

Abstract
The question of how natural selection affects asexual mutation rates has been considered since the 1930s, yet our understanding continues to deepen. The distribution of mutation rates observed in natural bacteria remains unexplained. It is well known that environmental constancy can favor minimal mutation rates. In contrast, environmental fluctuation (e.g., at period T) can create indirect selective pressure for stronger mutators: genes modifying mutation rate may “hitchhike” to greater frequency along with environmentally favored mutations they produce. This article extends a well-known model of Leigh to consider fitness genes with multiple mutable sites (call the number of such sites α). The phenotypic effect of such a gene is enabled if all sites are in a certain state and disabled otherwise. The effects of multiple deleterious loci are also included (call the number of such loci γ). The analysis calculates the indirect selective effects experienced by a gene inducing various mutation rates for given values of α, γ, and T. Finite-population simulations validate these results and let us examine the interaction of drift with hitchhiking selection. We close by commenting on the importance of other factors, such as spatiotemporal variation, and on the origin of variation in mutation rates.
THE question of how natural selection affects asexual mutation rates has been considered since as early as 1930 (Fisher 1930), yet understanding is still not complete.
Most nonneutral mutations are unconditionally deleterious. In asexuals, this tends to penalize alleles that cause elevated genomic mutation rates, because they are tightly linked to the deleterious mutations they cause. Theory tells us that a population that is not adapting will favor mutation rates approaching zero (Liberman and Feldman 1986).
In contrast, in an adapting population, nonzero mutation rates can be favored (Leigh 1970, 1973; Gillespie 1981; Ishii et al. 1989). If the environment and genotype are aligned such that genotypes with higher fitness are genetically “nearby” (e.g., a single mutation away), then alleles that increase mutation rate will tend to produce more of these beneficial mutations and “hitchhike” along with them to greater multiplicity. Laboratory studies have indeed shown that high mutation rates can be thus selected in an adapting population (Cox and Gibson 1974; Chao and Cox 1983; Mao et al. 1997; Sniegowski et al. 1997). Studies of natural isolates have found that “mutator” strains, whose mutation rates are 10–1000 times the mutation rates of most members of their species, are found at measurable frequencies (e.g., 1–14%) in many populations (Jyssum 1960; Gross and Siegel 1981; LeClerc et al. 1996; Matic et al. 1997). However, we lack a complete understanding of the dynamics that produce the observed frequency distributions of various mutator strengths.
Analysis by Leigh (1970) and Ishii et al. (1989) describes how hitchhiking in an alternating environment dictates the evolutionarily stable strategy (ESS) mutation rate in a simple two-locus model. One dimorphic locus experiences direct selection conditioned on the environment, which alternates between two states. A second locus affects the mutation rate of the first locus, but itself experiences no direct selection. Maynard Smith (Maynard Smith and Price 1973) added to the model of Leigh an ensemble of potential unconditionally deleterious mutations (call the number of such potential mutations γ).
In this article, we extend these analyses to consider an environmental locus comprising many mutable sites (call the number of sites α), to more realistically model a gene that is disabled in many configurations, but operational in only a single configuration. We combine the analytical result regarding a multisite environmental locus with the result of Maynard Smith, obtaining a single expression for the effects of a multisite environmental locus, together with an ensemble of potentially deleterious loci. This combination is significantly more realistic than Leigh's original model if the biological system of interest is, for example, a bacterium that must gain or lose functionality of a certain gene to adapt to one, or the other, of a pair of alternating environments, while simultaneously avoiding deleterious mutations. Such an adaptive challenge might be faced, for example, by human pathogens, as they hop from host to host.
In addition to the above analytical results, which apply to infinite populations, this article also describes computer simulations of finite populations. These finite-population simulations include both a multisite environmental locus and multiple potentially deleterious loci, allowing us to observe the interaction of drift with the indirect selective effects described above. Under certain conditions that violate the assumptions of our analytical model, drift overwhelms the indirect selective effects upon the mutator gene deriving from hitchhiking with the environmental locus. The prevalent mutation rates in such scenarios tend to be considerably lower than the analytically derived ESS mutation rates, because the effects of hitchhiking become vanishingly weak relative to mutational load and drift.
Much recent theoretical and simulation work has considered observed bacterial mutation rates in light of three forces: (1) hitchhiking with beneficial mutations (selecting for higher mutation rate); (2) mutational load (selecting for lower mutation rate); and (3) the cost of fidelity (selecting for higher mutation rate). The importance of these three forces is actively debated. The cost of fidelity is especially poorly understood—in particular, how exactly it might increase as the mutation rate decreases, in the context of extant natural mechanisms of DNA replication.
In this article, we demonstrate that, for biologically reasonable values of α and γ, mutation rates in the observed range for bacteria may be attained without the need to invoke a “cost of fidelity” (i.e., any direct selection on the mutator alleles themselves). This supports the possibility that selection due to the cost of fidelity is negligibly weak in the natural context, compared to the other two forces. Some recent articles have agreed (Johnson 1999) and some have disagreed (Sniegowski et al. 2000) with this suggestion.
Leigh's (1970) two-locus model essentially considers a balance of two indirect selective effects: (1) higher mutators are favored immediately after an environmental alternation, by indirect selection for higher mutation rates due to increased association with the newly favored allele; however, (2) lower mutators are favored during the wait to the next alternation, by indirect selection for lower mutation rates due to mutational load. The winning ESS mutation rate is a result of the competition between these two factors over the long term. (For an example of such a competition, please see supplemental plots SUPP1 and SUPP2 at http://www.genetics.org/supplemental/.)
Leigh indicated that, for his analysis to hold, T must be long enough to allow a complete substitution of the environmental allele at each environmental alternation. Ishii et al. (1989) show, more precisely, that complete allele substitution occurs for large values of the product sT, rather than for large T alone. Ishii further shows that uessT is in fact not constant but depends on sT. For high sT, Ishii agrees with Leigh that uessT = 1; however, as sT decreases below a certain threshold value, uessT gradually increases to a plateau of 1.6, in the case of periodic environmental alternation. For the case of random environmental alternation, Ishii finds that uessT = 1 for high sT; but that uessT decreases to zero for low sT (see Figure 2 of Ishii et al. 1989). In other words, the system obeys the simple rule uessT = 1 when sT is high; otherwise the behavior is more complex because complete substitution of the environmental allele is not guaranteed at each environmental alternation.
Figure 2.

Overview of all well-converged tuples for periodic environmental alternation. The tuples are divided into three classes: “high seT ” points, which generally obeyed Equation 6; “weak sd” points, for which potential deleterious mutations are present but weak; and “low seT” points, for which hitchhiking due to selection becomes vanishingly weak. The three classes of points are further analyzed in Figures 3–6. The black line indicates the prediction of Equation 6.
The impact of drift:
Both Leigh and Ishii considered infinite populations. Neither author remarked on the vanishing strength of indirect selection upon the mutator alleles as sT approaches zero and the completeness of allele substitution decreases. In an infinite population, and given an infinite number of repeated environmental alternations, the absolute strength of the indirect selection does not matter (only the relative strengths among the various mutator strains matter) in determining the ESS winning mutation rate. In finite populations, however, drift is present; therefore, as the absolute strength of the indirect selection decreases at vanishing sT, it is eventually overwhelmed by drift. In this article, simulations are used to determine the minimum value of sT required to produce indirect selection on the mutator alleles due to hitchhiking, in the presence of drift.
RESULTS
Analytical results:
Modeling multiple mutable sites at the environmental locus:
Leigh's simple environmental locus mutates from one allele to the other and back again at symmetric rates. In contrast, natural genes typically contain multiple mutable sites at which a mutation will cause the gene to be disabled. To restore functionality, the particular site that suffered an adverse mutation must experience a reverse mutation. (We neglect the possibility of compensatory mutation.) Therefore, the rates at which the gene is disabled, and reenabled, are not symmetric. Furthermore, once the gene is disabled by a single adverse mutation, it may continue to suffer further adverse mutations; the chance of an exact reversal of multiple adverse mutations then becomes slim. We wish to incorporate these effects into a model of hitchhiking. The derivation below borrows the general reasoning of Leigh (1970).
Consider an environmental gene represented by multiple sites that have the potential to experience adverse mutation; one of more such mutations will disable the gene and its resulting fitness effect. Let α be the number of such sites. Each site holds one of two states, labeled by the digits 1 (one) or 0 (zero). If all sites are in state 1, then the entire gene is in state A; however, if one or more of the sites are in state 0, then the entire gene is in state a. All sites independently mutate between states 1 and 0 at the symmetric rate u. As in Leigh's model, the environment alternates periodically between favoring state A and state a, at period T; fitnesses are 1 + s in the favored environment and 1 otherwise.
Consider the moment just before an alternation from environment A to a. The population is nearly all all-1 (all sites in state 1, meaning that the environmental gene is overall in state A). Due to immediately recent mutation, there is a fraction αu that are single-0 (a single site, of α total sites, is in state 0; the gene overall is in state a). Immediately after the alternation, all the all-1 individuals are quickly eliminated and the single-0 mutants survive. Over the T generations to the next alternation, these single-0 individuals lose a fraction αu per generation: u per generation are lost back to all-1 and quickly decrease, and (α − 1)u are transformed to double-0 (the double-0 mutants do not immediately die, but are “doomed” to die at the next alternation), for a total of αu lost per generation. At the moment of the next environmental alternation back to favoring A, there will be only a fraction u of the single-0 mutants that immediately switch to all-1 and survive. The double-0 mutants (and the smaller number of triple-0 mutants, etc.) cannot make the double (triple, etc.) switch to all-1 and quickly decrease. Over the next T generations, the all-1 mutants lose αu per generation to the single-0 state, which is now disfavored. Overall, following Leigh's method, this indicates that we must maximize (over a complete environmental cycle of two epochs A → a → A):
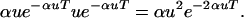 |
(1) |
Maximizing over u produces the following result for an environmental gene with multiple mutable sites capable of disabling the gene:
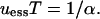 |
(2) |
The foregoing argument assumes that the population at the time of each environmental alternation is nearly pure and that a single generation of mutants seeds the population that takes over after the alternation; in fact, mutation–selection balance may be approached within each environmental epoch. This adds a factor of 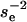 to Equation 1 but does not change the optimal value of u.
to Equation 1 but does not change the optimal value of u.
The addition of multiple unconditionally deleterious sites:
Maynard Smith (Maynard Smith and Price 1973) made a similar extension to Leigh's model, incorporating multiple potential sites for unconditionally (not dependent on the environment) deleterious mutations. If one calls the number of such sites γ, Maynard Smith maximized
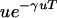 |
(3) |
to obtain
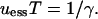 |
(4) |
Combined result:
Maynard Smith's result can be combined with that of Equation 2, to yield the following uess mutation rate for a genome with both a multisite environmental locus (α-sites) and multiple potential unconditionally deleterious sites (γ-sites). Maximizing the following over u,
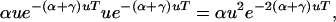 |
(5) |
produces the following combined result:
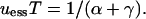 |
(6) |
This result holds both for infinite and (approximately) for finite populations, provided that sT is high enough to generate strong allele reversal at each environmental alternation. Below, we show by simulation of finite populations that, at low sT, drift overwhelms the indirect selective effects leading to this ESS mutation rate.
Simulation results:
We have created a population genetics simulator for finite populations of organisms with genomes of several loci, such as the two-locus genome defined by Leigh. The simulator resembles several simulators used recently to study mutation rates (Taddei et al. 1997; Tenaillon et al. 1999, 2000; Travis and Travis 2002, 2004). It employs discrete generations and additive fitness, and it cycles all individuals through the steps of (1) differential reproduction, (2) sampling down to the carrying capacity, and (3) mutation. The simulator explicitly models a population of individuals with genomes of multiple loci, each locus potentially containing multiple mutable sites.
Genome definition:
Our simulated genomes contain two classes of loci: fitness loci, which directly affect fitness, and mutator loci, which affect global mutation rate.
Fitness loci directly affect fitness and may be of two types: AND fitness loci and additive fitness loci. Table 1 contains one fitness locus of each type. The fitness of each genotype is initially set to the base fitness value indicated for each environment (A or a). Both types of fitness loci then contribute additively to this fitness. An AND fitness locus can be used to model a single gene that is operational only in one configuration of all its sites (all sites in state 1) and disabled in any other configuration (one or more of its sites in state 0). In Table 1, if all of the α-sites of the AND fitness locus are in the 1 state, it contributes (once) an additive fitness of se (or −se in environment a; this is the source of the dependence of fitness on the environment); otherwise, if one or more sites are in the 0 state, the entire locus is ignored. In contrast, the additive fitness locus in Table 1 models an ensemble of γ potential unconditionally deleterious mutations. It additively contributes the value −sd to the fitness of the genotype for each of its γ-sites that are in the 1 state for that genotype; sites in the 0 state are ignored.
TABLE 1.
A three-locus model
| Locus type | Site count | Strength | Wild-type forward rate | Wild-type reverse rate |
|---|---|---|---|---|
| Environment A: base fitness = 1.0, base mutation factor = mb | ||||
| AND fitness | α | se | u | u |
| Additive fitness | γ | −sd | u | u |
| Multiplicative mutator | mk | mr | 0 | 0 |
| Environment a: base fitness = 1.0 + se base mutation factor = mb | ||||
| AND fitness | α | −se | u | u |
| Additive fitness | γ | −sd | u | u |
| Multiplicative mutator | mk | mr | 0 | 0 |
The genome contains three loci. The first locus produces environmentally sensitive selection and can be disabled by a mutation at any of α sites. The second locus allows for γ potential deleterious mutations, which have no environmental dependence. The third locus controls the mutation rate of the sites in the other two loci: it will range from umb to 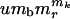 .
.
Mutator loci affect mutation rates, but not fitness. The genome in Table 1 has one multiplicative mutator locus, which contains mk dimorphic (states 1 or 0) sites. The state of the sites in this locus for a particular genotype determines the “mutation factor” of that genotype. The value of mi, the mutation factor for genotype i, is computed as mi = 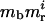 , where mb is the base mutation factor, mr is the multiplicative strength of each mutator site, and i indicates the number of mutator sites in the 1 state for that particular genotype (sites in the 0 state are ignored). The number i ranges from zero to mk for the genome in Table 1. The mutation factor of a genotype multiplicatively increases the specified “wild-type” mutation rates for all sites in that genotype (the per site wild-type mutation rate of the fitness loci in Table 1 is u and is symmetric); hence, mutator strains experience multiplied mutation rates at all sites, compared to the wild-type strain. In Table 1, the sites in the mutator locus do not mutate: their mutation rates are zero.
, where mb is the base mutation factor, mr is the multiplicative strength of each mutator site, and i indicates the number of mutator sites in the 1 state for that particular genotype (sites in the 0 state are ignored). The number i ranges from zero to mk for the genome in Table 1. The mutation factor of a genotype multiplicatively increases the specified “wild-type” mutation rates for all sites in that genotype (the per site wild-type mutation rate of the fitness loci in Table 1 is u and is symmetric); hence, mutator strains experience multiplied mutation rates at all sites, compared to the wild-type strain. In Table 1, the sites in the mutator locus do not mutate: their mutation rates are zero.
Estimating uess at many points in parameter space:
The simulator described above can perform a competition of multiple mutator strains, typically resulting in dominance by one strain of a particular mutation rate. To precisely estimate the optimal mutation factor, and hence the ESS per site mutation rate, for a particular genome and a particular set of parameters (e.g., T, se, sd, α, γ), we conduct multiple competitions of increasing resolution of the mutation factor (by decreasing the value mr from Table 1 and appropriately varying mb). This allows convergence to a precise estimate of the ESS mutation rate uess.
We created a total of 2400 (T, se, sd, α, γ) five-dimensional tuples (or “parameter sets”), by crossing five values of T (64, 256, 1024, 4096, and 16,384) by four values of se (6.25 × 10−4, 2.5 × 10−3, 0.01, and 0.04) by four values of sd (1.5625 × 10−4, 6.25 × 10−4, 2.5 × 10−3, and 0.01) by five values of α (1, 5, 10, 50, and 100) by six values of gamma (0, 1, 5, 10, 50, and 100).
For each of these 2400 tuples, we applied a recursive process (of up to three levels of recursion) to converge to an estimate uess at that point in the five-dimensional parameter space. (Please see supplemental plot SUPP3 at http://www.genetics.org/supplemental/ for an illustration of this process.) At each level of the convergence process for a particular tuple, we caused 18 mutator strains to compete. The strains used at the first level are separated from each other by a large mutation factor (i.e., mr is initially large); we “zoom in” for increasing precision as follows. The competition at a particular level for a particular tuple is repeated several times in distinct runs of the simulator, each with a different random seed. We declare a particular strain to be the winner of a run if it has been the most populous strain for a number of generations equal to the maximum of 20,000 and 10T. In addition, runs are terminated at 500,000 generations even if there is no winner. We require that at least four runs, within a maximum of 10 attempts, obtain a winning strain. If a particular level yields a tight enough distribution of winners (as specified below), then we zoom in for increasing precision: we start another competition of 18 new mutator strains by dividing the width of 2 steps from the previous level into 18 smaller steps, centering them around the average winning value from the previous level.
Periodic environmental alternation:
We first present simulation results using periodic environmental alternation at constant period T.
Overview of tuples that converged exceptionally:
The four values of se and the five values of T we used yielded eight distinct seT values (0.04, 0.16, 0.64, 2.56, 10.24, 40.96, 163.84, and 655.35). Figure 1 plots, by seT, the disposition of each of the 2400 tuples after convergence to an estimate of uess was attempted. The total number of tuples at each seT value is shown by the line marked “total.” The line marked “converged” indicates the count of well-converged tuples at each seT value. Well-converged tuples are those whose convergence process resulted in an estimate of uess (to some precision level), rather than terminating with an exceptional condition. Most tuples with seT values of ≥10.24 were well converged, because the indirect selection resulting from hitchhiking is quite strong at high seT. However, many tuples of lower seT terminated with an exceptional condition, yielding no estimate of uess for that tuple.
Figure 1.

Final disposition of all tuples under periodic alternation. The total number of tuples attempted at each seT value is indicated by the line marked “total.” The count of well-converged tuples at each seT value is indicated by the line marked “converged.” Tuples not converging but finishing with various exceptional conditions are also indicated.
One such exceptional condition is indicated in Figure 1 by the line labeled “not enough runs.” At a particular level of the convergence process, if we do not obtain four runs (within a maximum of 10 attempts) with a declared winner, that level of convergence terminates with the exceptional condition of not enough runs. If this occurs at the first level of the convergence process for that tuple, the entire tuple receives the not enough runs exception; we can make no estimate of uess for that tuple. If it occurs at a subsequent level (of a maximum of three levels), the convergence process is terminated, and the tuple receives a valid uessT value with appropriately larger error bars. Generally, this exception indicates that two or more strains were alternately dominating the population, often with one preferentially associated with environmental state A and another with state a. In Figure 1, this can be seen to sometimes occur for intermediate values of seT (2.56 and 10.24).
Occurrence of another exceptional condition is plotted by the line labeled “wide span.” It can happen that we do obtain a winner in at least four runs of a particular competition, but that those winners are spaced too widely apart (i.e., the lowest and highest winning strains were separated by more than two steps), indicating that it would be fruitless to seek a more precise estimate of uess. If this occurs at the first level of the convergence process, then we do not return any uess value at all and terminate with the exceptional condition of wide span; however, if it happens at a subsequent level, we do return a uess value, including appropriately larger error bars. As shown in Figure 1, this generally occurs at low values of seT, indicating that drift is playing a large role in the determination of the winning uess at these values.
Another exceptional termination condition is labeled “low”; this indicates that, at the first level of the convergence process, the lowest of the strains was the winner for at least one run. In this case, it is possible that our preset range of mutation rates did not go low enough to contain the true winning uess value; therefore we cannot continue the convergence process normally. Again, if this occurs past the first level of convergence, we do return a uess value and give it appropriately larger error bars. However, if this happens at the first level, we terminate exceptionally with the low condition. At the first level of the convergence process for each of the 2400 tuples, mb was 0.0019531, mr was 4, and and mk was 17. Since a base mutation rate of u = 7.45058 × 10−9 was used, this created 18 strains with per site mutation rates of from 1.45519 × 10−11 to 0.25 (the latter being an absurdly high mutation rate for natural organisms), separated by factors of 4. The low condition therefore indicates that a strain with per site mutation rate of 1.45519 × 10−11 won at least once, at the first level of convergence, for a given tuple.
Other exceptional termination conditions included “high” and “both high and low,” which are self-explanatory; however, these did not occur for any of the 2400 tuples.
Overview of well-converged tuples:
We now consider the 1321 of 2400 tuples that converged normally, yielding an estimate of uess to some level of precision. Figure 2 plots these 1321 points. For each tuple, the empirical estimate of uess multiplied by T is shown on the y-axis; the x-axis indicates the value of α + γ for the respective tuples. A black line plots the value of uessT = 1/(α + γ) as predicted by Equation 6. Error bars about each point indicate how closely the convergence process was able to estimate uessT for that tuple. (More precisely, the error bars indicate the highest and lowest uessT values of the four winning strains of the four trials at the lowest level of convergence attained by that tuple. Note that the presence of a tuple in Figure 2 implies that it was well converged.) Three separate classes of points are plotted:
“Weak sd” points are those with sd/se < 0.1, as well as γ > 0 (i.e., the genome did contain one or more potentially deleterious sites). The remaining tuples (those with stronger sd or no deleterious sites at all, i.e., γ = 0) are split into two further groups.
Points with seT < 10.24 are classified as “low seT.” This was subjectively determined to be the threshold below which most tuples did not adhere to the prediction of Equation 6.
Finally, the remaining points with seT ≥ 10.24 are labeled “high seT.” The high seT points generally match the prediction of Equation 6. The next several paragraphs discuss these three classes of points in detail; they illuminate some details of how a finite population diverges from the prediction of Equation 6 at the borderline between sufficiently and insufficiently complete substitution of the environmental allele at each environmental alternation. (i.e., in the range of seT = 10.24).
High seT tuples:
Of the 1321 well-converged tuples, 611 fell into the category “high seT” (i.e., seT ≥ 10.24, and not falling into the “weak sd” category, below). These are the points that are expected to match the prediction of Equation 6. Figure 3 plots the high seT points, separated by seT value. Of these 611 points, 496 (81%) matched the prediction of Equation 6 (i.e., their error bars span the black line). It can be seen from Figure 3 that the points that strayed furthest from the prediction were generally those with the relatively low seT = 10.24. Points with high (α + γ) are particularly prone to diverge when seT is low.
Figure 3.

High seT tuples for periodic alternation. The “high seT ” tuples from Figure 2 are plotted, separated by seT value. Those of lower seT value (seT = 10.24) tend to be the ones that stray from the prediction of Equation 6.
Weak sd tuples:
Of the1321 well-converged tuples, 512 fell into the category of weak sd (i.e., sd/se < 0.1 and γ > 0). These are points that have at least one (γ > 0) potentially deleterious site, but a low sd, relative to se. Figure 4 plots these points, separated by seT value. Some of these weak sd points, particularly those with moderate seT (seT = 10.24 or 40.96) or low seT, behave as if there are no potentially deleterious mutations (γ = 0) and nearly obey the relationship uessT = 1/α. For example, there are a number of points in a horizontal line around uessT = 1.2; for these points, α = 1 (α-value is not indicated on the plot). (We believe that the equality to 1.2 instead of 1.0 is congruent with the observation of Ishii et al. (1989) that uessT should gradually increase from 1.0 to 1.6 as seT decreased through intermediate values such as 10.24.) Similarly, there are other points in a horizontal line at uessT ≈ 0.2 =  , with α-values of 5 (α-value is not indicated on the plot), and points in a horizontal line at uessT ≈ 0.1 =
, with α-values of 5 (α-value is not indicated on the plot), and points in a horizontal line at uessT ≈ 0.1 = 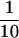 , with α-values of 10. There are also some tuples with both weak sd and low seT (Figure 4, red squares), which attain very low uessT values—see the next paragraph.
, with α-values of 10. There are also some tuples with both weak sd and low seT (Figure 4, red squares), which attain very low uessT values—see the next paragraph.
Figure 4.

Weak sd tuples for periodic alternation. The “weak sd” tuples (those for which deleterious mutations are present, but weak) from Figure 2 are plotted, separated by seT value. Those for which sd is weak and seT is intermediate (seT = 10.24) or low stray most frequently from the prediction of Equation 6. Weak sd tuples for which seT is intermediate often behave as if the deleterious locus is being ignored (γ = 0), appearing to approach the rule uessT = 1/α; see text.
Low seT tuples:
Of the 1321 well-converged tuples, 198 fell into the category “low seT” (seT < 10.24, and not falling into the weak sd category, above). Figure 5 plots these points separately by seT value. In general, the pattern is evident that adherence to Equation 6 drops off as seT decreases, particularly for higher values of (α + γ). Points of lower seT and/or higher (α + γ) all obtain low uessT values. Recall that Figure 5 plots only points that converged well; referring back to Figure 1, we note that, for tuples of low seT, many more points terminated with the exceptional conditions of low or wide span than converged well. This yields the overall picture that indirect selection due to hitchhiking with the environmental allele for tuples of low seT becomes negligibly weak: their low value of uessT is due to selection against mutational load and/or drift.
Figure 5.

Low seT tuples under periodic alternation. The “low seT ” tuples from Figure 2 are plotted, separated by seT value. Generally, adherence to Equation 6 decreases as seT decreases and/or (α + γ) increases.
Random (geometrically distributed) environmental alternation:
We also performed the convergence process for the same 2400 tuples under geometrically distributed random environmental alternation at average period T. The behavior of the 2400 tuples, as compared to the periodic case, differed in two important ways: first, we would place the subjective threshold between low and high seT at 40.96, as compared to 10.24 for periodic alternation. Second, for points of lower seT, there is not an abrupt transition from strong hitchhiking to no hitchhiking: a moderately strong hitchhiking effect can exist, producing moderate values of uessT. Figure 6 shows the low seT points in the random alternation case. (For space reasons, we do not show all the various plots for the random case. However, see supplemental plots SUPP4–SUPP8 at http://www.genetics.org/supplemental/, which plot the tuples in the random case in the same manner that Figures 1–5 plot the tuples in the periodic case.) If we compare Figure 6 to Figure 5 (the corresponding plot of low seT points for the periodic case), we see these novel intermediate values of uessT. Since the population is no longer subjected to a single period of environmental alternation, but rather to a geometric distribution of periods, we may expect strong hitchhiking during some epochs and no hitchhiking in others. The ratio of long-enough to short-enough epochs should gradually decrease with decreasing seT . In Figure 6, it is apparent that the higher seT tuples more closely approach adherence to Equation 6 than the lower ones. Moreover, although we do not plot them here, the high seT points (seT ≥ 40.96) in the stochastic case largely (302 out of 370 tuples, or 82%) did adhere to Equation 6.
Figure 6.

Low seT tuples under random environmental alternation. In the random alternation case, intermediate values of uessT appear for intermediate values of seT, indicating a gradual transition from the presence to the absence of hitchhiking. Compare low seT tuples under periodic alternation in Figure 5.
In summary:
If seT is high, producing strong hitchhiking, then uessT = 1/(α + γ) (Equation 6) is a good predictor of uessT. As sd becomes weaker, deleterious mutations begin to be ignored in the hitchhiking dynamic. As seT becomes weaker, the hitchhiking effect itself becomes so weak as to be overwhelmed by drift (with or without potentially deleterious mutations), and uessT is instead determined by selection against mutational load (if potentially deleterious mutations are present) and drift.
DISCUSSION
Relation to previous work:
The findings described here are consistent with the findings of previous theoretical studies and provide a context for generalizing some of them. For example, Taddei et al. (1997) and Tanaka et al. (2003) found that mutators could be selected for during periods of environmental change (i.e., at times when beneficial mutations were available) but might then disappear after all such mutations had been found. Our model, building on the work of Leigh (1970), Ishii et al. (1989), and Maynard Smith and Price (1973), places the findings of Taddei and Tanaka in the context of a longer time span including many episodes of environmental change and stasis. Moreover, by abstracting from the distributions of deleterious and lethal mutations of Taddei et al., we find by simulation that the rate of mutations at least approximately one-tenth as deleterious as the beneficial environmental mutation can be incorporated into the ESS rate by using the formula of Maynard Smith. We also clarify the relationship of drift to hitchhiking selection, which was not considered by Leigh, Ishii, and Maynard Smith.
Our findings also provide a context for the findings of Travis and Travis (2002), who found that mutator strains are most favored in regimes of intermediate environmental alternation frequency. In our model, this finding naturally arises because excessively rapid alternation (low T) for a given se produces “low seT,” making hitchhiking ineffective and leading to selection for lower mutation rates. Alternately, very slow alternation (high T) also leads to selection for reduced mutation rates due to increased back and deleterious mutations. This framework allows extrapolation of Travis and Travis' simulation findings; for example, we predict that as the selective value of the environmental locus (se) declines, environmental alternation periods (T) that were previously long enough to support a given mutator will become too short.
Comparison to observed natural mutation rates:
Observed baseline mutation rates in bacteria are low: Drake et al. (1998) estimate the per base pair, per generation, mutation rate to be ubp = 5.4 × 10−10 in Escherichia coli “well adapted to laboratory conditions.” A long-term laboratory study by Lenski found a slightly lower estimate of ubp = 1.44 × 10−10 for nonmutator populations (Lenski et al. 2003; Lenski 2004).
The simple model of Leigh (1970) would require a T > 109 to produce such rates; it clearly does not apply—1 billion generations is an unrealistically long wait between selective sweeps of beneficial mutations. Does Equation 6 attain more reasonable rates, given natural values of α, γ, and T?
Rough estimates of the values of γ and α in E. coli are as follows. Kibota and Lynch (1996) also consider E. coli in an artificially stable laboratory environment. They estimate the genomic rate of deleterious mutation to be ug = 2 × 10−4, as a lower bound. (The average deleterious fitness effect of the mutations they measured was 0.012 per mutation.) Since γ × ubp = ug (where ubp is the mutation rate per base pair of 5.4 × 10−10), we arrive at an estimate of γ ∼ 370,000 (as a lower bound). As for α, we assume that the disabling or the reenabling of a single protein is what is required to adapt to two alternating environments. If the length of the E. coli genome is 4.6 × 106 bp, and E. coli produces ∼4290 proteins, then an upper bound on the number of sites that could disable the average protein is ∼1000. Therefore we estimate α at 1000—a small fraction of γ.
Using these estimates of α and γ, a T of ∼5000 generations would be required to attain the low baseline mutation rates (5.4 × 10−10) found by Drake et al. (1998). It has been suggested that the turnover of E. coli populations in a human host occurs on a timescale of weeks to a month (Caugant et al. 1981). Similar carriage durations are reported for such bacteria as Streptococcus pneumoniae (Ekdahl et al. 1997) and Haemophilus influenzae (Faden et al. 1995), while Staphylococcus aureus (Scanvic et al. 2001) and Neisseria meningitidis (De Wals and Bouckaert 1985) have durations of the order of 5–10 months. While 1 month may represent ∼1500 bacterial generations in laboratory medium, one plausible estimate of the net doubling time for E. coli in a human host is ∼40 hr (Savageau 1989), so that 1 month may represent as few as 20 generations. Thus, it seems safe to say that many bacteria change human hosts with a timescale of 101–104 generations, only the very upper range of which approaches the order of 5000 generations predicted by Equation 6. Therefore, it appears that Equation 6 does not easily explain the low rates found by Lenski and Drake, at least for the potential source of repeated environmental variation represented by host-to-host transfer (however, see the next section, What combination of forces produces the low baseline mutation rate?).
What about bacteria “in the wild,” e.g., living within and between human hosts? These strains might be expected to experience significantly more fluctuating selection than strains maintained in the laboratory. Matic et al. (1997) collected 504 isolates of commensal and pathogenic E. coli from human hosts. They screened the strains for forward mutagenesis in the lacI gene, finding that nearly 90% of these strains were nonpapillating (generally indicating lower mutation rate—i.e., these are the nonmutators), having an average mutation rate to rifampicin resistance of 1 × 10−8/individual/generation, with low variance. Previously, Jin et al. (1988) counted all point mutations producing resistance to rifampicin, finding a total of 17. If we divide the rate of mutation to rifampicin resistance according to Matic by this number, we obtain a per base pair mutation rate of 5.9 × 10−10, approximately equal to that found by Drake. Therefore, although they were collected “from the wild,” 90% of all strains tested by Matic appear to maintain low rates in the range of those of Drake and Lenski. Matic found that the other 10% of the strains did produce papillation (tending to indicate higher mutation rate). These strains produced an average mutation rate to rifampicin resistance of 2.6 × 10−7, corresponding to a rate of 1.5 × 10−8/bp (comparable to the 100-fold mutators found by Lenski). What do we make of Matic's observations? Apparently, 90% of strains in the wild demonstrate the same mutation rate as strains that were intentionally maintained in a stable environment. There appear to be common features that pertain to both “stable” and “unstable” environments: the same baseline rate and typical rates for common mutators (i.e., the 100× mutator strains).
Two questions stand out.
What combination of forces produces the low baseline mutation rate?
Kimura (1967) introduced a hypothesized cost of fidelity to explain why bacterial mutation rates do not decrease to zero. The cost of fidelity would exert an upward force on mutation rates. Kimura and others, e.g., Sniegowski et al. (2000), have assumed that this force was in balance with the opposing downward force of mutational load. Instead, our model suggests that a balance between hitchhiking selection and mutational load could be responsible for setting the optimum; in other words, the invocation of a cost of fidelity may not be required. However as we note above, something seems to be depressing observed mutation rates lower than our model predicts (given our estimates of α, γ, and T and assuming strong hitchhiking under high seT). Therefore, in addition to the problem of explaining why mutation rates are nonzero, it is also difficult to explain why they are as low as they are.
Remaining within the confines of our model for the moment, for Equation 6 to produce lower rates, either γ or T would have to increase (since α cannot become much larger relative to γ). Taking a T ∼ 500 generations, in the middle range of plausible estimates given above, and α and γ as above, Equation 6 yields uess of 5.4 × 10−9—∼10 times higher than Drake's rate. Since the number of base pairs in E. coli is only ∼4.6 × 106, it would be unlikely that γ could increase 10 times from our previous estimate of 370,000.
As for T, we do note that Travis and Travis (2004), investigating spatial variation using a two-patch model in which the environmental alternation of the patches was at the same frequency but out of phase, found that this provided a sort of “refuge” for higher mutators, due to some individuals migrating between the patches and effectively experiencing a higher T. Whether such effects could produce the missing 10-fold difference is beyond our scope here. Nonetheless, the complexity observed in Travis and Travis' (2004) study of selection on mutation rate with only two populations suggests that a variety of interesting phenomena may appear in such models. Bacteria in human hosts may be highly compartmentalized. Much work remains to be done to understand how the present model can be generalized to understand the evolution of mutation rates in such metapopulations.
But what if the model's assumption of high seT were relaxed? First of all, let us assume that the random alternation case, rather than the periodic case, better resembles the distribution of T between selective sweeps of adaptive mutations experienced by bacteria. Recall from Figure 6 that certain tuples in the random alternation case do strongly converge to uessT values that are 10- and 100-fold less than that predicted by Equation 6. These are the higher of the values in the low seT category; therefore we call them “intermediate seT .” For these cases, selection on the environmental allele is not quite strong enough to produce a complete substitution of alleles within the duration of many epochs. If the intermediate seT case applies, then the low rates observed by Drake could be produced within the confines of our current model (with the high seT assumption relaxed).
Why does environment seem not to affect the low baseline mutation rate?
The second question that arises from Matic's observed distribution of mutation rates is: Why do bacteria in the wild (presumably a strongly fluctuating environment) maintain the same low baseline rate found in the laboratory? The simplest explanation is that Drake's and Lenski's strains were not in the laboratory long enough for a new ESS mutation rate to dominate; they simply reflect the wild ESS rate. For example, Lenski's populations were well documented for 20,000 generations at the time of publication in 2003 (Lenski et al. 2003), but what happened to the inoculating strain prior to this is less clear. It had been in the laboratory since at least 1966, but, for example, might have been frozen for much of that time. In our simulations, it was easy to construct cases in which an ESS mutation rate would not fix for tens or hundreds of thousands of generations—for example, when T is very long, as would be expected in a stable environment.
Genetic accessibility of mutation rates:
Another important influence upon the distributions of mutation rates found in nature could be the genetic accessibility of various mutator strengths. Obviously, the theoretical ESS mutation rate cannot come to dominate a population if no strain with that rate arises in the first place. Consider that only a subset of the genes in the E. coli genome have an influence on mutation rate. All possible point mutations to these genes produce a finite set of distinct mutator strains (and most such mutations simply produce unviable strains). One might imagine a graph in which the nodes represent the common mutator strains of E. coli genetically near to a wild type, and the edge weights represent the transition (e.g., mutation) probabilities among the strains. This graph may constitute a hidden genetic context for the evolution of mutators. When a beneficial mutation is available (i.e., within a single point mutation or so, due to the confluence of environmental and genetic factors), the population may temporarily crowd into higher mutation rate strains via hitchhiking. However, when beneficial mutations are not available, the population will tend to retreat into nearby lower mutation rate strains. (One article making a simulation study of this pursuit of mutation–selection balance was Taddei et al. 1997, which allowed forward and reverse mutation between a wild type and a single mutator type.) The indirect selection felt by the various mutator strains may well be described by Equations 5 and 6, but any individual does not have complete flexibility as to what mutation rates its progeny may adopt next; these are dictated by the graph, which evolves only over the very long term. Several mutational “hops” back and forth through relatively favored and disfavored mutation rates may be required to attain the optimum rate. Such hopping, perhaps in the face of drift, may take time—and moreover any “optimum” rate may be ephemeral. Therefore, the distribution of mutator strengths observed in natural bacteria probably cannot be fully explained by considering only environmental influence, without any genetic influence. Much work remains to be done in this area.
Interplay between mutation rate and population size:
Finally, we mention a subtlety of the model that depends on effective population size. In our derivation of Equations 5 and 6, we assumed that each mutator strain had an ample number of both the favored and disfavored alleles, meaning that exponential growth could begin immediately after the environmental alternation to a new favored allele. Therefore, we neglected any effect due to a delay in “discovery” of the favored allele by each strain or delay due to its struggle against loss by drift. (Recall that Leigh's original model considered infinite populations, so this was not a concern.) Such delays would tend to decrease the effectiveness of hitchhiking at low mutation rates relative to the reciprocal of the effective population size. In addition, they would also decrease the penalty due to deleterious mutations. Further discussion is beyond our scope, but this effect is discussed in Tanaka et al. (2003).
In summary, we believe that we have furthered the understanding of the indirect selection experienced by mutators due to hitchhiking and deleterious mutation. A full and detailed understanding of the distribution of mutation rates observed in bacteria, however, may require the consideration of genetic constraints as well, and the nature of these constraints is not currently well understood. In addition, we believe that further complication will be uncovered by models of temporally and spatially complex environments.
Acknowledgments
We thank Richard Moxon and Christopher Bayliss, of Oxford, for helpful discussions throughout the course of this work. We thank several anonymous reviewers of the manuscript in progress for important comments and suggestions. M.P. thanks his wife for her support of this work. This work was supported by National Institutes of Health grant 1R21AI055825, to principal investigator Marc Lipsitch.
References
- Caugant, D. A., B. R. Levin and R. K. Selander, 1981. Genetic diversity and temporal variation in the E. coli population of a human host. Genetics 98: 467–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao, L., and E. C. Cox, 1983. Competition between high and low mutating strains of Escherichia coli. Evolution 37: 125–134. [DOI] [PubMed] [Google Scholar]
- Cox, E. C., and T. C. Gibson, 1974. Selection for high mutation rates in chemostats. Genetics 77: 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wals, P., and A. Bouckaert, 1985. Methods for estimating the duration of bacterial carriage. Int. J. Epidemiol. 14: 628–634. [DOI] [PubMed] [Google Scholar]
- Drake, J. W., B. Charlesworth, D. Charlesworth and J. F. Crow, 1998. Rates of spontaneous mutation. Genetics 148: 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl, K., I. Ahlinder, H. B. Hansson, E. Melander, S. Molstad et al., 1997. Duration of nasopharyngeal carriage of penicillin-resistant Streptococcus pneumoniae: experiences from the South Swedish Pneumococcal Intervention Project. Clin. Infect. Dis. 25: 1113–1117. [DOI] [PubMed] [Google Scholar]
- Faden, H., L. Duffy, A. Williams, D. A. Krystofik and J. Wolf, 1995. Epidemiology of nasopharyngeal colonization with nontypeable Haemophilus influenzae in the first 2 years of life. J. Infect. Dis. 172: 132–135. [DOI] [PubMed] [Google Scholar]
- Fisher, R. A., 1930. The Genetical Theory of Natural Selection. Clarendon Press, Oxford.
- Gillespie, J. H., 1981. Mutation modification in a random environment. Evolution 35: 468–476. [DOI] [PubMed] [Google Scholar]
- Gross, M. D., and E. C. Siegel, 1981. Incidence of mutator strains in Escherichia coli and coliforms in nature. Mutat. Res. 91: 107–110. [DOI] [PubMed] [Google Scholar]
- Ishii, K., H. Matsuda, Y. Iwasa and A. Sasaki, 1989. Evolutionarily stable mutation rate in a periodically changing environment. Genetics 121: 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, D. J., W. A. Walter and C. A. Gross, 1988. Characterization of the termination phenotypes of rifampicin-resistant mutants. J. Mol. Biol. 202: 245–253. [DOI] [PubMed] [Google Scholar]
- Johnson, T., 1999. Beneficial mutations, hitchhiking and the evolution of mutation rates in sexual populations. Genetics 151: 1621–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyssum, K., 1960. Observations on two types of genetic instability in Escherichia coli. Acta Pathol. Microbiol. Scand. 48: 113–120. [DOI] [PubMed] [Google Scholar]
- Kibota, T. T., and M. Lynch, 1996. Estimate of the genomic mutation rate deleterious to overall fitness in E. coli. Nature 381: 694–696. [DOI] [PubMed] [Google Scholar]
- Kimura, M., 1967. On the evolutionary adjustment of spontaneous mutation rates. Genet. Res. 9: 23–34. [Google Scholar]
- LeClerc, J. E., B. Li, W. L. Payne and T. A. Cebula, 1996. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274: 1208–1211. [DOI] [PubMed] [Google Scholar]
- Leigh, Jr., E. G., 1970. Natural selection and mutability. Am. Nat. 104: 301–305. [Google Scholar]
- Leigh, Jr., E. G., 1973. The evolution of mutation rates. Genetics 73: 1–18. [PubMed] [Google Scholar]
- Lenski, R. E., 2004. Phenotypic and genomic evolution during a 20,000-generation experiment with the bacterium Escherichia coli. Plant Breed. Rev. 24: 225–265. [Google Scholar]
- Lenski, R. E., C. L. Winkworth and M. A. Riley, 2003. Rates of DNA sequence evolution in experimental populations of Escherichia coli during 20,000 generations. J. Mol. Evol. 56: 498–508. [DOI] [PubMed] [Google Scholar]
- Liberman, U., and M. W. Feldman, 1986. Modifiers of mutation rate: a general reduction principle. Theor. Popul. Biol. 30: 125–142. [DOI] [PubMed] [Google Scholar]
- Mao, E. F., L. Lane, J. Lee and J. H. Miller, 1997. Proliferation of mutators in a cell population. J. Bacteriol. 179: 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matic, I., M. Radman, F. Taddei, B. Picard, C. Doit et al., 1997. Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science 277: 1833–1834. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J., and G. R. Price, 1973. The logic of animal conflict. Nature 246: 15–18. [Google Scholar]
- Savageau, M., 1989. Escherichia coli habitats, cell types, and molecular mechanisms of gene control. Am. Nat. 122: 732–744. [Google Scholar]
- Scanvic, A., L. Denic, S. Gaillon, P. Giry, A. Andremont et al., 2001. Duration of colonization by methicillin-resistant Staphylococcus aureus after hospital discharge and risk factors for prolonged carriage. Clin. Infect. Dis. 32: 1393–1398. [DOI] [PubMed] [Google Scholar]
- Sniegowski, P. D., P. J. Gerrish and R. E. Lenski, 1997. Evolution of high mutation rates in experimental populations of E. coli. Nature 387: 703–705.9192894 [Google Scholar]
- Sniegowski, P. D., P. J. Gerrish, T. Johnson and A. Shaver, 2000. The evolution of mutation rates: separating causes from consequences. BioEssays 22: 1057–1066. [DOI] [PubMed] [Google Scholar]
- Taddei, F., M. Radman, J. Maynard-Smith, B. Toupance, P. H. Gouyon et al., 1997. Role of mutator alleles in adaptive evolution. Nature 387: 700–702.9192893 [Google Scholar]
- Tanaka, M. M., C. T. Bergstrom and B. R. Levin, 2003. The evolution of mutator genes in bacterial populations: the roles of environmental change and timing. Genetics 164: 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon, O., B. Toupance, H. Le Nagard, F. Taddei and B. Godelle, 1999. Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics 152: 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon, O., H. Le Nagard, B. Godelle and F. Taddei, 2000. Mutators and sex in bacteria: conflict between adaptive strategies. Proc. Natl. Acad. Sci. USA 97: 10465–10470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis, E. R., and J. M. Travis, 2004. Mutators in space: the dynamics of high-mutability clones in a two-patch model. Genetics 167: 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis, J. M., and E. R. Travis, 2002. Mutator dynamics in fluctuating environments. Proc. R. Soc. Lond. Ser. B Biol. Sci. 269: 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]