

Abstract
Field bean (Dolichos lablab) contains a single isoform of PPO (polyphenol oxidase) – a type III copper protein that catalyses the o-hydroxylation of monophenols and oxidation of o-diphenols using molecular oxygen – and is a homotetramer with a molecular mass of 120 kDa. The enzyme is activated manyfold either in the presence of the anionic detergent SDS below its critical micellar concentration or on exposure to acid-pH. The enhancement of kcat upon activation is accompanied by a marked shift in the pH optimum for the oxidation of t-butyl catechol from 4.5 to 6.0, an increased sensitivity to tropolone, altered susceptibility to proteolytic degradation and decreased thermostability. The Stokes radius of the native enzyme is found to increase from 49.1±2 to 75.9±0.6 Å (1 Å=0.1 nm). The activation by SDS and acid-pH results in a localized conformational change that is anchored around the catalytic site of PPO that alters the microenvironment of an essential glutamic residue. Chemical modification of field bean and sweet potato PPO with 1-ethyl-3-(3-dimethylaminopropyl)carbodi-imide followed by kinetic analysis leads to the conclusion that both the enzymes possess a core carboxylate essential to activity. This enhanced catalytic efficiency of PPO, considered as an inducible defence oxidative enzyme, is vital to the physiological defence strategy adapted by plants to insect herbivory and pathogen attack.
Keywords: acid-pH, carboxy group, hydrodynamic radius, polyphenol oxidase, SDS, turnover
Abbreviations: BCIP, 5-bromo-4-chloroindol-3-yl phosphate; CMC, critical micellar concentration; EDAC, 1-ethyl-3-(3-dimethylaminopropyl)carbodi-imide; GME, glycine methyl ester hydrochloride; GOD, glucose oxidase; NBT, Nitro Blue Tetrazolium; PFPA, pentafluoropropionic acid; PPO, polyphenol oxidase; RP-HPLC, reversed-phase HPLC; TBC, tertiary butyl catechol; Tos-Phe-CH2Cl, tosylphenylalanylchloromethyl ketone
INTRODUCTION
Polyphenol oxidase (PPO; EC 1.10.3.1), a member of type III copper proteins, catalyses ortho-hydroxylation of monophenols (cresolase activity) and oxidation of ortho-diphenols to ortho-di-quinones (catecholase activity) at the expense of molecular oxygen. The resulting highly reactive quinones autopolymerize to form brown polyphenolic catechol melanins, a process thought to protect damage to plants from pathogens and insects [1–3]. The enzyme can be found not only in different fungal, mammalian, avian and insect species but also in a variety of plant species [4]. In plants, PPO is located in the chloroplast thylakoid membranes and often exists in multiple forms. An unusual and intriguing characteristic of the enzyme is its ability to exist in either a latent and/or an active form [5,6]. PPO can be released from latency or activated by acid and base shock [7], detergents [4,8,9], urea [10] and proteases [11,12]. SDS as an activating agent is intriguing because very few enzymes are known to be activated by SDS in contrast with the many that are inactivated by it. Kenten [7] has reported that the activation of crude broad bean leaf PPO by SDS occurred below 1 mM SDS. Extending these observations further, Swain et al. [10] and Robb et al. [13] observed that this activation process is reversible and that prolonged incubation in the presence of SDS leads to a loss of activity. Laveda et al. [14] demonstrated the total reversibility of the SDS activation of latent peach PPO by SDS entrapment with cyclodextrins. Wittenberg and Triplett [15] have characterized Xenopus laevis tyrosinase and show that the activation is related to enzyme stabilization. SDS activation of a purified broad bean leaf PPO is found to alter both its enzymatic and physical characteristics, suggesting a limited conformational change, due to binding of small amounts of SDS, which could induce or initiate the activation of latent PPO [4]. Studies to date on the activation phenomenon of PPO only advocate that it is due to a limited conformational change. However, convincing structural and experimental details of the conformational changes at the molecular level are yet to be elucidated.
PPO, purified from field bean (Dolichos lablab) of molecular mass 120 kDa, is a tetramer and exhibits no cresolase activity [16]. Although purified PPO exhibits no monophenolase activity, in the presence of catalytic quantities of a diphenol, it exhibits monophenolase activity [17]. Field bean PPO, like other PPOs, also exhibits latency and is activated by exposure to acid-pH or in the presence of SDS below the CMC (critical micellar concentration) [18]. The present study is aimed at understanding and correlating at the molecular level the conformational changes that accompany this activation. We have employed size-exclusion chromatography, electrophoretic mobility, fluorescence spectroscopy and CD to investigate the structure and conformation of PPO following activation by acid-pH and SDS treatment. These studies have demonstrated that the activation by either SDS or acid-pH results in a distinct increase in the Stokes radius accompanied by localized secondary and tertiary structural changes, leading to an elevated kcat and a marked shift in the pH optimum from 4.5 to 6.0. Further, these conformational changes are similar for both the acid-pH- and SDS-activated PPOs, suggesting a common feature in the activation phenomena.
MATERIALS AND METHODS
Field bean (D. lablab) seeds were purchased from the local market. TBC (tertiary butyl catechol) was from Merck (Germany). Sequanal grade SDS was obtained from Pierce Chemical (U.S.A.). PFPA (pentafluoropropionic acid), acrylamide, BIS (N,N′-methylene bisacrylamide), tropolone, Tos-Phe-CH2Cl (tosyl-phenylalanylchloromethyl ketone; ‘TPCK’)–trypsin, catechol, glutaraldehyde, sodium deoxycholate, GME (glycine methyl ester hydrochloride), EDAC [1-ethyl-3-(3-dimethylaminopropyl)-carbodi-imide], BCIP (5-bromo-4-chloroindol-3-yl phosphate), NBT (Nitro Blue Tetrazolium) and analytical gel-filtration molecular mass markers were obtained from Sigma (St. Louis, MO, U.S.A.). All the other chemicals used were of the purest grade commercially available.
Enzyme purification
PPO was purified from field bean (D. lablab) seeds as reported in [16]. Protein concentration was determined by the dye binding method of Bradford [19]. BSA was used as a standard.
Enzyme assay
PPO was assayed spectrophotometrically at 25±1 °C using a Shimadzu UV–Visible spectrophotometer Model 1601 at 400 nm with TBC. The assay mixture consisted of 0.9 ml of 0.05 M sodium acetate/0.05 M acetic acid buffer (pH 4.5) (pH 6.0 for activated PPO), 0.1 ml of 0.4 M TBC and 10–100 μg of enzyme. The quinone formed was measured at 400 nm (molar absorption coefficient ϵ400 1150 M−1·cm−1). One unit of enzyme activity is defined as the amount of enzyme that produces 1 μmol of tert-butylquinone per minute under the assay conditions.
Activation of PPO
PPO was pre-incubated at various concentrations of SDS prepared in 25 mM Tris/HCl (pH 7.0) containing 1.2% (w/v) NaCl for 30 min, following which PPO was assayed at pH 6.0 as described above. Relative activity was plotted against SDS concentration. Acid-pH activation of PPO was performed by pre-incubating PPO in McIlvaine buffer (0.1 M citric acid and 0.2 M disodium phosphate, pH 2.5 or 4.0). PPO activity was measured at pH 6.0 periodically as described above.
Determination of pH optimum and stability
PPO activity as a function of pH was determined using the substrate TBC. McIlvaine buffer in the pH range 2.5–8.0 was used to determine the pH optimum. A plot of relative activity versus pH was employed to obtain the pH optimum. The stability of native and activated PPO as a function of pH was evaluated by pre-incubating PPO at pH 2.5, 4.0 and 7.0. At periodic intervals, aliquots of PPO were removed and residual activity was assayed in 0.05 M sodium acetate buffer (pH 4.5 or 6.0) for the native and activated forms respectively.
Inhibition by tropolone
The kinetics of tropolone inhibition was carried out by pre-incubating native and activated PPO for 3 min at 27 °C in the presence of tropolone (0.2–2.0 μM). Residual PPO activity was determined at various TBC concentrations. The mode of inhibition was determined from the Lineweaver–Burk plot at constant tropolone concentrations. The inhibition constant Ki was determined from the Dixon plot.
HPLC size-exclusion chromatography
Stokes radii measurements were carried out on a TSK gel G2000 SWXL (7.8 mm×30 cm; 5 μm) column, using a Waters HPLC system, equipped with a 1525 binary pump and Waters 2996-photodiode array detector. The column was pre-equilibrated with 0.1 M sodium phosphate buffer (0.1 M disodium hydrogen phosphate and 0.1 M monosodium dihydrogen phosphate; pH 7.0) containing 0.1 M Na2SO4 at a flow rate of 0.5 ml/min. A set of proteins whose molecular mass and Stokes radii are known were used to construct the calibration curve of log R values versus migration rate.
Fluorescence studies
Fluorescence measurements were recorded on a Shimadzu RF 5000 spectrofluorophotometer using a 10 mm path length quartz cell, at 27 °C. For measuring the intrinsic fluorescence, the protein was excited at 280 nm, and the emission spectrum was recorded between 300 and 400 nm. Appropriate blanks were used for baseline correction of fluorescence intensity. Fluorescence quenching by acrylamide data was analysed using the general form of the Stern–Volmer equation.
CD studies
CD measurements were carried out using a Jasco J-810 automatic recording spectropolarimeter fitted with a xenon lamp and calibrated with +d-10-camphor sulphonic acid. Dry nitrogen was purged continuously before and during the experiment. The measurements were made at 25 °C. The path length of the cell used was 1 mm in the far-UV region and 10 mm in the near-UV region. The scan speed was 20 nm/min and spectra were taken as an average of three scans. The results were expressed as the mean residual ellipticity [θ]MRW obtained from the relation [θ]=100×θobs/(lc), where θobs is the observed ellipticity in degrees. The mean residual ellipticity [θ]MRW was calculated using a value of 115 for mean residue mass of PPO, c is the concentration in grams per litre and l is the length of the light path in centimetres. The values obtained were normalized by subtracting the baseline recorded for the buffer under similar conditions.
Limited proteolysis
Native and activated forms of PPO were subjected to limited proteolysis using Tos-Phe-CH2Cl–trypsin (2:100, w/w) at pH 8.2 for 60 min at 37 °C. The proteolysis was arrested by incubating the reaction mixture for 20 min in a boiling-water bath and then freeze-dried. The tryptic digest was analysed by RP-HPLC (reversed-phase HPLC) using a Waters Symmetry Shield C18 column (4.6 mm×150 mm; 5 μm) on a Waters HPLC system equipped with a 1525 binary pump and Waters 2996 photodiode array detector in a water/acetonitrile gradient containing 0.1% PFPA.
Dot-blot analysis
Aliquots of PPO (10 μg) were immobilized on a nitrocellulose membrane and probed with PPO antibodies raised in New Zealand White rabbits and detected using a mixture of BCIP and NBT in substrate buffer (0.1 M Tris, pH 9.5, containing 0.5 M NaCl and 5 mM MgCl2).
Cross-linking using glutaraldehyde
Glutaraldehyde (25%, w/v) was added to aliquots of both native and activated PPO (8 μg) to a final concentration of 10% and incubated at 27 °C for 5 min. Quenching of cross-linking was achieved by addition of glycine to a final concentration of 97 mM. After 20 min incubation, 3 μl of aq. 10% sodium deoxycholate was added. The pH of the reaction was lowered to approx. 2.5 by addition of orthophosphoric acid (85%, v/v), which resulted in precipitation of the cross-linked protein. After centrifugation (8000 g, 4 °C, 20 min), the precipitate was re-dissolved in 0.1 M Tris/HCl (pH 8.0) containing 1% SDS and 50 mM dithiothreitol and heated at 90–100 °C. Samples were analysed by SDS/PAGE [10% T (total acrylamide concentration) and 2.7% C (degree of cross-linking)].
Modification of carboxyl residue
The importance of carboxylate group of glutamate in the activity of PPO was investigated by reaction with EDAC [20]. All inactivation experiments were performed at 25 °C with field bean and sweet potato PPO in 0.05 M sodium acetate buffer (pH 4.8) with 275 mM GME and 0–300 mM EDAC. EDAC and GME were dissolved in water immediately before use and inactivation was initiated by the addition of EDAC. A control experiment of enzymes and the nucleophile GME in buffer was run simultaneously and corresponded to 100% activity for both field bean and sweet potato PPO. Aliquots were removed for determination of residual activity at 10 min intervals and the pseudo first-order rate constants for inactivation were determined. The inactivation kinetics was fitted to the equation: log (% residual activity)=−kit, where ki is the pseudo first-order inactivation rate constant for a given concentration of EDAC and t is time of inactivation. The inactivation order (n) was calculated from the equation: log ki=n log [inactivator]+log ki, where ki is the second-order inactivation constant.
Thermal inactivation studies
The loss of enzyme activity as a function of temperature was studied for the native and activated forms of PPO. Native PPO and the activated forms were incubated for 15 min in their respective buffers at different temperatures ranging from 25 to 90 °C. After cooling to 4 °C, the residual activity was measured at 25±1 °C as described earlier. The midpoint of thermal inactivation, Tm, at which the activity was diminished by 50%, was calculated from the plot of percentage residual activity versus temperature.
Kinetics of thermal inactivation of native, SDS-activated and acid-pH-activated PPOs were studied, using a constant temperature bath, at desired temperatures. Aliquots of enzyme, removed at periodic intervals, were subjected to assay after cooling in an ice bath. The residual activity was measured as a percentage of initial activity. From the semi-logarithmic plot of residual activity as a function of time, the inactivation rate constants (kr) were calculated. The apparent half-life values were estimated and energy of activation (Ea) was calculated from the slopes of the Arrhenius plots. Thermodynamic functions were calculated according to the following relationships:
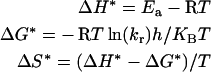 |
(h is the Planck constant, KB is the Boltzmann constant, R is gas constant and kr is rate constant.)
RESULTS
Activation of PPO by acid-pH
The effect of pH on the activation of PPO was evaluated by incubating the purified PPO in buffers of pH 2.5, 4.0 and 7.0. At periodic intervals, aliquots were assayed for PPO using TBC as substrate. The results indicate that PPO activation occurs at pH 2.5 as well as 4.0. However, the activation was more rapid at pH 2.5, reaching a maximum in 30 min. At pH 4.0, although a similar maximal steady-state rate was achieved, the time taken to reach this maximum was between 2 and 4 h. The activity of PPO, after exposure to pH 2.5 for 30 min, is 9.6±0.27×104 units/mg. PPO, activated at pH 2.5 although activated rapidly, was unstable, losing 97% of its activity in 24 h. The activity of native PPO assayed at pH 6.0 is 5.9±0.24×103 units/mg, which is less than 10% of the activated forms. PPO activated by exposure to pH 2.5 for 10 min was used as ‘acid-pH-activated PPO’ in all further studies. The activation of PPO and the subsequent decline in the activity suggest that conformational changes take place in the enzyme upon exposure to a pH shock. Reversal of this activation was possible by exposure to pH 7.0 [18].
To confirm that the activation and subsequent decrease in activity are indeed due to conformational changes, attempts were made to induce similar changes in the PPO by means other than pH shock, by treatment with anionic detergent SDS.
Effect of SDS on PPO activity
The effect of increasing SDS concentration on the activity of purified field bean PPO is represented in Figure 1. PPO activity increased with increasing SDS concentration. The most effective concentration of SDS was 1.25 mM where the measured activity was 1.32±0.2×105 units/mg. At SDS concentrations below 0.3 mM, the increase in activity is 15–20% of native PPO. However, a very sharp linear increase in PPO activity from 0.3 to 1.25 mM with half the maximum activation at 0.6 mM SDS was observed. The maximum steady-state rate was achieved with 1.25 mM SDS. A further increase in the SDS concentration led to a near linear decrease in the PPO activity. Maximum activation was achieved in 30 min, further incubation with SDS led to a decrease in PPO activity (Figure 1, inset). On the basis of these observations, a concentration of 1.25 mM SDS at pH 7.0 for 30 min was used to activate PPO in all further studies.
Figure 1. Effect of SDS concentration on field bean PPO activity.

PPO was incubated in 25 mM Tris/HCl (pH 7.0) containing 1.2% NaCl at indicated concentrations of SDS for 30 min and then assayed at pH 6.0 using TBC as substrate. Native PPO activity assayed at pH 4.5 using TBC in the absence of SDS is considered as 100%. Inset: effect of incubation time on SDS activation of field bean PPO activity.
pH optimum
The effect of pH on the optimal activity of SDS- and acid-pH-activated PPO was evaluated using McIlvaine buffer (pH 2.5–7.5 and Tris/HCl, pH 8.0). The pH optimum of the SDS-activated PPO was 6.0 with a relatively high activity between pH 5.5 and 6.5 (Figure 2). PPO activity of the acid-pH-activated form also indicated a pH optimum of 6.0, above which the activity decreased. At pH 4.5, the SDS- and pH-activated forms of PPO exhibit only approx. 10% of the maximum activity. In contrast, the native PPO exhibits maximum activity at pH 4.5 for the oxidation of TBC (Figure 2). These results suggest that field bean PPO exists in two forms, a native form with a pH optimum of 4.5 and an activated form with a pH optimum of 6.0. The similar shifts in the pH optimum for both SDS- and acid-pH-activated PPO suggest similarities in the activation. This change in the pH optimum could be related to the displacement of a sensitive pKa value of the enzyme. All further assays of the activated PPO were performed at pH 6.0.
Figure 2. Effect of pH on the activity of field bean PPO.

Native PPO in 25 mM Tris/HCl (pH 7.0) containing 1.2% NaCl (●), SDS-activated PPO (▲) and acid-pH-activated PPO (▼). PPO assays using TBC as the substrate were performed in 0.1 M McIlvaine buffer (pH 2.5–7.5) and 25 mM Tris/HCl (pH 8.0 and 8.5) as described in the Materials and methods section.
Determination of Km and Vmax
Activation of an enzyme occurs either due to an increase in the Vmax or a decrease in the Km or both. These parameters were therefore evaluated for field bean PPO before and after activation. Km and Vmax for PPO following activation were determined at pH 6.0 using TBC. The kinetic parameters were calculated from the double reciprocal plots, which showed no indication of non-linearity. The kinetic parameters Km and Vmax computed from nonlinear fitting of the data to the Michaelis–Menten equation were similar to those obtained from the double reciprocal plots. These parameters are compared with the kinetic parameters of native PPO determined at pH 4.5 (Table 1). Both SDS and acid-pH treatment caused a very small decrease in the Km of PPO for its phenolic substrate TBC. The results revealed that the binding affinity towards TBC increased only marginally for both the activated forms. The SDS- and acid-pH-activated forms show remarkably higher Vmax values towards the oxidation of TBC (Table 1). The overall catalytic efficiency (kcat) increased 6.5- and 5.8-fold for the SDS- and acid-pH-activated forms respectively. The Ki values for competitive inhibition by tropolone of the activated PPOs were far lower than that of native PPO (Table 1). The activation energy (Ea) for the oxidation of TBC catalysed by native PPO was 135.2 kJ/mol, whereas that for SDS-activated PPO was 118.9 kJ/mol.
Table 1. Kinetic parameters of native, SDS-activated and acid-pH-activated PPO.
| Properties | Native PPO | SDS-activated PPO | Acid-pH-activated PPO |
|---|---|---|---|
| pH optimum | 4.5 | 6.0 | 6.0 |
| Km (TBC; mM) | 4.2±0.2 | 3.9±0.3 | 3.6±0.04 |
| Vmax (×105 units/mg) | 1.97±0.25 | 12.03±0.29 | 9.77±0.24 |
| Ki(tropolone) (×10−7 M) | 5.7±0.2 | 1.8±0.04 | 3.6±0.22 |
| Vmax/Km (kcat)×103 | 0.47 | 3.1 | 2.7 |
| Ea (kJ/mol) | 135.2±0.29 | 118.9±0.20 | 91.6±0.25 |
Chemical modification of carboxylate
A significant shift in the pH optima from 4.5 to 6.0 is observed for the SDS- and acid-pH-activated PPO (Figure 2). This shift is in the ionization range of carboxy groups implicating them in catalysis. Klabunde et al. [21] in their three-dimensional structure of a PPO from Ipomea batatas imply that Glu236 functions as a general base/acid in the catalysis of diphenol oxidation. An examination of plant PPO sequences (Swiss-Prot release) and a multiple alignment indicates that this glutamic residue is conserved in all plant PPOs (Figure 3). The amino acid-modifying agent EDAC specific to the carboxylic groups of glutamate and aspartate was used to verify the presence of glutamic residue at the active site of PPO. Incubation with GME alone had no effect on the enzyme activity of field bean PPO and sweet potato PPO. The enzymes were fully stable in the presence of EDAC alone. The lack of inhibition by either of the reagents supports the direct involvement of the carboxylic groups, indicating that the inactivation measured is not due to cross-linking of other amino acids. A kinetic analysis of the inactivation of field bean and sweet potato PPO with various concentrations of EDAC was carried out (Figures 4A and 4B). The semi-logarithmic plots of residual enzyme activity at various EDAC concentrations versus time were linear for both field bean PPO and sweet potato PPO, indicating that the inactivation followed pseudo first-order kinetics. A plot of the first-order inactivation rate constant (ki) against EDAC concentration was also linear (Figures 4A and 4B insets). The second-order rate constants of field bean and sweet potato PPO were 0.099±0.02 and 0.105±0.03 M−1·min−1 respectively. These results indicate the occurrence of an irreversible complex between EDAC and PPO. A plot of log ki versus log [EDAC] yielded a slope of 1.25±0.21 (r>0.99) and 1.10±0.12 (r>0.99) for field bean and sweet potato PPO respectively. The stoichiometry of the inactivation reaction is near 1.0 with respect to EDAC, indicating that a single carboxylate group is essential to both field bean PPO and sweet potato PPO activities. The concentrations of EDAC needed for inhibition (20–300 mM) were relatively high when compared with other studies. It could be partly due to the high level of glutamic acid and aspartic acid in field bean and sweet potato PPOs [16,21]. Similar conditions were used for the modification of glutathione transferase (100 mM) [22], oxalate oxidase (150 mM) [23] and α-1,4-glucan lyase (200 mM) [24].
Figure 3. Multiple alignment of plant PPO sequences.

Multiple amino acid sequence alignment of the catalytic region of PPOs. The invariant glutamic and tryptophan residues are indicated by down arrows. The multiple alignment was generated using Multalin (http://www.expasy.org). The PPO sequences were retrieved from UniprotKB/Swiss-Prot release and from http://www.ncbi.nlm.nih.gov. Numbers on the left side are accession numbers of the protein. The amino acid numbering is with respect to that of sweet potato PPO [21].
Figure 4. Inactivation of PPO by EDAC and GME.

(A) Field bean PPO and (B) sweet potato PPO. PPO was modified at different concentrations of EDAC (■, 20 mM; ●, 50 mM; ▲, 100 mM, ▼, 140 mM; ◆, 200 mM; ◇, 300 mM) and 275 mM GME in 50 mM sodium acetate buffer (pH 4.8) at 25 °C. Inactivation was quenched at the indicated times and the residual PPO activity was determined using TBC as substrate. Insets: plot of pseudo first-order inactivation rate constant as function of EDAC concentration. The slopes represent the second-order inactivation rate constant.
Subunit assembly and molecular dimensions of PPO
The effect of SDS- and acid-pH-induced activation on the molecular dimensions of field bean PPO was evaluated by size-exclusion studies on a TSK-G2000 SWXL column. The results are summarized in Figure 5. Native PPO of molecular mass approx. 120 kDa eluted as a single symmetrical peak with a retention time of 13.39 min. In contrast, the SDS- and acid-pH-activated forms elute much earlier with a retention time of 11.37 min (Figure 5A, panels 2 and 3). The retention time of 13.39 min corresponds to the tetrameric form of native PPO [16]. This decrease in the retention time of the activated PPOs as compared with native PPO could be due to either aggregation or enhancement in the hydrodynamic radius.
Figure 5. Evaluation of the molecular dimensions of field bean PPO.

(A) Size-exclusion chromatography profile of field bean PPO on a TSK gel G2000 SWXL (7.8 mm×30 cm; 5 μm) column, using a Waters HPLC system equipped with Waters 2996-photodiode array detector set at 230 nm. The column was pre-equilibrated with 0.1 M NaPi buffer (pH 7.0) containing 0.1 M Na2SO4 at a flow rate of 0.5 ml/min. Panel 1: native PPO; panel 2: SDS-activated PPO, eluted in a buffer containing 1.25 mM SDS; and panel 3: acid-pH-activated PPO, eluted in 100 mM glycine/HCl (pH 2.5). Inset: determination of Stokes radius. The proteins used to construct the calibration curve were cytochrome c (17.0 Å), carbonic anhydrase (21.2 Å), BSA (33.9 Å) and thyroglobulin (79.9 Å). (B) SDS/PAGE (10% T and 2.7% C) profile of PPO. Native and activated PPOs were subjected to SDS/PAGE under reducing conditions, followed by Coomassie Brilliant Blue staining. Lane 1: native PPO; lane 2, SDS-activated PPO; lane 3, acid-activated PPO. Position of molecular mass markers (97.0, 66.0, 45.0, 30.0, 20.1 and 14.4 kDa) is denoted by arrows. RT, retention time.
Light scattering measurements of PPO at 325 nm revealed a decrease in the absorbance (A) with increasing SDS concentration. These results advocate that the decrease in elution volume of the activated forms is not due to aggregation. Furthermore, SDS/PAGE of the activated forms show a similar subunit size of approx. 30 kDa, indicating that they still exist as tetramers of 30 kDa (Figure 5B).
The hydrodynamic radii (Stokes radius) of native and activated PPO were determined by the method of Uversky [25]. A set of proteins of known molecular mass and Stokes radii were used to construct the calibration curve. The hydrodynamic radius for native PPO was 49.1±2 Å (1 Å=0.1 nm; Figure 5A, inset). The hydrodynamic radius of SDS- and acid-pH-activated PPO increased to 75.9±0.6 Å. These results evidence that the decrease in the retention volume of the activated PPOs is due to an enhanced hydrodynamic radii (Figure 6A). The observed increase in the hydrodynamic radius could be due to either swelling of the molecules or due to a partial unfolding upon SDS/acid-pH treatment. The PPO that eluted at 11.37 min cross-reacts with anti-PPO (results not shown).
Figure 6. CD spectra of field bean PPO.

(A) Far-UV CD spectra. (·······) Native PPO in 50 mM Tris/HCl buffer (pH 7.0); (–·–·–·–) SDS-activated PPO; (–––) pH 2.5-activated PPO; and (– – –) pH 4.5-activated PPO. (B) Near-UV CD spectra. (–––) Native PPO in 50 mM Tris/HCl buffer (pH 7.0); (–·–·–·–) SDS-activated PPO; (– – –) acid-pH-activated PPO (pH 2.5); and (·······) acid-pH-activated PPO (pH 4.5). The measurements were carried out on a Jasco J-810 automatic spectropolarimeter at 25 °C. The path length of the cell used was 1 mm in the far-UV region and 10 mm in the near-UV region. The scan speed was 20 nm/min and spectra were taken as an average of three scans.
Glutaraldehyde cross-linking studies were carried out to study the effect of SDS and acid-pH activation on the subunit configuration of PPO. For the cross-linked forms of native and activated PPO, only the tetrameric configuration was observed (results not shown), suggesting that neither SDS nor acid-pH activation brought about any change in the quaternary structure.
SDS and acid-pH activation induce partial unfolding: intrinsic fluorescence
If the activation of PPO by SDS and acid-pH caused a conformation change, the intrinsic fluorescence of Tyr/Trp is likely to be affected. The emission spectra of native PPO had an emission maximum at 330 nm, suggesting that the tryptophan residues were buried in a predominantly hydrophobic milieu shielded from the solvent. The emission intensity of the PPO was found to decrease upon activation with either SDS or acid-pH, with no shift in the emission maximum. The intrinsic fluorescence spectra of the SDS-activated PPO were quenched to a greater extent than the acid-activated PPO. This quenching of fluorescence increased with increasing SDS concentrations. These results further strongly evidence a conformational change, which probably occurs during the initial binding and activation by SDS. Binding of SDS beyond that required for maximum activity (1.25 mM) quenches the fluorescence and reflects gross conformational changes due to inactivation. Collisional quenching experiments with acrylamide were performed to assess the accessibility of tryptophan residues in the SDS-activated form of PPO and corresponding Stern–Volmer constants (Ksv) were calculated. Stern–Volmer plots for SDS-activated PPO show increasing slopes with increasing SDS concentrations, suggesting that the tryptophans in the activated forms are more accessible. Native PPO had a lower Stern–Volmer constant, approx. 3.86±0.4 M−1. The higher Stern–Volmer constant, 4.26±0.5 M−1, following SDS activation is indicative of a slightly disrupted tertiary structure with tryptophans that are more exposed to the solvent. The higher Ksv value is also consistent with a more open conformation of SDS-activated PPO. These results taken together with the fluorescence quenching implicate a subtle yet measurable change in the tertiary structure of PPO upon activation with SDS.
Far-UV CD was used to examine the secondary structure of the native and activated forms of PPO (Figure 6A). The far-UV CD spectra of native PPO indicated the presence of 29% helix, consistent with the helix content reported for PPOs [21]. The far-UV CD spectra of the SDS- and acid-pH-activated forms showed a decrease in the intensity of the negative band at 208 and 222 nm, suggesting changes in the secondary structure. Secondary structure analysis indicated a decreased α-helical content for both the SDS- and acid-pH-activated PPO.
The near-UV CD spectrum of the native PPO reveals positive ellipticity peaks centred at 284 and 291 nm. This indicates a rigid asymmetric environment of the aromatic residues. The ellipticity values at these wavelengths decreased upon SDS activation and activation at pH 4.5 (Figure 6B). In contrast, the spectrum of PPO activated at pH 2.5 showed a shift with high positive ellipticity centred at 280 nm and an increased ellipticity at 291 nm. The positive ellipticity at 291 nm is attributable to tryptophan's environment. The differential changes observed at this wavelength are indicative of a change in the tryptophan environment as a result of the loss of tertiary structure. The loss in tertiary structure is apparently of a higher magnitude for the PPO activated at pH 2.5, compared with the SDS-activated form.
Limited proteolysis
Limited proteolysis has been effectively used to monitor protein surface regions, ligand-induced conformational changes and protein folding as well as unfolding. The vulnerability of a protein to degradation by a protease is governed by conformational parameters one of which is accessibility. Figure 7 shows the susceptibility of native and the activated PPOs to trypsin digestion as studied by RP-HPLC. An extensive proteolytic degradation of the activated PPOs occurs as evidenced by the increased number of peptide fractions in the digests of activated PPO (Figure 7, panels B and C). The presence of multiple peptide peaks eluting early in the acetonitrile gradient is suggestive of increased protease accessibility, reckoned by a more open conformation and/or partial unfolding. The resistance to proteolytic digestion by native PPO points to a more compact conformation as compared with the SDS- or acid-pH-activated PPO.
Figure 7. RP-HPLC profile of the tryptic digest of field bean PPO.

Proteolysis was performed using Tos-Phe-CH2Cl–trypsin (2%, w/w) at pH 8.2 at 37 °C. The tryptic digest was analysed by RP-HPLC using a Waters Symmetry Shield C18 column (4.6 mm×150 mm; 5 μm) and water/acetonitrile gradient containing 0.1% (v/v) PFPA. Peptides were detected at 230 nm. Panel A: native field bean PPO; panel B, SDS-activated field bean PPO; and panel C, acid-pH-activated field bean PPO. RT, retention time.
Thermal stability of activated PPO
Structural changes induced by either SDS or acid shock (pH 2.5) were further assessed by monitoring the effect of temperature (25–95 °C) on the catecholase activity of activated PPO (Figure 8). Native PPO was apparently more thermostable than the activated forms. The activity of the native enzyme remained stable between 20 and 60 °C beyond which the enzyme lost activity rapidly probably due to denaturation. In contrast, the activated forms were more thermolabile with loss in activity observed at approx. 40 °C. The loss in activity of the acid-pH-activated form was gradual as compared with the SDS-activated form. The greater thermostability of native PPO can be attributed to the compact conformation as implicated by the smaller Stokes radius (Figure 5A). Native, SDS-activated and pH 2.5-activated PPO exhibit a Tm of 70.4, 60.7 and 51.7 °C respectively. These observations suggest that the increased thermolability upon activation is probably due to the partial unfolding as evidenced by the changes in secondary and tertiary structure (Figure 6). The semi-logarithmic plots of residual activity versus incubation time at different temperatures are characterized by single straight lines of r>0.98. Therefore the denaturation process can be attributed to a single exponential decay, for both the native and activated forms (Figure 8). The semi-logarithmic plots also indicate that the thermal inactivation of PPO native and activated forms follows first-order kinetics. The Arrhenius plots for irreversible denaturation, natural logarithm of kr versus reciprocal of the absolute temperature are linear (r>0.99) for native and activated PPOs in the temperature range evaluated. The calculated Arrhenius activation energy (Ea) for thermal inactivation is 167.2, 84.4 and 56.3 kJ/mol for native, SDS-activated and acid-pH-activated PPO respectively. The half-life of native PPO at 60 °C was 85 min, whereas that of the SDS-activated PPO was 4-fold lower. Concurrently, the activation energy decreased by 83.6 kJ/mol with an increase in entropy (ΔS*) from −199.5 to +31.9 J·K−1·mol−1. The decrease in Ea and increase in ΔS* are greater for the acid-pH-activated PPO, indicating that it is more thermolabile than the SDS-activated form (Figure 8, Table 2). The dramatic change in ΔS* suggests that the transition to the activated state presents a more disordered structure than the native PPO. The significant changes in the entropy and the difference in the slopes of the Arrhenius plots indicate that the activation of PPO is due to a conformational change as evidenced earlier by the significant changes in the hydrodynamic radii, intrinsic fluorescence and CD spectra and limited proteolysis data.
Figure 8. Kinetics of thermal inactivation of field bean seed PPO.

(A) Native PPO incubated in 25 mM Tris/HCl (pH 7.0) containing 1.2% NaCl. ●, 60 °C; ○, 65 °C; ■, 70 °C; and □, 75 °C. (B) SDS-activated PPO incubated in 25 mM Tris/HCl (pH 7.0) containing 1.25 mM SDS. ●, 50 °C; ○, 55 °C; ■, 60 °C; and □, 65 °C. (C) Acid-pH-activated PPO incubated in 25 mM glycine/HCl (pH 2.5). ○, 40 °C; ●, 45 °C; □, 50 °C; and ■, 55 °C. Samples were incubated at the desired temperatures under respective conditions. Aliquots of the enzyme were removed at the indicated time intervals and assayed using TBC as the substrate. (D) Arrhenius plots of PPO inactivation. ▲, Native PPO; ▼, SDS-activated PPO; and ●, acid-pH-activated PPO.
Table 2. Thermal inactivation parameters of native, SDS-activated and acid-pH-activated PPO.
| PPO | Ea (kJ/mol) | Tm ( °C) | Incubation temperature ( °C) | Half-life (min) | Inactivation rate constant (kr×10−3 s−1) | ΔG* (kJ/mol) | ΔH* (kJ/mol) | ΔS* (J·K−1·mol−1) |
|---|---|---|---|---|---|---|---|---|
| Native | 167.2 | 70.4 | ||||||
| 60 | 85 | 3.3 | 97.9 | 164.2 | 199.5 | |||
| 65 | 54 | 5.7 | 97.4 | 164.2 | 197.4 | |||
| 70 | 24 | 11.8 | 97.4 | 164.2 | 194.9 | |||
| 75 | 10 | 30.7 | 96.2 | 164.2 | 195.7 | |||
| SDS-activated | 84.4 | 60.7 | ||||||
| 50 | 47 | 7.27 | 92.8 | 81.9 | 34.4 | |||
| 55 | 28 | 13.9 | 92.4 | 81.9 | 32.8 | |||
| 60 | 21 | 22.7 | 92.4 | 81.9 | 31.9 | |||
| 65 | 10 | 28.7 | 92.8 | 81.5 | 33.2 | |||
| Acid-pH-activated | 56.3 | 51.7 | ||||||
| 40 | 31 | 5.9 | 90.3 | 66.4 | 77.3 | |||
| 45 | 20 | 10.3 | 90.3 | 66.4 | 76.4 | |||
| 50 | 14 | 13.7 | 91.0 | 65.9 | 77.3 | |||
| 55 | 8 | 30.0 | 90.3 | 65.9 | 74.3 |
DISCUSSION
Most enzymes, with a few exceptions, are inactivated when exposed to extremes of pH and anionic detergent like SDS, one of the exceptions being PPO [26–28]. It is well documented that plant PPOs are activated by a plethora of physical and chemical treatments [4]. The molecular mechanism by which SDS binding or acid shock mediates activation of plant PPOs is yet unknown. Most models invoke some form of allosterism based on observations that detergent binding often results in a conformational change in proteins. Kenten [7] observed SDS-induced activation of a crude preparation of broad bean PPO and attributed this to a limited conformational change. Extending these studies to homogeneous preparations of broad bean PPO, Moore and Flurkey [4] suggest that at the levels of SDS needed for activation, a minor conformational change may occur, which leads to opening or unlocking of the active site, resulting in an enhanced enzyme activity. Using a homogeneous preparation of field bean PPO that exists as a single isoform, we have investigated the biochemical and structural changes that are associated with PPO activation by SDS and acid shock. In plants, PPO functions as an inducible antinutritive defence against herbivore insects and pathogen attack. During herbivore insect-feeding, PPO is mixed with endogenous polyphenolic substrates and the resulting quinones alkylate essential amino acids of the insect dietary protein, making them nutritionally unavailable to the insect [2]. Therefore any increase in PPO levels or activity would be very useful to the plant in its defence against herbivory.
Homogeneous field bean PPO, as evidenced by the release of a single N-terminal asparagine, exists as a single isoenzyme of molecular mass approx. 120 kDa [16] and is a homotetramer of approx. 30 kDa. A marked increase in field bean PPO activity occurred after exposure to either pH 2.5 or 4.5 when compared with pH 7.0. This activation process at pH 2.5 was transient, which decreased significantly after 30 min. Activation of field bean PPO also occurred in the presence of SDS (Figure 1). The concentration of SDS required for half the maximal activation is 0.6 mM, similar to that reported for plant PPOs [4,29,30]. The SDS concentration of 1.25 mM required for maximum PPO activity is below the determined CMC of SDS [18]. The activation of field bean PPO increased linearly with the SDS concentration up to 1.25 mM and decreased thereafter (Figure 1). Moore and Flurkey [4] observed that broad bean leaf PPO was activated by SDS in a sigmoidal manner below the CMC and attributed it to a co-operative interaction between SDS binding and activation. The activity of Vicia faba leaf tyrosinase was also enhanced 6-fold by exposure to pH 3.0 for 2 min followed by neutralization [11]. In contrast, the amount of SDS needed to activate a Xenopus tyrosinase was related to the maximum number of detergent monomers in solution [15].
In the present study, the activation by either SDS or acid-pH introduced similar changes in the Vmax values with little change in the Km for TBC (Table 1). The changes in the kinetic parameters are more obvious when the catalytic efficiency, kcat, of the activated PPOs is compared with native field bean PPO. The activation by SDS decreased the Ea of TBC oxidation, thereby enhancing the catalytic efficiency. SDS was a better activating agent than acid-pH treatment as evidenced by the higher kcat value (Table 1). This increase in catalytic efficiency in both the SDS and acid-pH activations can be attributed to the increase in the catalytic power similar to SDS-activated potato leaf PPO [31] and table beet PPO [32]. This increased PPO turnover (kcat) by acid activation can be seen as a mechanism that significantly enhances the production of quinones, which are implicated in cross-linking of insect dietary proteins. Mild denaturing conditions have led to activation in Vic. faba tyrosinase, while stronger denaturing conditions caused irreversible loss in enzyme activity [11].
The increase in the catalytic efficiency of field bean PPO is associated with a shift in the pH optimum from 4.5 to 6.0 (Figure 2). The activity of native field bean PPO at pH 6.0 is 10% of the activated form (Figure 2). The low pH optimum of 3.5 reported for a broad bean leaf PPO was abolished in the presence of SDS [4]. A latent mushroom tyrosinase is reported to show no activity at the pH optimum of the SDS-activated form [33]. The observed shift in the pH optima of field bean PPO (Figure 2) could be related to the displacement of sensitive pKa values at the catalytic site. Displacement of the pH optimum of Vic. faba leaf PPO towards higher pH values as SDS concentration increased has been reported [4]. Jimenez and Garcia-Carmona [34] have opined that among the binding centres for SDS, there was one responsible for pH dependence of PPO activity, the dissociation constant being 0.52 mM. The pH optimum of 4.5 for native PPO reflects the role of a carboxy group at the active site. In the crystal structure of sweet potato PPO, Klabunde et al. [21] observe that Glu236 is hydrogen-bonded to a solvent molecule close to the dimetal active site, which donates a proton and thereby functions as a general base/acid in the oxidation reaction of diphenols. Robert et al. [35] hypothesize that the acidic pKa of palmito PPO refers to an acidic amino acid at the active centre. Interestingly, in this study, the multiple alignment of several plant PPO protein sequences also showed that a glutamic residue, corresponding to that of Glu236 of sweet potato PPO, is invariant in all plant PPOs, implicating a vital role for this residue in catalysis (Figure 4). Chemical modification of glutamic residues of field bean and sweet potato PPO resulted in the abolition of catecholase activity, following pseudo first-order kinetics (Figure 4). The observed stoichiometry of near 1 for the inactivation of sweet potato and field bean PPO by EDAC is implicative of a single core carboxylate involved in catalysis. The shift in the pH optimum from 4.5 to 6.0 upon activation can therefore be explained by perturbations in the ionization constant of the crucial γ-carboxy group of glutamic residue, as a consequence of changes in the microenvironment caused by the observed conformational changes. The pKa values of sensitive groups can often be substantially perturbed from the normal values and are even more perturbed in the enzyme–substrate complex. The pKa of Glu270 of carboxypeptidase [36] and Glu35 of lysozyme [37] titrates with anomalously high pKa values of 7.0 and 6.5 respectively.
No change in the molecular mass, subunit dissociation or protein aggregation was noticed in the activated PPOs when examined by SDS/PAGE (Figure 5B). Glutaraldehyde cross-linking and light scattering at 325 nm also indicated that upon activation, the field bean PPO maintained the same quaternary structure. The activation of field bean PPO however was accompanied by a substantial increase in the hydrodynamic size. The Stokes radius increased from 49.1±2 to 75.9±0.6 Å. Similar electrophoretic mobilities in native PAGE of the activated PPOs suggest that the charge to mass ratio remained unaltered (results not shown). The increase in the hydrodynamic radii of the activated field bean PPO can thus be attributed to a more ‘open conformation’ in and around the active site. This structural change thereby unlocks the catalytic site for easy access by the substrates, resulting in the increased turnover of PPO (Table 1). A similar increase in the Stokes radii of the SDS- and acid-pH-activated PPO is suggestive of similar conformational changes between the two forms (Figure 5A, inset). The increase in the Stokes radius of a broad bean PPO was previously speculated to arise from a conformational change [27,38]. Moore and Flurkey [4] interpreted the small increase in the size of broad bean leaf PPO to be due to the bound SDS molecules. The activation and kinetic behaviour of a latent thylakoid-bound grape (Vitis vinifera) PPO was interpreted in terms of a pH-induced slow transition, which did not affect the active site of the enzyme [39,40]. Moore and Flurkey [4] by sedimentation co-efficient measurements found little or no differences in the molecular mass of broad bean leaf PPO either in the presence or absence of SDS.
The far-UV CD spectrum of field bean PPO indicated 29% helical content for native PPO (Figure 6). The decrease in α-helical content of SDS- and acid-pH-activated PPO suggests subtle changes in secondary structure upon activation. Near-UV CD studies also showed a loss in tertiary structure upon SDS/acid-pH activation (Figure 6B). However, a significantly higher loss of secondary structure occurred at pH 2.5. The subtle differences observed at 291 nm relate to a change in the tryptophan environment (Figure 6B). The striking similarity between the changes in the far-UV CD spectrum of SDS- and acid-pH-activated field bean PPO points to similar perturbations in the secondary structure. These results taken together with the decreased intrinsic fluorescence and increased tryptophan accessibility to collisional quenching are in accordance with a partial unfolding of field bean PPO, leading to a more open conformation that enhances catalysis. The extensive proteolysis of SDS/acid-pH-activated PPO by trypsin also supports a more unfolded or open conformation for the activated forms under these conditions (Figure 7), thereby increasing their susceptibility to proteolytic digestion. Akhtar and Bhakuni [41] have also noticed that the more open conformation of GOD (glucose oxidase) rendered it more susceptible to proteolytic digestion than the compact deglycosylated GOD.
Data on changes in the tertiary structure of plant PPOs following activation by SDS/acid-pH are yet to be reported; therefore this is the first study. The near-UV and far-UV CD data (Figure 6) pointed to a localized change stemming from the fact that the CD spectra retained their original shape with minor alterations in intensity. Given that the observed conformational changes are localized and not global as evidenced by CD data, it is possible to speculate that these changes are centred in and about the catalytic site. The increased catalytic efficiency (Kcat), shift in the pH optimum caused by a change in the microenvironment of glutamic residue and increased inhibition by tropolone (Table 1) further strengthen the fact that the conformational change involves the catalytic region of the enzyme. In addition to the conserved glutamic residue, the presence of a conserved and invariant tryptophan (Figure 4) in the dimetal catalytic CuB site of PPOs [21], together with the observed increase in solvent accessibility of the SDS- and acid-pH-activated field bean PPO, supports the localized conformational change anchored around this site. Acyl-carrier protein, a small acid protein, adopts a partially unfolded structure at neutral pH, but has a tight fold at acidic pH [42]. The presence of both secondary (Figure 6A) and tertiary structures (Figure 6B) indicated that the activated field bean PPOs are not in the molten globule conformation.
In order to probe the effects of these observed conformational changes, thermal stability of the native PPO versus activated PPO was evaluated. The decrease in the Tm for the activated field bean PPO suggests decreased thermal stability of the enzyme caused by the partial unfolding under these conditions as compared with the more compact native PPO. The thermal stability curves showed a relatively higher stability of native field bean PPO in comparison with the activated field bean PPO (Figure 8). The semi-logarithmic plots of residual activity versus incubation time characterized by a single exponential decay (Figure 8) suggest that the same thermal inactivation mechanism operates for the native and activated forms. The observed single exponential decay for field bean PPO further evidences its homogeneity as reported by us previously [16]. The biphasic nature of heat inactivation isotherms of PPO isolated from different sources has been explained by the presence of more than one isoenzyme in the preparation [35]. Effectively, a biphasic rate of inactivation most likely points to the enzyme being heterogeneous. The calculated thermodynamic parameters over the temperature range studied for native field bean PPO indicated a negative ΔS, whereas the activated PPOs exhibited a positive value. These results further indicated that native PPO presents a more ordered structure than the acid-pH- or SDS-activated PPO and SDS-activated PPO was more ordered than acid-pH-activated PPO. These results further support the fact that the localized secondary and tertiary structural perturbations that accompany SDS/pH activation culminated in a partially unfolded thermolabile PPO.
Conclusion
The partial unfolding of field bean PPO induced by SDS or acid-pH leads to the opening or unblocking of the active site, thereby accelerating the oxidation of the o-diphenols and enhancing the catalytic efficiency severalfold. This conformational change alters the microenvironment of a core glutamic residue at the active site, resulting in a shift in the pKa value of the carboxy group as reflected by the shift in pH optimum and chemical modification of an essential carboxylate. Whether this structural change is a mimic of the in vivo regulatory mechanism of activation upon insect or pathogen attack is yet unknown. PPO, an inducible plant defence protein against insect herbivory, is considered as a defence-related antinutritive oxidative enzyme. The effect of high PPO levels against insect herbivory is proposed to reside in the propensity of PPO-generated o-quinones to covalently modify and cross-link dietary proteins during feeding, resulting in reduced insect nutrition and performance [2]. Overexpression and high levels of PPO in transgenic Poplar leaves have been demonstrated to enhance resistance to herbivory by caterpillars (Malacosoma disstria) [43]. Wounding of plant tissue by either insect herbivory or mechanical injury evokes a jasmonic acid burst, consequently lowering the pH, which is amplified by insect feeding [2]. Elicitors such as salicylic acid and oxalic acid induce defence-related enzyme activities such as peroxidase and PPO against Alternaria alternata, a fungal pathogen [44]. Such acidic milieu would activate PPO, resulting in high activity and enhanced turnover. Cipollini et al. [45] have demonstrated increased PPO, peroxidase, N-acetylglucoaminidase and trypsin inhibitor activity of Arabidopsis thaliana and reduced insect growth following jasmonic acid treatment. Therefore the activation and labile nature of field bean PPO could be an adaptive defence strategy to maximize the supply of PPO-mediated o-quinones to covalently modify proteins and ensure a decreased nutrition to the feeding predator. This activation is therefore critical to the in vivo physiological defence mechanism postulated for plant PPOs.
Acknowledgments
We are grateful to Dr V. Prakash (Director, CFTRI, Mysore, India) for advice and useful suggestions during the course of this investigation. We also thank Mr P.S. Kulashekhar for help in the editing of this paper. We thank Dr S.A. Singh (Department of Protein Chemistry and Technology, CFTRI) for help in CD measurements. S.R.K. is the recipient of a Senior Research Fellowship from Council of Scientific and Industrial Research (New Delhi, India).
References
- 1.Dervall B. J. Phenolase and pectic enzyme activity in the chocolate spot disease of beans. Nature (London) 1961;189:311. [Google Scholar]
- 2.Kessler A., Baldwin I. T. Plant responses to insect herbivory: the emerging molecular analysis. Annu. Rev. Plant Biol. 2002;53:299–328. doi: 10.1146/annurev.arplant.53.100301.135207. [DOI] [PubMed] [Google Scholar]
- 3.Pierpoint W. S. o-Quinones formed in plant extracts. Their reactions with amino acids and peptide. Biochem. J. 1969;112:609–616. doi: 10.1042/bj1120609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore B. M., Flurkey W. H. Sodium dodecyl activation of a plant polyphenol oxidase. J. Biol. Chem. 1990;265:4982–4988. [PubMed] [Google Scholar]
- 5.Mayer A. M., Harel E. Polyphenol oxidases in plants – review. Phytochemistry. 1979;18:193–215. [Google Scholar]
- 6.Mason H. S. Oxidases. Annu. Rev. Biochem. 1965;34:595–634. doi: 10.1146/annurev.bi.34.070165.003115. [DOI] [PubMed] [Google Scholar]
- 7.Kenten R. H. Latent phenolase in extracts of broad bean (Vicia faba L.). 1. Activation by anionic wetting agents. Biochem. J. 1958;68:244–251. doi: 10.1042/bj0680244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugumaran M., Nellaiappan K. Lysolecithin – a potent activator of prophenol oxidase from the hemolymph of the lobster, Homarus americanas. Biochem. Biophys. Res. Commun. 1991;176:1371–1376. doi: 10.1016/0006-291x(91)90438-d. [DOI] [PubMed] [Google Scholar]
- 9.Jiang H., Shi C., Xie Y., Xu X., Liu Q. Activation of tobacco leaf polyphenol oxidase by sodium dodecyl sulfate. Indian J. Biochem. Biophys. 2003;40:350–353. [PubMed] [Google Scholar]
- 10.Swain T., Mapson L. W., Robb D. A. Activation of Vicia faba (L.) tyrosinase as effected by denaturing agents. Phytochemistry. 1966;5:469–482. [Google Scholar]
- 11.King R. S., Flurkey W. H. Effects of limited proteolysis on broad bean polyphenol oxidase. J. Sci. Food Agric. 1987;41:231–240. [Google Scholar]
- 12.Robinson S. P., Dry I. B. Broad bean leaf polyphenol oxidase is a 60-kilodalton protein susceptible to proteolytic cleavage. Plant Physiol. 1992;99:317–323. doi: 10.1104/pp.99.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robb D. A., Mapson L. W., Swain T. Activation of the latent tyrosinase of broad bean. Nature (London) 1964;201:503–504. [Google Scholar]
- 14.Laveda F., Nuñez-Delicado E., García-Carmona F., Sánchez-Ferrer A. Reversible sodium dodecyl sulfate activation of latent peach polyphenol oxidase by cyclodextrins. Arch. Biochem. Biophys. 2000;379:1–6. doi: 10.1006/abbi.2000.1838. [DOI] [PubMed] [Google Scholar]
- 15.Wittenberg C., Triplett E. L. A detergent-activated tyrosinase from Xenopus laevis. II. Detergent activation and binding. J. Biol. Chem. 1985;260:12542–12546. [PubMed] [Google Scholar]
- 16.Paul B., Gowda L. R. Purification and characterization of a polyphenol oxidase from the seeds of field bean (Dolichos lablab) J. Agric. Food Chem. 2000;48:3839–3846. doi: 10.1021/jf000296s. [DOI] [PubMed] [Google Scholar]
- 17.Gowda L. R., Paul B. Diphenol activation of the monophenolase and diphenolase activities of field bean (Dolichos lablab) polyphenol oxidase. J. Agric. Food Chem. 2002;50:1608–1614. doi: 10.1021/jf010913s. [DOI] [PubMed] [Google Scholar]
- 18.Paul B. Ph.D. thesis. India: University of Mysore; 2000. Purification and structural characterization of a polyphenol oxidase from field bean (Dolichos lablab) [DOI] [PubMed] [Google Scholar]
- 19.Bradford M. M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 20.Hoare D. G., Koshland D. E. A method for the quantitative modification and estimation of carboxylic acid groups in proteins. J. Biol. Chem. 1967;242:2447–2453. [PubMed] [Google Scholar]
- 21.Klabunde T., Eicken C., Sacchettini J. C., Krebs B. Crystal structure of plant catechol oxidase containing a dicopper center. Nat. Struct. Biol. 1998;5:1084–1090. doi: 10.1038/4193. [DOI] [PubMed] [Google Scholar]
- 22.Xia C., Meyer D. J., Chen H., Reinemer P., Huber R., Ketterer B. Chemical modification of GSH transferase P1-1 confirms the presence of Arg-13, Lys-44 and one carboxylate group in the GSH-binding domain of the active site. Biochem. J. 1993;293:357–362. doi: 10.1042/bj2930357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kotsira V. P., Clonis Y. D. Chemical modification of barley root oxalate oxidase shows the presence of a lysine, a carboxylate, and disulfides, essential for enzyme activity. Arch. Biochem. Biophys. 1998;356:117–126. doi: 10.1006/abbi.1998.0764. [DOI] [PubMed] [Google Scholar]
- 24.Nyvall P., Pedersen M., Kenne L., Gacesa P. Enzyme kinetics and chemical modification of α-1,4-glucan lyase from Gracilariopsis sp. Phytochemistry. 2000;54:139–145. doi: 10.1016/s0031-9422(00)00059-5. [DOI] [PubMed] [Google Scholar]
- 25.Uversky V. N. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins, which denature through the molten globule. Biochemistry. 1993;32:13288–13298. doi: 10.1021/bi00211a042. [DOI] [PubMed] [Google Scholar]
- 26.Flurkey W. H. Polyphenol oxidase in higher plants immunological detection and analysis of in vitro translation products. Plant Physiol. 1986;81:614–618. doi: 10.1104/pp.81.2.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lerner R. H., Mayer A. M., Harel E. Evidence for conformational changes in grape catechol oxidase. Phytochemistry. 1972;11:2415–2421. [Google Scholar]
- 28.Wang J., Constabel C. P. Biochemical characterization of two differentially expressed polyphenol oxidases from hybrid poplar. Phytochemistry. 2003;64:115–121. doi: 10.1016/s0031-9422(03)00159-6. [DOI] [PubMed] [Google Scholar]
- 29.Kenten R. H. Latent phenolase in extracts of broad bean (Vicia faba L.) leaves. Biochem. J. 1957;67:300–307. doi: 10.1042/bj0670300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chazarra S., Cabanes J., Escribano J., Garcia-Canovas F. Partial purification and characterization of latent polyphenol oxidase in iceberg lettuce (Lactuca sativa) J. Agric. Food Chem. 1996;44:984–988. [Google Scholar]
- 31.Sanchez-Ferrer A., Laveda F., Garcia-Carmona F. Substrate-dependent activation of latent potato leaf polyphenol oxidase by anionic surfactants. J. Agric. Food Chem. 1993;41:1583–1586. [Google Scholar]
- 32.Escribano J., Cabanes J., Garcia-Carmona F. Characterization of latent polyphenol oxidase in table beet: effect of sodium dodecyl sulfate. J. Sci. Food Agric. 1997;73:34–38. [Google Scholar]
- 33.Espin J. C., Wichers H. J. Activation of a latent mushroom (Agaricus bisporus) tyrosinase isoform by sodium dodecyl sulfate (SDS). Kinetic properties of the SDS-activation isoform. J. Agric. Food Chem. 1999;47:3518–3525. doi: 10.1021/jf981275p. [DOI] [PubMed] [Google Scholar]
- 34.Jimenez M., Garcia-Carmona F. The effect of sodium dodecyl sulfate on polyphenol oxidase. Phytochemistry. 1996;42:1503–1509. [Google Scholar]
- 35.Robert C. M., Cadet F. R., Rouch C. C., Pabion M., Richard-Forget F. Kinetic study of the irreversible thermal deactivation of palmito (Acanthophoeix rubra) polyphenol oxidase and effect of pH. J. Agric. Food Chem. 1995;43:1143–1150. [Google Scholar]
- 36.Petra P. H., Neurath H. Modification of carboxyl groups in bovine carboxypeptidase A. II. Chemical identification of a functional glutamic acid residue and other reactive groups. Biochemistry. 1971;10:3171–3177. [PubMed] [Google Scholar]
- 37.Parsons S. M., Raftery M. A. Ionization behavior of the catalytic carboxyls of lysozyme effects of ionic strength. Biochemistry. 1972;11:1623–1629. doi: 10.1021/bi00759a013. [DOI] [PubMed] [Google Scholar]
- 38.Lerner H. R., Mayer A. M. Stokes' radius changes of solubilized grape catechol oxidase. Phytochemistry. 1975;14:1955–1957. [Google Scholar]
- 39.Valero E., Garcia-Carmona F. pH-induced kinetic co-operativity of a thylakoid-bound polyphenol oxidase. Biochem. J. 1992;286:623–626. doi: 10.1042/bj2860623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valero G., Garcia-Carmona F. Hysteresis and cooperative behavior of a latent plant polyphenol oxidase. Plant Physiol. 1992;98:774–776. doi: 10.1104/pp.98.2.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akhtar S. M., Bhakuni V. Alkaline treatment has contrasting effects on the structure of deglycosylated and glycosylated forms of glucose oxidase. Arch. Biochem. Biophys. 2003;413:221–228. doi: 10.1016/s0003-9861(03)00127-9. [DOI] [PubMed] [Google Scholar]
- 42.Park S. J., Kim J., Son W., Lee B. J. pH-induced conformational transition of. H pylori acyl carrier protein: insight into the unfolding of local structure. J. Biochem. (Tokyo) 2004;35:337–346. doi: 10.1093/jb/mvh041. [DOI] [PubMed] [Google Scholar]
- 43.Wang J., Constabel P. C. Polyphenol oxidase over expression in transgenic Populus enhances resistance to herbivory by forest tent caterpillar (Malacosoma disstria) Planta. 2004;220:87–96. doi: 10.1007/s00425-004-1327-1. [DOI] [PubMed] [Google Scholar]
- 44.Tian S., Wan Y., Qin G., Xu Y. Induction of defense responses against Alternaria rot by different elicitors in harvested pear fruit. Appl. Microbiol. Biotechnol. 2005;13:1–6. doi: 10.1007/s00253-005-0125-4. [DOI] [PubMed] [Google Scholar]
- 45.Cipollini D., Enright S., Traw M. B., Bergelson J. Salicylic acid inhibits jasmonic acid-induced resistance of Arabidopsis thaliana to Spodoptera exigua. Mol. Ecol. 2004;13:1643–1653. doi: 10.1111/j.1365-294X.2004.02161.x. [DOI] [PubMed] [Google Scholar]