation 2b, which yields a linear model. Here we applied their approach on the individual level. In contrast to the previous estimators, this procedure obtains 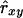 and
and 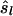 simultaneously. However, the weighted log-linear model (WLM) estimator required substantial computing time and yielded poor results (not presented), which seemed to originate from the log-transformation when Sxy,l = 1.
simultaneously. However, the weighted log-linear model (WLM) estimator required substantial computing time and yielded poor results (not presented), which seemed to originate from the log-transformation when Sxy,l = 1.
Category 2:
Estimators based on two-gene and four-gene coefficients of identity:
The second category of estimators is based on the relationship between relatedness and two-gene and four-gene coefficients of identity in “noninbred” populations (Lynch and Ritland 1999),
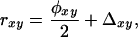 |
(12) |
where φxy is the probability that, at a certain locus, a single allele in individual x is IBD to a single allele in individual y, and Δ is the probability that both alleles in individual x are IBD to both alleles in individual y (φ and Δ are denoted Δ8 and Δ7 in Lynch and Walsh 1998). In the following, we summarize the estimators of Lynch and Ritland (1999) and Wang (2002), which are based on Equation 12. Beware of a typo in Lynch and Ritland (1999) and Wang (2002), which reads φ = 0.25 instead of φ = 0.5 for half-sibs (Falconer and Mackay 1996).
Lynch and Ritland:
Lynch and Ritland (1999) proposed an asymmetrical estimator that is now commonly used. Their estimator is based on regression of genotype probabilities of the one individual on the genotype of the other individual of a pair. A symmetrical multilocus estimator is obtained as the weighted arithmetic mean over loci, taking the average of the reciprocal multilocus estimates,
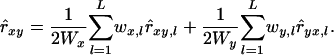 |
(13) |
The locus-specific estimator 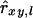 has as denominator
has as denominator 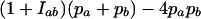 , where pa is the frequency of allele a at locus l and, as in Equation 1, Iab = 1 when alleles a and b of individual x are identical and 0 otherwise. Consequently, a division by 0 occurs when pa = pb = 0.5 and Iab = 0, and the Lynch and Ritland (L&R) estimator performs poorly at biallelic loci due to rounding errors at allele frequencies close to 0.5. We solved this problem by combining the product
, where pa is the frequency of allele a at locus l and, as in Equation 1, Iab = 1 when alleles a and b of individual x are identical and 0 otherwise. Consequently, a division by 0 occurs when pa = pb = 0.5 and Iab = 0, and the Lynch and Ritland (L&R) estimator performs poorly at biallelic loci due to rounding errors at allele frequencies close to 0.5. We solved this problem by combining the product 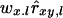 in Equation 13 into a single term, yielding the estimator
in Equation 13 into a single term, yielding the estimator
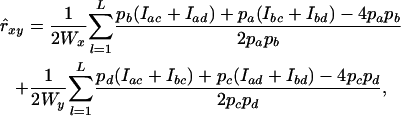 |
(14) |
where Wx and Wy are the sums of all weighting factors wx,l and wy,l, respectively. (See Lynch and Ritland 1999 for details). Following Toro et al. (2002), we used estimated allele frequencies in Equation 14.
Wang (2002):
Using Equation 12, Wang (2002) developed an estimator that takes into account the uncertainty of estimated allele frequencies. Briefly, the approach of Wang consists of the following. First, for a single locus, joint probabilities of observing a pair of genotypes are expressed as a function of φ and Δ. Subsequently, resulting expressions are solved for φ and Δ, by treating genotype probabilities as known observations. Next, solutions for φ and Δ are substituted into Equation 12, giving an estimate for rxy,l. Finally, a multilocus estimate is obtained by using weighted least squares, where weights are obtained assuming that φ and Δ = 0. Further details are in Wang (2002). We implemented Wang's estimator using his Fortran code available at http://www.zoo.cam.ac.uk/ioz/software.htm.
Category 3:
The estimator of Queller and Goodnight:
Queller and Goodnight (1989) (Q&G) developed an estimator that was originally designed for estimating average relatedness between populations, instead of individuals. However, it can be modified to obtain a pairwise asymmetric estimator for individuals, which is commonly used nowadays (Lynch and Ritland 1999; Toro et al. 2002; Wang 2002; Milligan 2003). With Q&G, relatedness of individual x with individual y at locus l is
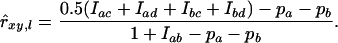 |
(15) |
A number of alternative implementations of Equation 15 are possible. We obtained relatedness by averaging the reciprocal estimates over L loci,
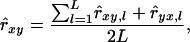 |
(16) |
where L is the number of loci. For biallelic loci, Equation 15 is undefined when individual x is heterozygous, because it results in a division by zero. The Q&G estimator was therefore omitted with biallelic loci.
Simulated populations:
To compare estimators, populations with several discrete generations were simulated. The following two sections describe the standard population and five alternatives. Table 2 summarizes population parameters.
TABLE 2.
Simulated standard population and alternatives
| Alleles | Loci | Generations | Structureb | Capacitye | |
|---|---|---|---|---|---|
| Alternativea | 2 | 10 | 5 | Panmictic | 100 |
| 5 | 20 | 10 | Structured Ac | 10 | |
| 2–8 | 50 | 15 | Structured Bd | ||
| 10 | 100 | 20 | |||
| 2–18 | |||||
| Unique |
Values for the standard population are in italics.
Input parameters were varied one at a time, and other parameters were as in the standard population.
The panmictic population had 10 male and 50 female parents until generation 10. The structured population had 10 male and 50 female parents until generation 5, after which it split into two subpopulations.
Ninety individuals were sampled from the subpopulation bred from 10 male and 50 female parents, and 10 were sampled from a subpopulation bred from 8 male and 40 female parents.
Ten individuals were sampled from the subpopulation bred from 10 male and 50 female parents, and 90 were sampled from a subpopulation bred from 8 male and 40 female parents.
Capacity denotes the number of individuals that can be conserved.
Standard population:
The standard population was panmictic and was bred from a base generation of 10 male and 50 female founders. Twenty marker loci were simulated. Each locus had a random number of alleles (n), ranging from 2 through 8. At each locus, alleles were sampled with a probability of 1/n for each allele, so that, on average, alleles at a particular locus had the same frequency in the base generation. Alleles were codominant, autosomal, unlinked, neutral, without mutation, and followed Mendelian inheritance.
Ten discrete generations of 400 individuals were bred, using random mating and selection of 10 male and 50 female individuals as parents of the next generation. The last generation consisted of 100 individuals, which were genotyped for all 20 loci. Relatedness between all pairs of individuals was estimated from the marker data, for each of the estimators described above. In addition, relatedness between individuals was calculated from the pedigree, using the tabular method (Emik and Terrill 1949), and was considered to be the true value. Finally, quality of estimators was assessed by comparing estimated with pedigree relatedness, using both statistical and diversity criteria (see below).
Alternatives:
The effect of the following five variables on quality of estimators was investigated (Table 2): (a) the number of alleles per locus in the base generation; (b) the number of loci; (c) the average level of relatedness in the current generation, by varying the number of generations simulated; (d) a structured population; and (e) a limitation to the number of individuals that could be used in a conservation program, which was either all 100 or only the genetically most important 10 (see Diversity criterion). Alternative d was included to investigate quality of estimators in structured populations. The structured population had 10 male and 50 female parents until generation 5, after which it split into two subpopulations, of which one was bred with 8 male and 40 female parents and the other with 10 male and 50 female parents. Two final generations of 100 individuals were simulated, and 90 individuals were sampled from one and 10 from the other population or vice versa. Alternative e resembles the situation in practice, where conservation funds are limited. For each alternative, one parameter was varied at a time, and other parameters were as in the standard population. One hundred replicates were run per alternative, and results were averaged over replicates.
Criteria:
Two types of criteria were used: (1) statistical criteria that compared estimated with pedigree relatedness and (2) a diversity criterion that measures the genetic variation conserved by using an estimator in conservation strategies.
Statistical criteria:
Four statistical criteria were used: (1) the average bias, being the difference between average estimated relatedness and average pedigree relatedness (bias); (2) the regression coefficient of estimated relatedness on pedigree relatedness (β1), which is a measure for bias in the estimated differences in relatedness among pairs of individuals; (3) the regression coefficient of pedigree relatedness on estimated relatedness (β2), which indicates whether estimated relatedness yields an “unbiased” prediction of pedigree relatedness, which is important in practice because conservation decisions are based on the estimates, not on the true values; and (4) the correlation between estimated and pedigree relatedness (ρ), which measures the proportion of the variance in pedigree relatedness explained by the estimator. Relatedness of individuals with themselves was excluded from the calculation of these criteria.
Diversity criterion:
Although statistical criteria are informative for the quality of estimators, they do not directly reveal the amount of genetic diversity conserved by using an estimator in practice. In addition to statistical criteria, therefore, we develop a criterion that evaluates the genetic diversity conserved when selection decisions are based on estimated relatedness.
In this section we argue that relatedness is a key factor in conservation. An important aspect in conservation genetics is to minimize inbreeding levels and maximize genetic diversity (Ballou and Lacy 1995; Frankham et al. 2002). Here we interpret genetic diversity as additive genetic variance, for the following reasons. Fisher's fundamental theorem of natural selection (Fisher 1958), stating that the rate of increase in fitness equals the additive genetic variance of relative fitness, shows that adaptive potential of populations should be measured by their additive genetic variance for fitness. In random mating populations, additive genetic variance in generation t for any trait equals
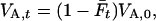 |
(17) |
where 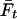 is the average inbreeding level in the population in generation t, measured relative to the base generation, and VA,0 is the additive genetic variance in the base generation (Falconer and Mackay 1996). With random mating, the inbreeding level in the next generation,
is the average inbreeding level in the population in generation t, measured relative to the base generation, and VA,0 is the additive genetic variance in the base generation (Falconer and Mackay 1996). With random mating, the inbreeding level in the next generation, 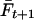 , equals the average coancestry of the current population and thus half the average relatedness of the current population (r = 2f). Thus, maximizing genetic diversity and minimizing inbreeding in generation t + 1 is identical to minimizing relatedness in generation t. In conclusion, therefore, conservation decisions within a species should aim at minimizing the average additive genetic relatedness in that species. Consequently, our diversity criterion measures the efficiency of estimators when the objective is to minimize average relatedness in a group of individuals.
, equals the average coancestry of the current population and thus half the average relatedness of the current population (r = 2f). Thus, maximizing genetic diversity and minimizing inbreeding in generation t + 1 is identical to minimizing relatedness in generation t. In conclusion, therefore, conservation decisions within a species should aim at minimizing the average additive genetic relatedness in that species. Consequently, our diversity criterion measures the efficiency of estimators when the objective is to minimize average relatedness in a group of individuals.
With random mating, average relatedness in the next generation is given by Meuwissen (1997),
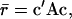 |
(18) |
where c is a vector of proportional contributions of individuals to the next generation, so that elements of c sum to one, and A is a matrix of additive genetic relatedness between all individuals, including relatedness of individuals with themselves. Average relatedness among parents, and thus the inbreeding level in the next generation, can be decreased or increased by varying the contributions of individuals (c). Thus average relatedness can be minimized by finding an optimum contribution vector 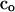 that minimizes c′Ac, which is given by
that minimizes c′Ac, which is given by
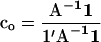 |
(19) |
(Meuwissen 1997; Eding et al. 2002), where 1 is a column vector of ones. The matrix of additive genetic relationships has to be estimated from marker data. The amount of genetic diversity conserved by using estimated optimal contributions ( 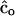 ) will depend on the estimator used. To obtain estimated optimum contributions, we substituted the matrix of pedigree relatedness by the matrix of estimated relatedness (
) will depend on the estimator used. To obtain estimated optimum contributions, we substituted the matrix of pedigree relatedness by the matrix of estimated relatedness ( 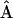 ) in Equation 19. When negative contributions were obtained, the most negative contribution was set to zero and optimal contributions were recalculated, until all contributions were nonnegative. In alternative e the lowest contribution was set to zero and optimal contributions were recalculated, until all contributions were nonnegative or only 10 contributions were left.
) in Equation 19. When negative contributions were obtained, the most negative contribution was set to zero and optimal contributions were recalculated, until all contributions were nonnegative. In alternative e the lowest contribution was set to zero and optimal contributions were recalculated, until all contributions were nonnegative or only 10 contributions were left.
We evaluated the result on two scales. On the first scale, the diversity criterion equals the proportion of additive genetic variance conserved relative to the base generation,
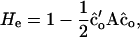 |
(20) |
which is derived by combining Equations 17 and 18. Note that, in Equation 20, A refers to relatedness calculated from the pedigree. With random mating, He equals expected heterozygosity in a population with estimated optimum contributions of individuals, expressed as a proportion of heterozygosity in the base generation. On the second scale, the diversity criterion equals the number of founders (Nge) that would have the same average coancestry (and thus the same additive genetic variance) as the population obtained using estimated optimum contributions. Average coancestry among N founders = 1/(2N), so that
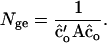 |
(21) |
Caballero and Toro (2000) referred to Nge as the number of founder genome equivalents. Equation 21 is an expression on the scale of effective population size, since it equals 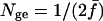 .
.
In contrast to the statistical criteria, relatedness of individuals with themselves was included in 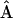 and was estimated by using y = x in the relevant expressions for
and was estimated by using y = x in the relevant expressions for 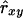 .
.
RESULTS
Comparison of estimators on the standard population:
Table 3 gives results for the standard population. Average pedigree relatedness in the simulated standard population in the 10th generation was 0.282. With fM and Q&G, average estimated relatedness deviated considerably from the pedigree average, as reflected by bias. Bias depends on the definition of the base population, which is essentially arbitrary (see discussion). Bias, therefore, is not an important quality criterion and is not presented further.
TABLE 3.
Comparison of estimators in the standard population
| Estimator | Biasa | β1b | β2c | ρd | Hee | Ngef |
|---|---|---|---|---|---|---|
| Pedigree | 0 | 1 | 1 | 1 | 0.86 | 3.69 |
| fM | 0.43 | 0.76 | 0.40 | 0.55 | 0.84 | 3.10 |
| UCS | −0.02 | 1.02 | 0.27 | 0.52 | 0.84 | 3.08 |
| WCS | −0.08 | 1.04 | 0.32 | 0.57 | 0.84 | 3.17 |
| WEDS | 0.10 | 0.94 | 0.35 | 0.57 | 0.84 | 3.15 |
| L&R | −0.24 | 1.01 | 0.35 | 0.60 | 0.85 | 3.33 |
| Wang | −0.28 | 1.16 | 0.28 | 0.57 | 0.84 | 3.17 |
| Q&G | −0.96 | 1.66 | 0.15 | 0.50 | 0.82 | 2.82 |
Results are averages of 100 replicates. Standard errors of results were ≤0.01.
Estimated relatedness minus pedigree relatedness.
The regression of estimated relatedness on pedigree relatedness.
The regression of pedigree on estimated relatedness.
The correlation between estimated relatedness and pedigree relatedness.
The expected heterozygosity with estimated optimum contributions, Equation 19.
The number of founder genome equivalents with optimum contributions, Equation 20.
The regression of estimated relatedness on pedigree relatedness (β1) was close to one for most estimators, except for fM and Q&G. Results indicate a relationship between bias and β1, showing that β1 is underestimated when bias is positive. The fM estimator performed best for the regression of pedigree on estimated relatedness (β2), but in all cases β2 was substantially less than one. The correlation between estimated and pedigree relatedness (ρ) ranged from 0.50 (Q&G) to 0.60 (L&R), indicating that differences between estimators are relatively small. When pedigree information was known, the use of optimum contributions maintained 3.69 founder genome equivalents (Nge). Application of the estimators maintained between 2.82 (Q&G) and 3.33 (L&R) founder genome equivalents, which are 76 and 90% of the maximum value obtained with known pedigree. When quality of estimators is judged by the correlation and the number of founder genome equivalents, the following order is obtained: L&R performs best, followed by the group of WCS, WEDS, and Wang; next comes fM; then UCS; and finally Q&G.
Number of alleles:
Table 4 summarizes results for different numbers of alleles per locus. The number of distinct alleles in the current generation was reduced by almost 90% when alleles in the base generation were unique, whereas no reduction was observed when the base generation had only 2 alleles per locus. As expected, the correlation between estimated and pedigree relatedness increased with the number of alleles. The benefit of increasing the number of alleles was smaller when there were already many alleles. On average, the correlation increased by 50% when the number alleles increased from 2 to 5, whereas the correlation increased by 16% when the number of alleles increased from 5 to 10. The L&R estimator had the highest correlation for all schemes considered. WEDS, WCS, and Wang showed nearly identical correlations.
TABLE 4.
Correlation between pedigree and estimated relatedness for a varying number of alleles in base populations
| No. alleles: | 2 | 2–8 | 5 | 2–18 | 10 | 120 | |
|---|---|---|---|---|---|---|---|
| Average no.: | 2.00 | 4.56 | 4.71 | 6.71 | 7.45 | 13.3 | |
| Estimator | % alleles left: | 100 | 91 | 94 | 67 | 75 | 11 |
| fM | 0.37 | 0.54 | 0.57 | 0.62 | 0.66 | 0.77 | |
| UCS | 0.37 | 0.52 | 0.57 | 0.60 | 0.65 | 0.77 | |
| WCS | 0.37 | 0.56 | 0.58 | 0.65 | 0.67 | 0.79 | |
| WEDS | 0.38 | 0.57 | 0.59 | 0.65 | 0.67 | 0.79 | |
| L&R | 0.41 | 0.63 | 0.65 | 0.71 | 0.73 | 0.81 | |
| Wang | 0.35 | 0.57 | 0.58 | 0.65 | 0.67 | 0.79 | |
| Q&G | —a | 0.49 | 0.52 | 0.60 | 0.65 | 0.78 |
Results are averages of 100 replicates. Standard errors of results were ≤0.01.
Q&G is not applicable to biallelic loci.
It follows from Tables 3 and 4 that Q&G and UCS had the poorest results, whereas WCS and WEDS had nearly identical results. This trend was observed in all alternatives. No further results, therefore, are presented for UCS, Q&G, and WCS.
Number of loci:
Table 5 summarizes results for schemes with different numbers of loci. The number of loci did not affect the regression coefficient of estimated relatedness on pedigree relatedness (β1). In contrast, the regression coefficient of pedigree on estimated relatedness (β2) increased considerably when the number of loci increased, but still deviated clearly from unity. The correlation increased by ∼30% when going from 10 to 20 loci, by ∼30% when going from 20 to 50 loci, and by ∼14% when going from 50 to 100 loci. The L&R estimator showed the highest correlation and maintained the most genetic variation, whereas fM showed the lowest correlation and maintained the least genetic variation. WEDS performed slightly better than Wang, but differences were small.
TABLE 5.
Comparison of estimators for a varying number of loci
| Estimator | β1a | β2b | ρc | Nged | |
|---|---|---|---|---|---|
| 10 loci | Pedigree | 1 | 1 | 1 | 3.70 |
| fM | 0.76 | 0.23 | 0.42 | 2.86 | |
| WEDS | 0.92 | 0.21 | 0.44 | 2.93 | |
| L&R | 1.04 | 0.23 | 0.49 | 3.34 | |
| Wang | 1.15 | 0.17 | 0.43 | 2.83 | |
| 20 loci | Pedigree | 1 | 1 | 1 | 3.69 |
| fM | 0.76 | 0.39 | 0.55 | 3.10 | |
| WEDS | 0.94 | 0.35 | 0.57 | 3.17 | |
| L&R | 1.03 | 0.39 | 0.63 | 3.50 | |
| Wang | 1.16 | 0.28 | 0.57 | 3.06 | |
| 50 loci | Pedigree | 1 | 1 | 1 | 3.67 |
| fM | 0.75 | 0.68 | 0.72 | 3.30 | |
| WEDS | 0.95 | 0.58 | 0.74 | 3.35 | |
| L&R | 1.02 | 0.60 | 0.78 | 3.55 | |
| Wang | 1.16 | 0.46 | 0.73 | 3.26 | |
| 100 loci | Pedigree | 1 | 1 | 1 | 3.70 |
| fM | 0.76 | 0.90 | 0.82 | 3.44 | |
| WEDS | 0.97 | 0.73 | 0.84 | 3.48 | |
| L&R | 1.02 | 0.73 | 0.86 | 3.59 | |
| Wang | 1.16 | 0.59 | 0.83 | 3.39 |
Results are averages of 100 replicates. Standard errors of results were ≤0.01.
The regression of estimated relatedness on pedigree relatedness.
The regression of pedigree on estimated relatedness.
The correlation between estimated relatedness and pedigree relatedness.
The number of founder genome equivalents with optimum contributions, Equation 20.
Average level of relatedness:
As expected, an increase in the number of generations increased pedigree relatedness and decreased the number of alleles surviving from the base to the current generation. Performance of estimators decreased in correspondence with the decreasing number of alleles (results not shown). Apart from an effect via the number of alleles, there was no effect of the level of relatedness on performance of estimators.
Structured populations:
Table 6 summarizes results for the structured population for two sampling schemes. In scheme A, 90 individuals were sampled from the subpopulation bred from 10 and 50 parents and 10 from the subpopulation from 8 and 40 bred parents. In scheme B, the sampling of individuals was reversed. With scheme A, average relatedness was 0.26 and average inbreeding was 0.13. On average, correlations between estimated and pedigree relatedness had the same level as in the standard population. When judged by the correlation, estimators performed equally well. When judged by the number of founder genome equivalents, however, Wang showed poorest results and L&R highest, indicating that ranking of estimators depends on the criterion used. With scheme B, average relatedness was 0.38 and average inbreeding was 0.19. In contrast to the panmictic standard population and scheme A, L&R had the lowest correlation and lowest founder genome equivalents, whereas WEDS had the highest founder genome equivalents.
TABLE 6.
Comparison of estimators in structured populations
| Structured Aa | Structured Bb | |||||||
|---|---|---|---|---|---|---|---|---|
| Estimator | β1c | β2d | ρe | Ngef | β1c | β2d | ρe | Ngef |
| Pedigree | 1 | 1 | 1 | 4.23 | 1 | 1 | 1 | 3.82 |
| fM | 0.75 | 0.43 | 0.57 | 3.52 | 0.74 | 0.65 | 0.70 | 3.22 |
| WEDS | 0.90 | 0.38 | 0.58 | 3.58 | 0.86 | 0.54 | 0.68 | 3.25 |
| L&R | 0.88 | 0.39 | 0.59 | 3.84 | 0.65 | 0.47 | 0.55 | 2.96 |
| Wang | 1.15 | 0.31 | 0.59 | 3.45 | 1.13 | 0.42 | 0.69 | 3.13 |
Results are averages of 200 replicates (instead of 100). Standard errors of results were ≤0.01 (except for Nge).
Ninety individuals were sampled from the subpopulation bred from 10 male and 50 female parents, and 10 were sampled from a subpopulation bred from 8 male and 40 female parents.
Ten individuals were sampled from the subpopulation bred from 10 male and 50 female parents, and 90 were sampled from a subpopulation bred from 8 male and 40 female parents.
The regression of estimated reletedness on pedigree relatedness.
The regression of pedigree on estimated relatedness.
The correlation between estimated and pedigree relatedness.
The number of founder genome equivalents with optimum contributions, Equation 20. Standard errors of results were ≤0.02.
Use in conservation:
Table 7 shows the number of founder genome equivalents conserved in sets of either 10 or all 100 individuals, having either optimal or equal contributions of individuals, and for the panmictic standard population or a structured population.
TABLE 7.
Number of founder genome equivalents in sets of either 10 or 100 individuals, in a panmictic or structured population
| Panmictic | Structured Aa | Structured Bb | ||||
|---|---|---|---|---|---|---|
| Individuals in set | 100 | 10 | 100 | 10 | 100 | 10 |
| Pedigree | 3.69 | 3.17 | 4.23 | 3.52 | 3.82 | 3.39 |
| Equalb | 3.56c | 2.78d | 3.85c | 2.89d | 2.61c | 2.17d |
| fM | 3.12 | 2.88 | 3.52 | 3.20 | 3.22 | 3.03 |
| WEDS | 3.17 | 2.90 | 3.58 | 3.21 | 3.25 | 3.02 |
| L&R | 3.51 | 2.91 | 3.84 | 3.13 | 2.96 | 2.62 |
| Wang | 3.07 | 2.87 | 3.45 | 3.18 | 3.13 | 2.97 |
Results are averages of 200 replicates (instead of 100). Standard errors of results were ≤0.02.
Ninety individuals were sampled from the subpopulation bred from 10 male and 50 female parents, and 10 were sampled from a subpopulation bred from 8 male and 40 female parents.
Ten individuals were sampled from the subpopulation bred from 10 male and 50 female parents, and 90 were sampled from a subpopulation bred from 8 male and 40 female parents.
All 100 individuals have equal contributions to the set.
Ten random individuals have equal contributions to the set.
In the standard population, the number of founder genome equivalents conserved using optimal contributions calculated from pedigree relatedness was only a little higher than when using equal contributions (3.69 vs. 3.56). In standard populations, variation in relatedness among pairs of individuals is relatively small, and the benefit of using optimum contributions is limited when the set contains all individuals. Surprisingly, when all 100 individuals were included, the use of optimum contributions based on estimated relatedness conserved fewer founder genomes than equal contributions did. Hence, conservation strategies based on estimated relatedness of limited accuracy can actually reduce the genetic variation conserved, instead of increasing it. When sets consisted of only 10 individuals, sets of optimum contributions always had higher founder genome equivalents than sets with equal contributions.
In the structured population, the number of founder genome equivalents conserved using optimal contributions calculated from pedigree relatedness was higher than when using equal contributions with scheme A (4.23 vs. 3.85) and substantially higher with scheme B (3.82 vs. 2.61), indicating that optimizing contributions is more important in structured than in standard populations When only 10 individuals were included in the set, the use of optimal contributions calculated from estimated relatedness always conserved more founder genomes then the use of equal contributions. Scheme B always conserved more founder genomes, irrespective of the number of individuals in the set, illustrating the importance of the sampling procedure.
Differences between estimators are in agreement with results in Tables 3–6. The L&R estimator performed best in the standard population, whereas fM and WEDS performed best in the panmictic structured population.
DISCUSSION
We investigated quality of relatedness estimators in simulated populations with many generations of pedigree. The estimators UCS and Q&G showed lowest accuracy. Differences among fM, WCS, WEDS, Wang, and L&R were relatively small, and ranking of estimators depended on the population structure. In contrast to previously published results (Wang 2002), the L&R estimator clearly performed better than the Wang estimator in panmictic populations. The WEDS and fM estimators performed best in structured populations. The difference between UCS and WCS showed that weighting the impact of loci plays a significant role in relatedness estimation. Average level of relatedness in the population did not affect quality of estimators. When interest is not in conservation, but merely in point estimates for relatedness between pairs of individuals, quality of estimators may be judged by the correlation between true and estimated relatedness. When judged by the correlation, L&R performs best in panmictic populations and WEDS, fM, and Wang in structured populations. Fernandez et al. (2005) argued that minimizing simple molecular coancestry (fM) is the optimum way to maximize diversity, which would imply that other relatedness estimators are redundant. Our results show, however, that there is a clear benefit of using more sophisticated relatedness estimators (see, e.g., L&R vs. fM in Table 5). In structured populations, sets of estimated optimum contributions had in most cases more diversity than sets of equal contributions of individuals. Surprisingly, in panmictic populations, sets of optimum estimated contributions sometimes had less diversity than sets with equal contributions of individuals, showing that estimates of relatedness can be useful in conservation programs, but should be used with caution.
L&R vs. Wang:
In contrast to results presented by Wang (2002), the L&R estimator performed consistently better than the Wang estimator in panmictic populations, irrespective of the numbers of alleles and loci. We identified three reasons for this discrepancy:
We have used a modified version of the L&R estimator that avoids the numerical rounding errors that may occur when pa = pb = 0.5 or when estimates “blow up” when they approach this value. Wang (2002) noted this problem, but did not correct for it. We observed that the L&R estimator improved considerably when calculating the product of relatedness and weight in a single step (Equation 14). However, with the exception of biallelic loci, L&R also performed better than Wang when relatedness and the weight were calculated separately, indicating that this cannot be the only source of differences.
Wang (2002) presented results only for close relatives of a single type at a time, nonrelatives, full-sibs, or half-sibs. In reality, however, pedigree relatedness is unknown so that it is impossible to a priori distinguish between different types of relatives. Pedigree relatedness will take many distinct values, since all real populations have many generations of pedigree. It is not possible, therefore, to judge performance of the Wang estimator in general populations from results presented in Wang (2002). In our study, we considered populations with general relationships and evaluated estimators by the correlation between pedigree and estimated relatedness, without a priori distinguishing between categories of relatives.
Wang (2002) observed that the L&R estimator performed better for “unrelated” individuals (i.e., not sibs or parent–offspring), which will be the majority even in small populations. In contrast to current belief, therefore, we find the L&R estimator to be superior to the Wang estimator in panmictic populations. Furthermore, the L&R estimator is substantially simpler.
Bias and base population:
For most estimators, average estimated relatedness differed substantially from average pedigree relatedness, but the difference (bias) was unrelated to the accuracy (ρ) of estimators, illustrating that the choice of a base population is arbitrary in a panmictic population. Bias depends on the way estimators define the base population or, in other words, on how they divide the average observed similarity into a proportion due to IBD vs. a proportion due to AIS. The L&R and Wang estimators set the probability of AIS equal to the expected homozygosity in the current population, which implicitly defines the current generation as the base population. Average estimated relatedness is therefore close to zero for Wang and L&R, bias is negative, and many estimates are negative. Negative estimates may seem confusing, because relatedness equals twice the probability that alleles are IBD, which cannot be negative by definition. However, negative estimates can easily be scaled to positive values using an equation similar to Equation 8, which solves the interpretation problem (see also Eding and Meuwissen 2003). Alternatively, relatedness may be interpreted as a measure of additive genetic covariance between individuals, in which case below average values indicate individuals with dissimilar breeding values.
Regression of estimated relatedness on pedigree relatedness:
The regression coefficient of estimated relatedness on pedigree relatedness (β1) is a measure of bias. Unbiasedness, i.e., 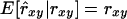 , requires that β1 = 1. (Note that the criterion bias refers to average relatedness, whereas β1 refers to pairs of individuals.) There was a clear relationship between bias and β1; positive bias was accompanied by underestimation of β1 (Table 3). This result is due to the population genetic relationship between absolute differences among coancestries of pairs of individuals and the average coancestry level of a population. Equation 3 illustrates this phenomenon. Positive bias, i.e., overestimation of sl, reduces absolute differences between coancestries because similarities are scaled by 1 − sl. Alternatively, the relationship can be understood by considering coancestry as a function of generation number (t),
, requires that β1 = 1. (Note that the criterion bias refers to average relatedness, whereas β1 refers to pairs of individuals.) There was a clear relationship between bias and β1; positive bias was accompanied by underestimation of β1 (Table 3). This result is due to the population genetic relationship between absolute differences among coancestries of pairs of individuals and the average coancestry level of a population. Equation 3 illustrates this phenomenon. Positive bias, i.e., overestimation of sl, reduces absolute differences between coancestries because similarities are scaled by 1 − sl. Alternatively, the relationship can be understood by considering coancestry as a function of generation number (t), 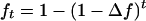 , which is a function starting at zero at t = 0 and asymptoting to 1 when t → ∞ (Falconer and Mackay 1996). At low values of ft the function is steep and differences in coancestries within a generation are large, whereas at high ft the function is flat and differences are small. Thus the relationship between bias and β1 is a direct consequence of standard population genetic theory, and estimators that are consistent with population genetic theory will always show this relationship.
, which is a function starting at zero at t = 0 and asymptoting to 1 when t → ∞ (Falconer and Mackay 1996). At low values of ft the function is steep and differences in coancestries within a generation are large, whereas at high ft the function is flat and differences are small. Thus the relationship between bias and β1 is a direct consequence of standard population genetic theory, and estimators that are consistent with population genetic theory will always show this relationship.
Regression of pedigree on estimated relatedness:
The regression coefficient of pedigree on estimated relatedness (β2) may be interpreted as the reciprocal of a usual measure of unbiasedness, i.e., 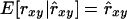 requires that β2 = 1. (Note that rxy is treated as a random variable here.) In conservation practice, selection of breeding individuals relies on estimated relatedness; pedigree relatedness is unknown. To avoid overestimation of the genetic diversity conserved, it is important that estimated relatedness is an unbiased predictor of pedigree relatedness, which requires that β2 = 1. However, β2 =
requires that β2 = 1. (Note that rxy is treated as a random variable here.) In conservation practice, selection of breeding individuals relies on estimated relatedness; pedigree relatedness is unknown. To avoid overestimation of the genetic diversity conserved, it is important that estimated relatedness is an unbiased predictor of pedigree relatedness, which requires that β2 = 1. However, β2 = 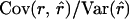 = 1 requires that
= 1 requires that 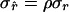 , indicating that estimates should have lower variance than pedigree values. As a consequence,
, indicating that estimates should have lower variance than pedigree values. As a consequence, 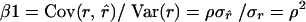 . Therefore, when β2 = 1, β1 must equal the square of the correlation between pedigree and estimated relatedness. Consequently, irrespective of the estimator used, β1 = β2 = 1 can be attained only when ρ = 1, which requires data on many loci. All estimators had values for β2 substantially <1 (Table 3), indicating that the amount of genetic diversity conserved will be overestimated when selecting least-related individuals on the basis of estimated relatedness.
. Therefore, when β2 = 1, β1 must equal the square of the correlation between pedigree and estimated relatedness. Consequently, irrespective of the estimator used, β1 = β2 = 1 can be attained only when ρ = 1, which requires data on many loci. All estimators had values for β2 substantially <1 (Table 3), indicating that the amount of genetic diversity conserved will be overestimated when selecting least-related individuals on the basis of estimated relatedness.
To investigate the effect of β2 on the number of founder genome equivalents conserved, we rescaled relatedness estimates to obtain β2 = 1. First, we derived an empirical relationship between β2 and the amount of information and next regressed estimated relatedness to its mean, using predicted β2. The empirical prediction of β2 was
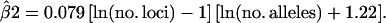 |
(22) |
For the WEDS estimator, Equation 22 explained 99% of the variation in β2 observed in the schemes analyzed (Table 2). We regressed relatedness estimates to their mean using 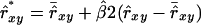 , which was applied separately to relatedness between individuals and to relatedness of individuals with themselves. Finally, Nge was calculated using
, which was applied separately to relatedness between individuals and to relatedness of individuals with themselves. Finally, Nge was calculated using 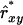 instead of
instead of 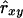 . Results showed a clear increase in Nge, in particular in the panmictic population with a conservation capacity of 100 individuals (Table 8 vs. Table 7). Furthermore, as indicated by the Nge values for equal vs. estimated optimal contributions, the use of
. Results showed a clear increase in Nge, in particular in the panmictic population with a conservation capacity of 100 individuals (Table 8 vs. Table 7). Furthermore, as indicated by the Nge values for equal vs. estimated optimal contributions, the use of 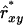 almost completely removed the loss of diversity that occurred when using
almost completely removed the loss of diversity that occurred when using 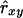 with limited accuracy. Those results show that, when conservation decisions are based on estimated relatedness, the reverse of unbiasedness, i.e.,
with limited accuracy. Those results show that, when conservation decisions are based on estimated relatedness, the reverse of unbiasedness, i.e., 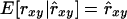 , may be more important than the usual definition,
, may be more important than the usual definition, 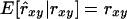 . Regression of relatedness estimates to their mean will be particularly relevant when the amount of marker information differs between individuals, in which case individuals with little information would be selected too often because they have higher variance of their estimates.
. Regression of relatedness estimates to their mean will be particularly relevant when the amount of marker information differs between individuals, in which case individuals with little information would be selected too often because they have higher variance of their estimates.
TABLE 8.
Number of founder genome equivalents in sets of either 10 or 100 individuals, in a panmictic or structured population after a priori β2 correction
| Panmictic | Structured Aa | Structured Bb | ||||
|---|---|---|---|---|---|---|
| Individuals in set | 100 | 10 | 100 | 10 | 100 | 10 |
| Pedigree | 3.69 | 3.17 | 4.23 | 3.52 | 3.82 | 3.39 |
| Equalb | 3.56 | 2.78 | 3.85 | 2.89 | 2.61 | 2.17 |
| fM | 3.47 (11) | 2.93 (2) | 3.93 (12) | 3.24 (1) | 3.45 (7) | 3.09 (2) |
| WEDS | 3.49 (10) | 2.93 (1) | 3.95 (10) | 3.25 (1) | 3.41 (5) | 3.06 (1) |
| L&R | 3.58 (2) | 2.93 (1) | 3.93 (2) | 3.19 (2) | 2.89 (−2) | 2.69 (3) |
| Wang | 3.45 (12) | 2.91 (1) | 3.92 (14) | 3.23 (2) | 3.36 (7) | 3.00 (1) |
Results are averages of 200 replicates (instead of 100). Standard errors of results were ≤0.02. Numbers in parentheses show percentage change due to β2 correction.
Ninety individuals were sampled from the subpopulation bred from 10 male and 50 female parents, and 10 were sampled from a subpopulation bred from 8 male and 40 female parents.
Ten individuals were sampled from the subpopulation bred from 10 male and 50 female parents, and 90 were sampled from a subpopulation bred from 8 male and 40 female parents.
As expected, the correlation between pedigree and estimated relatedness was not affected by regressing estimates to the mean. Consequently, for the purpose of conservation, the correlation between pedigree and estimated relatedness is not the optimal criterion for quality of an estimator, since results in Table 8 are clearly better than those in Table 7. A criterion such as the number of founder genome equivalents, which directly reflects the amount of diversity conserved, is to be preferred for conservation purposes.
Although Equation 22 was obtained using the WEDS estimator, results in Tables 3, 5, and 6 show that the relationship between β2 and the numbers of alleles and loci is nearly identical for the L&R estimator and very similar for fM. Equation 22 is, therefore, not restricted to the WEDS estimator, but useful in general. Equation 22 is a simple but rather crude two-step method to regress estimates to their mean value depending on the amount of information. A statistically more appropriate method is to treat relatedness as a random, instead of fixed, variable when estimating relatedness. However, such models involve the estimation of the variance of relatedness, which may not be trivial.
Diversity criterion with nonrandom mating and selection:
The diversity criterion used in this study relates to the additive genetic variance in an unselected random-mating population; i.e., 1 − c′Ac equals the additive genetic variance in the sampled population, expressed as a proportion of that in the founder population, assuming that the sampled population is generated by random mating and that there has been no selection between the founder and current generation. Most actual populations, however, undergo either natural or artificial selection and show nonrandom mating, which raises questions about the utility and generality of our criterion. In our opinion, however, the additive genetic variance under random mating and no selection is a useful measure for diversity, also when the actual population is selected or shows nonrandom mating. The reasoning is as follows. By definition, the additive genetic variance is the variance of the breeding values. In diploids, this variance is composed of two components: (1) the additive genetic variance with Hardy–Weinberg and linkage equilibrium, sometimes referred to as the genic variance (Wei et al. 1996), which depends solely on the allele frequencies, and (2) a deviation from the genic variance that depends on the way in which alleles at all loci are combined within individuals. This deviation is due to nonrandom mating causing deviations from Hardy–Weinberg equilibrium and to mutation, selection, and drift causing linkage disequilibrium. Part of the total linkage disequilibrium is generated by selection in the short term and is not related directly to linkage, but occurs between any two loci affecting the selected trait. It is, therefore, also known as gametic-phase disequilibrium (Bulmer 1971).
In principle, deviations from Hardy–Weinberg and gametic-phase equilibrium are transient, in contrast to changes in allele frequency and linkage disequilibrium due to tight linkage. With two sexes, Hardy–Weinberg equilibrium is restored in two generations of random mating. Positive deviations from Hardy–Weinberg equilibrium, e.g., due to obligatory selfing, increase the additive genetic variance, but it is unclear what value to attribute to such additional variance, since utilization of it involves between-family selection causing rapid loss of diversity. Furthermore, when selection ceases, the gametic phase disequilibrium asymptotes quickly to zero (Bulmer 1971). Although natural selection will never cease, it probably generates little gametic-phase disequilibrium because components of fitness have low heritability. Hence, gametic-phase disequilibrium is mainly a phenomenon of artificial selection. Thus, in the long run, it is mainly the genic variance that represents true genetic diversity originating from the allelic variety. Transient components of the additive genetic variance should not be included in a diversity criterion. In our opinion, therefore, a diversity criterion based on additive genetic variance in an idealized population is still useful when real populations deviate from that situation.
Population structure:
Populations in need of conservation predominantly have fragmented structures. In agriculture, species are generally composed of breeds and relatedness within breeds is much higher than that between breeds. Within rare breeds, fragmentation (over different countries, for example) is common as well (FAO 2000). This is logical because many domestic species are kept in herds and breeding programs are often organized nationally. Similarly, populations in zoos frequently descend from groups from founders derived from different locations (see EAZA in Situ Conservation Database: http://www.eaza.net). Furthermore, human-induced habitat loss and fragmentation are recognized as the primary causes of loss of biodiversity (Ballou and Lacy 1995; Frankham et al. 2002). Hence, structured populations are the rule and panmictic populations the exception.
Results of the structured population with scheme B in Table 6 and results of Toro et al. (2002) indicate that estimators based on two- and four-gene coefficients of identity are sensitive to the population structure . This result is probably because the basic relationship underlying these estimators (Equation 12) is valid only in the absence of inbreeding. For example, the maximum value for relatedness in Equation 12 is 1, whereas in inbred populations, relatedness of an individual with itself is 1 + F, which has a maximum of 2. Furthermore, in the derivation in Wang (2002), the genotype pairs AiAi–AiAi and AiAj–AiAj are grouped into a single category that has a similarity value of 1 (according to the definition in Wang 2002), which is correct on the basis of Equation 12. For example, in the hypothetical situation that founder alleles are unique, both genotypes have φ = 0, Δ = 1, and r = 1. However, from a population genetic point of view, those genotype pairs are clearly different: AiAi–AiAi has f = 1 and r = 2, whereas AiAj–AiAj has f = 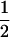 and r = 1.
and r = 1.
When noting that the basic equation underlying the L&R and the Wang estimators is invalid with inbreeding, the good performance of those estimators in inbred panmictic populations seem surprising at first. However, as argued above, the definition of a base population is arbitrary with a panmictic population. Occurrence of inbreeding, therefore, does not present a problem with random mating, because inbreeding coefficients can be shifted to approximately zero by redefining the base population. The L&R and Wang estimators “remove” inbreeding by determining the probability that alleles are AIS on the basis of observed allele frequencies, which defines the base population to be equal to the current population. The same would happen if sl in Equation 3 were set to the currently expected homozygosity, as in Ritland (1996). In a structured population, however, removing inbreeding by using the current population as the base population causes negative (true) coancestries between individuals in different subpopulations, which is theoretically incorrect. In structured populations, therefore, inbreeding cannot be removed by shifting the base population. We believe that the inability to fully remove inbreeding is the basis of the poor performance of the L&R estimator in scheme B of the structured population. The high number of founder genome equivalents of L&R with scheme A in Table 7 is probably because scheme A resembles a panmictic population, since the low number of sampled individuals from the high-drift population is in balance with a low contribution of diversity in this sample. The β2 correction hardly improves founder genome equivalents for L&R, whereas it improves all other estimators (Table 8).
Our results show that benefits of using relatedness estimates in conservation programs are substantially larger in structured than in panmictic populations (Table 7; Fernandez et al. 2005). What is needed in practice, therefore, is an estimator that can be applied to general populations. The WEDS estimator is based on: (1) the relationship between relatedness and coancestry (r = 2f) and (2) the relationship between molecular similarity and coancestry (Equation 2a). Both relationships are valid irrespective of the population structure (Lynch 1988; Falconer and Mackay 1996) and provide the theoretical basis for an estimator of both within- and between-population relatedness (Eding and Meuwissen 2001).
We obtained the WEDS estimator using a simple statistical approach, in which expected similarity was equated to observed similarity (Equation 3). Good results of the L&R estimator in panmictic populations indicate that estimators can be improved by using a more advanced statistical approach, such as conditional probabilities of observing genotypes, rather than similarity values. Hence, a promising approach to develop an estimator that performs better in both panmictic and structured populations is to follow the statistical approach of Lynch and Ritland (1999), but using r = 2f and the similarity definition of Equation 1 as a starting point (see also Toro et al. 2002).
Use in conservation practice:
The benefit of optimal contributions based on relatedness estimators in conservation programs depends on the population structure and on the breeding capacity available for conservation, e.g., expressed as the size of a population that can be conserved (N). The use of optimal contributions based on relatedness estimates that are regressed to their mean always maintains more diversity than applying equal contributions (Table 8), when populations are structured, which is common for populations in need of conservation. Even when they are panmictic and there is a limited breeding capacity (e.g., N = 10), optimal contributions based on relatedness estimates are beneficial. With panmictic populations and large capacity, it is equally good or better to use equal instead of optimal contributions (Table 8, N = 100), although this is seldom the case in conservation.
When using Equation 22 to regress estimates to their mean value, fM and WEDS are overall the best estimators. They are robust with respect to population structure, which is important when it is unknown whether the population is truly panmictic.
More information and partial Fortran code can be found at http://www.geneticdiversity.net/estimators.html.
Acknowledgments
We thank Sipke Joost Hiemstra of the Centre for Genetic Resources (CGN) for his thorough comments on previous versions of this manuscript. We also thank Jinliang Wang for making the Fortran code of his estimator available on the internet. This work was financed by the Ministry of Agriculture, Nature and Food Quality through the CGN, The Netherlands.
APPENDIX
From Equation 6, weights are 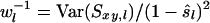 . For each locus l, we find the variance of the observed similarity as
. For each locus l, we find the variance of the observed similarity as 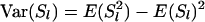 , where
, where 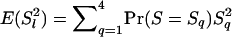 and
and 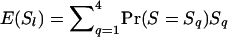 , where Sq takes values 0,
, where Sq takes values 0, 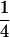 ,
, 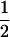 , and 1 for q = 1–4, and Pr(S = Sq) denotes the probability of observing S = Sq on locus l, which depends on the allele frequencies. (We drop subscript l for brevity.) Let Ai–Ak denote distinct alleles, pi be the frequency of allele Ai in the current population, and n be the number of alleles at locus l. Then, for each locus,
, and 1 for q = 1–4, and Pr(S = Sq) denotes the probability of observing S = Sq on locus l, which depends on the allele frequencies. (We drop subscript l for brevity.) Let Ai–Ak denote distinct alleles, pi be the frequency of allele Ai in the current population, and n be the number of alleles at locus l. Then, for each locus,
- Category S = 1 consists of the genotypes AiAi–AiAi, so that
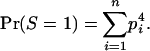
(A1) - Category S =
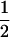 consists of the genotypes AiAi–AiAj and AiAj–AiAj, so that
consists of the genotypes AiAi–AiAj and AiAj–AiAj, so that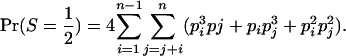
(A2) - Category S =
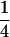 consists of the genotypes AiAj–AiAk, so that
consists of the genotypes AiAj–AiAk, so that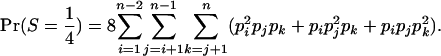
(A3)
Note that S = 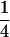 requires at least three distinct alleles. Finally,
requires at least three distinct alleles. Finally,
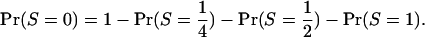 |
(A4) |
Substitution of the probabilities in the above equations for 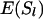 ,
, 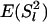 ,
, 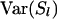 , and
, and 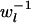 gives the weights for locus l.
gives the weights for locus l.
References
- Ballou, J. D., and R. C. Lacy, 1995. Identifying genetically important individuals for management of genetic variation in pedigreed populations, pp. 76–111 in Population Management for Survival and Recovery: Analytical Methods and Strategies in Small Population Conservation, edited by J. D. Ballou, M. E. Gilpin and T. J. Foose. Columbia University Press, New York.
- Blouin, M. S., 2003. DNA-based methods for pedigree reconstruction and kinship analysis in natural populations. Trends Ecol. Evol. 18: 503–511. [Google Scholar]
- Bulmer, M. G., 1971. The effect of selection on genetic variability. Am. Nat. 105: 201–211. [Google Scholar]
- Caballero, A., and M. A. Toro, 2000. Interrelations between effective population size and other pedigree tools for the management of conserved populations. Genet. Res. 75: 331–343. [DOI] [PubMed] [Google Scholar]
- Eding, H., and T. H. E. Meuwissen, 2001. Marker-based estimates of between and within population kinships for the conservation of genetic diversity. J. Anim. Breed. Genet. 118: 141–159. [Google Scholar]
- Eding, H., and T. H. E. Meuwissen, 2003. Linear methods to estimate kinships from genetic marker data for the construction of core sets in genetic conservation schemes. J. Anim. Breed. Genet. 120: 289–302. [Google Scholar]
- Eding, H., R. Crooijmans, M. A. M. Groenen and T. H. E. Meuwissen, 2002. Assessing the contribution of breeds to genetic diversity in conservation schemes. Genet. Sel. Evol. 34: 613–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emik, L. O., and C. E. Terrill, 1949. Systematic procedures for calculating inbreeding coefficients. J. Hered. 40: 51–55. [DOI] [PubMed] [Google Scholar]
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics. Longmans Green, Harlow, Essex, UK.
- FAO, 2000. World Watch List for Domestic Animal Diversity. FAO Publications, Rome.
- Fernandez, J., B. Villanueva, R. Pong-Wong and M. A. Toro, 2005. Efficiency of the use of pedigree and molecular marker information in conservation programs. Genetics 170: 1313–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R. A., 1958. The Genetical Theory of Natural Selection. Dover, New York.
- Frankham, R., J. D. Ballou and D. A. Briscoe, 2002. Introduction to Conservation Genetics. Cambridge University Press, Cambridge, UK.
- Hamilton, W. D., 1964. The genetical evolution of social behaviour. II. J. Theor. Biol. 7: 17–52. [DOI] [PubMed] [Google Scholar]
- Henderson, C. R., 1984. Application of Linear Models in Animal Breeding. University of Guelph, Guelph, Ontario, Canada.
- Jacquard, A., 1983. Heritability—one word, 3 concepts. Biometrics 39: 465–477. [PubMed] [Google Scholar]
- Li, C. C., and D. G. Horvitz, 1953. Some methods of estimating the inbreeding coefficient. Am. J. Hum. Genet. 5: 107–117. [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., 1988. Estimation of relatedness by DNA fingerprinting. Mol. Biol. Evol. 5: 584–599. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and K. Ritland, 1999. Estimation of pairwise relatedness with molecular markers. Genetics 152: 1753–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Malécot, G., 1948. Les Mathématiques de l'Hérédité. Masson, Paris.
- Meuwissen, T. H. E., 1997. Maximizing the response of selection with a predefined rate of inbreeding. J. Anim. Sci. 75: 934–940. [DOI] [PubMed] [Google Scholar]
- Milligan, B. G., 2003. Maximum-likelihood estimation of relatedness. Genetics 163: 1153–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas, F. W., and C. Smith, 1983. Increased rates of genetic change in dairy cattle by embryo transfer and splitting. Anim. Prod. 36: 341–353. [Google Scholar]
- Queller, D. C., and K. F. Goodnight, 1989. Estimating relatedness using genetic-markers. Evolution 43: 258–275. [DOI] [PubMed] [Google Scholar]
- Ritland, K., 1996. Estimators for pairwise relatedness and individual inbreeding coefficients. Genet. Res. 67: 175–186. [Google Scholar]
- Toro, M., C. Barragan, C. Ovilo, J. Rodriganez, C. Rodriguez et al., 2002. Estimation of coancestry in Iberian pigs using molecular markers. Conserv. Genet. 3: 309–320. [Google Scholar]
- Toro, M. A., C. Barragan and C. Ovilo, 2003. Estimation of genetic variability of the founder population in a conservation scheme using microsatellites. Anim. Genet. 34: 226–228. [DOI] [PubMed] [Google Scholar]
- Wang, J. L., 2002. An estimator for pairwise relatedness using molecular markers. Genetics 160: 1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, M., A. Caballero and W. G. Hill, 1996. Selection response in finite populations. Genetics 144: 1961–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1978. Evolution and the Genetics of Populations. University of Chicago Press, Chicago.