

Abstract
Ferritin is a ubiquitously distributed iron-binding protein. Cell culture studies have demonstrated that ferritin plays a role in maintenance of iron homoeostasis and in the protection against cytokine- and oxidant-induced stress. To test whether FerH (ferritin H) can regulate tissue iron homoeostasis in vivo, we prepared transgenic mice that conditionally express FerH and EGFP (enhanced green fluorescent protein) from a bicistronic tetracycline-inducible promoter. Two transgenic models were explored. In the first, the FerH and EGFP transgenes were controlled by the tTACMV (Tet-OFF) (where tTA and CMV are tet transactivator protein and cytomegalovirus respectively). In skeletal muscle of mice bearing the FerH/EGFP and tTACMV transgenes, FerH expression was increased 6.0±1.1-fold (mean±S.D.) compared with controls. In the second model, the FerH/EGFP transgenes were controlled by an optimized Tet-ON transactivator, rtTA2S-S2LAP (where rtTA is reverse tTA and LAP is liver activator protein), resulting in expression predominantly in the kidney and liver. In mice expressing these transgenes, doxycycline induced FerH in the kidney by 14.2±4.8-fold (mean±S.D.). Notably, increases in ferritin in overexpressers versus control littermates were accompanied by an elevation of IRP (iron regulatory protein) activity of 2.3±0.9-fold (mean±S.D.), concurrent with a 4.5±2.1-fold (mean±S.D.) increase in transferrin receptor, indicating that overexpression of FerH is sufficient to elicit a phenotype of iron depletion. These results demonstrate that FerH not only responds to changes in tissue iron (its classic role), but can actively regulate overall tissue iron balance.
Keywords: ferritin H, iron homoeostasis, kidney, tetracycline, tissue-specific expression, transferrin receptor
Abbreviations: CMV, cytomegalovirus; Dox, doxycycline; EGFP, enhanced green fluorescent protein; FerH, ferritin H; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; H/E, haematoxylin and eosin; IRE, iron responsive element; IRP, iron regulatory protein; LAP, liver activator protein; SN, substantia nigra; tTA, tet transactivator protein; rtTA, reverse tTA; TNF, tumour necrosis factor; TfR, transferrin receptor; UTR, untranslated region
INTRODUCTION
Ferritin is a ubiquitously expressed protein that stores cellular iron in a non-reactive form and thus plays a central role in the regulation of iron homoeostasis. The protein comprises 24 subunits of two types, H and L, which are the products of separate genes with separable functions. FerH (ferritin H) has a ferroxidase activity that oxidizes Fe(II) to Fe(III) [1], while FerL is associated with iron nucleation and protein stabilization [2]. The ratio of H to L subunits in the ferritin protein can affect its properties, including the ability to take up iron [3–5], to affect proliferation [6] and to reduce the impact of oxidant- and cytokine-induced stress [7,8].
Ferritin is regulated at multiple levels in response to different stimuli: at the translational level, ferritin is regulated by IRPs (iron regulatory proteins), which bind to an element [the IRE (iron responsive element)] in the 5′-UTR (5′-untranslated region) of ferritin and repress its translation (reviewed in [9,10]). IRPs also bind to IREs in the 3′-UTR of TfR1 (transferrin receptor 1), where they function to stabilize TfR mRNA [9]. Since iron decreases the activity of IRP1 and destabilizes IRP2, the net effect of excess iron is to increase iron storage via ferritin and to decrease iron transport by TfR1; conversely, the effects of iron restriction are decreased ferritin and increased TfR1 expression [9]. Ferritin is thus a key component of an intrinsic homoeostatic mechanism used by cells to restore iron balance when confronted with excess or insufficient iron.
In addition to its role in normal iron homoeostasis, ferritin plays a critical role in mitigation of inflammatory and oxidant stress by reducing the participation of iron in free-radical-generating reactions (see [11] for a review). The impact of FerH subunit induction in vitro has been assessed previously in several studies (reviewed in [11a]). Cells overexpressing FerH have slower growth rates [6], and increased resistance to oxidant stress [6,12,13]. These attributes are dependent on functional ferroxidase activity in the FerH subunit [6].
Increases in ferritin have been reported in numerous pathophysiological settings, including ischaemia/reperfusion injury, atherosclerosis, cancer and neurodegenerative disease [14]. It has been hypothesized that the increase in ferritin in these diverse contexts is elicited as a physiological cytoprotective response, designed to sequester iron and limit its participation in the oxygen free-radical-generating reactions that contribute to the pathology of these diseases. However, direct assessments of the ability of ferritin either to regulate iron homoeostasis or to serve such a cytoprotective function in vivo have been limited. In one study, FerH overexpression in the dopanergic SN (substantia nigra) neurons of the mouse brain was associated with reduction in markers of oxidant stress following treatment of mice with the Parkinson's-inducing agent 1-methyl-4-phenyl-1,2,3,6-tetrapyridine [15].
In order to assess the role of FerH in vivo, we have constructed a transgenic mouse model in which FerH can be expressed conditionally in a variety of tissues. We report that induction of FerH in vivo results in a phenotype of tissue-specific iron depletion. Further, our results indicate for the first time that FerH can act as a potent regulator of iron metabolism in tissues, and that these effects can dominate normal homoeostatic mechanisms.
EXPERIMENTAL
Development of a transgenic construct
A 2.4 kb BamHI/EcoRI fragment of a genomic clone of the murine FerH gene [7] was mutagenized using PCR to delete the first cytosine base in the CAGUG stem loop region of the IRE. This reduces the ability of the IRE to bind IRP over 100-fold [16]. This fragment was cloned into the Pvu2 site of the pBI tet expression vector (Clontech, Palo Alto, CA, U.S.A.) through blunt-ended ligation to yield the pTetMFhEGFP vector shown in Figure 1. This vector also contains the EGFP (enhanced green fluorescent protein) gene in the opposite orientation to the FerH gene, thus enabling tetracycline or Dox (doxycycline) regulation of both FerH and EGFP.
Figure 1. Bicistronic transgenic construct.

The transgenic construct that provides for tet-regulated transcription of both the murine FerH and EGFP genes was prepared as described under the Experimental section and is depicted as a linearized sequence. Note the central position of the bicistronic promoter, and the mutation in the IRE of the FerH gene that removes the transgenic ferritin from iron-based translational feedback regulation. SV40, simian virus 40; TRE, tetracycline responsive element.
Transient transfections and Western-blot analysis
HeLa Tet-OFF cells (Clontech) were transfected with pTetMFhEGFP using Lipofectamine™ (Invitrogen), cultured for 24 h and treated with or without 2 μg/ml Dox (Sigma) for 72 h before harvesting cells for Western-blot analysis. Antibodies used were from Biosource (FerH), Alpha Diagnostic International (San Antonio, TX, U.S.A.) (TfR type 1) and Research Diagnostics (Flanders, NJ, U.S.A.) [EGFP and GAPDH (glyceraldehyde-3-phosphate dehydrogenase)]. Immunoreactive proteins were detected using the PicoWest chemiluminescent solution (Pierce), according to the manufacturer's instructions. Developed X-ray films were quantified using the UnScanIt program (Silk Scientific, Orem, UT, U.S.A.). Equivalent loading and transfer were confirmed by staining with Ponceau S or by Western-blot analysis with GAPDH.
Generation of single and double transgenic mice
Pronuclear microinjections into fertilized eggs of FVB/N mice were performed at the University of Michigan Transgenic Animal Model Core using standard methods [17]. The FerH transgene was detected by PCR using one primer in the FerH sequence (5′-ACTGGTCACTGAGGCAGTGCATG-3′) and one in the vector β-globin poly AAA sequence (5′-GAGGGTCCATGGTGATACAAGGG-3′). EGFP was detected using the primers 5′-TCTCGTTGGGGTCTTTGCTCAGGG-3′ and 5′-AACATCCTGGGGCACAAGCTGGAG-3′. Single transgenic mice are designated FerHmIRE/TetO7/EGFP in the present study. Mice were treated using approved methods according to the guidelines established by our institutional animal care and use committee.
To generate double transgenic mice, FerHmIRE/TetO7/EGFP mice were bred with tTACMV Tet-OFF transactivator (where tTA and CMV are tet transactivator protein and cytomegalovirus respectively) [18], or rTALAP-1 [19] (which carries the rtTA2S-S2LAP Tet-ON gene; where rtTA is reverse tTA) reverse transactivator, strains. tTACMV Tet-OFF mice (Jackson Laboratories, Bar Harbor, ME, U.S.A.; stock number 003271) were backcrossed one generation into the FVB background before crossing to the FerHmIRE/TetO7/EGFP mice. The tTACMV transgene was detected as described (http://jaxmice.jax.org/pub-cgi/protocols/protocols.sh?objtype=protocol&protocol_id=502) and double transgenics were identified through multiple PCR screens. Double transgenic FerHmIRE/TetO7/EGFP×tTaCMV Tet-OFF mice are referred to as ‘CMVFerH’ mice in the present study. rTA LAP-1 mice (now distributed by the European Mouse Mutant Archive, http://www.emmanet.org) were backcrossed similarly into the FVB background before crossing to FerHmIRE/TetO7/EGFP mice. The transgene was detected by PCR as described in [21]. FerHmIRE/TetO7/EGFP×rTA LAP-1 double transgenic mice are referred to as ‘LAPFerH’ mice in the present study.
Dox administration
Doxycycline hyclate (Sigma; 2 mg/ml) was administered into drinking water containing 5% (w/v) sucrose for 2 weeks. Water containing 5% sucrose was also provided to Dox-free control animals. When mice were treated with Dox for 8 weeks, Dox was added to the solid diet at a final concentration of 2 g/kg.
Visualization of whole animal EGFP fluorescence
EGFP expression was visualized at necropsy using a fibre optic epi-configured light source with 470/40 nm (excitation) filter set, and a 515 nm (emission) viewing plate (Lightools Research, Encinitas, CA, U.S.A.). Images were acquired digitally.
Blood iron measurements
Blood was collected at necropsy and analysed for iron using the ferrozine method [22]. Haematocrits were measured in micro-haematocrit tubes (Fisher).
Creatinine assay
Creatinine levels in urine were determined from urine taken at the time of killing using a Creatine assay kit (Cayman Chemical, Ann Arbor, MI, U.S.A.). Samples and standards were evaluated in triplicate. Results are reported as μg/ml.
IRP activity assay
Cytosolic extracts for IRP assay were prepared essentially as described in [23]. The interaction between the IRP and its IRE binding site was analysed in a gel shift assay [24] using an IRE RNA probe prepared from a BamHI linearized pST18hFh-IRE plasmid template (obtained as a gift from Dr Prem Ponka, McGill University, Montreal, ON, Canada).
Histological evaluation of tissues
Following killing and gross inspection of mouse carcasses, portions of the kidney, spleen, liver, heart and small intestine were fixed in 4% (w/v) neutral buffered paraformaldehyde overnight. Tissues were trimmed, imbedded in paraffin, cut at 6 μm, stained with H/E (haematoxylin and eosin) and assessed by light microscopy.
RESULTS
Development of a transgenic construct
To attain conditional tissue-specific expression of the FerH transgene, we used the tet system (reviewed in [25,26]). In this system, the transgene is cloned downstream of a minimal promoter fused to seven tet operator sequences, where it remains transcriptionally silent until recognized by a tet transactivator, which is provided as a second independent transgene. The activity of the transactivator is regulated by tetracycline or its analogue Dox: depending on the transactivator used, it is either activated (Tet-ON) or repressed (Tet-OFF) in the presence of Dox [18]. In our experiments, mouse FerH was used as the primary transgene, with the following modifications. First, in order to express the FerH protein irrespective of cellular iron levels, we mutated the IRE of the murine FerH transgene to remove the transgene from known translational control [16]. Secondly, to readily discern cells that express transgenic FerH, we used an expression plasmid designed for bicistronic expression of FerH and EGFP. Expression of EGFP is easily visualized in living animals and in tissues using illumination with blue light [27]. The transgenic FerH construct, pTetMFhGFP, is depicted in Figure 1.
Evaluation of the FerH transgenic construct in tissue culture and production of transgenic founders
This bicistronic transgenic FerH vector was transiently transfected into HeLatTA Tet-OFF cells to test its ability to drive overexpression of both the FerH and the EGFP proteins. As expected, the EGFP gene was strongly expressed in the absence of Dox, but was not expressed following Dox addition (Figure 2). Endogenous human FerH gene (upper band) was present in all the cells, regardless of Dox. In contrast, the murine FerH transgene (lower band), like the EGFP gene, was strongly regulated by Dox (Figure 2). These results indicated that the transgene construct was capable of mediating ferritin overexpression even in cells with high endogenous levels of ferritin, and that this expression was appropriately regulated by Dox. This construct was injected into FVB mouse embryos, and potential FerHmIRE/TetO7/EGFP founders were identified. Founders were then bred with mice carrying tTA genes.
Figure 2. Construct expression is Dox regulated in vitro.
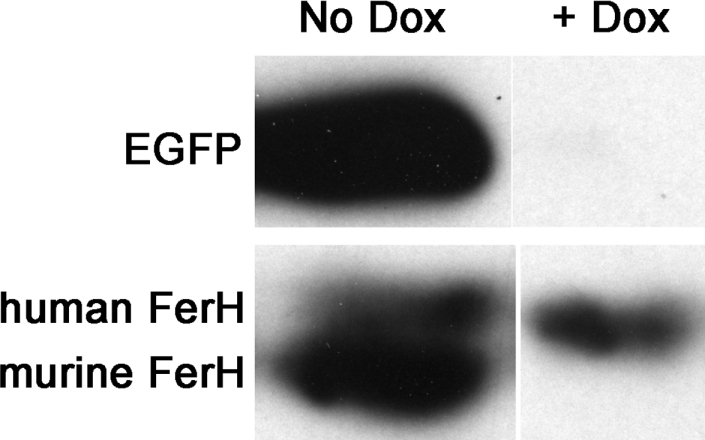
HeLa Tet-OFF cells were transiently transfected with pTetMFhEGFP, and either treated with Dox (lane 2) or left untreated (lane 1). Lysates were analysed for ferritin content by Western blotting. Note that the endogenous human FerH (upper band) is unaffected by Dox, while the transgenic murine FerH (lower band) is completely restricted by Dox treatment.
Phenotype of the CMVFerH mouse in skeletal-muscle tissue
Initially, FerHmIRE/TetO7/EGFP founder mice were crossed with mice containing a tTA Tet-OFF transactivator, in which the tet activator protein is driven by the broadly active CMV promoter (tTACMV). This transactivator theoretically drives expression of tet-responsive genes in a wide array of tissues [18]. We found that EGFP fluorescence was primarily evident in the skeletal muscle and the omentum adjacent to the spleen (Figures 3A and 3B). To determine the degree to which FerH was being expressed in these tissues, fluorescent muscle was harvested from three double transgenic mice and three controls and evaluated by Western blotting. Results, depicted in Figure 3(C), indicate that FerH expression is increased by 6.0±1.1-fold (mean±S.D.) in skeletal muscle from the double transgenic CMVFerH mice relative to wild-type or either of the single transgenic mice. As expected, EGFP expression only occurred in the CMVFerH mice. CMVFerH mice displayed no behavioural abnormalities or other signs of toxicity, and were indistinguishable from their control littermates up to the time of killing at 3 months.
Figure 3. tTA enables enhanced FerH and EGFP expression in CMVFerH double transgenic mice.

A mouse carrying the FerHmIRE/TetO7/EGFP transgene construct alone (A) and a mouse carrying both the FerHmIRE/TetO7/EGFP transgene construct and the tTACMV Tet-OFF transactivator transgene (B) were killed and their abdominal contents were visualized by laparotomy. EGFP expression was visualized as described in the Experimental section. As expected, no fluorescence was observed in the absence of the transactivator (A). Small arrows denote EGFP expression in bands of skeletal muscle (M) in the tail and leg. Strong expression in the mesentery/splenic omentum (O) is also denoted. (C) Expression of FerH and EGFP in skeletal muscle was detected by Western blotting using GAPDH as a loading control. Lanes 1, 2 and 3 respectively depict skeletal-muscle samples from wild-type, and single transgenic tTACMV and FerHmIRE/TetO7/EGFP mice. Lanes 4–6 depict protein isolates from double transgenic CMVFerH mice.
Phenotype of the LAPFerH mouse in kidney tissue
To assess the ability of tet transactivators to control expression of the FerH transgene in different tissues, we crossed the FerHmIRE/TetO7/EGFP mice to mice carrying the newly optimized Tet-ON rtTA2S-S2 transgene, which mediates very tightly controlled tet-responsive gene expression [21,28]. This transgene was under control of the LAP (liver activator protein) promoter, which directs primarily renal expression of the reverse transactivator in this mouse line [19]. Double transgenic progeny were treated with Dox and killed after 2 weeks. Whole-body fluorescent examination (Figure 4) (panel A versus panel B), revealed easily detectable EGFP fluorescence in the kidney, and low levels of fluorescence in the liver. The region of the kidney that was fluorescent (panel C versus panel D) was the cortex, consistent with the report by Gallagher et al. [19].
Figure 4. rtTA2S-S2LAP controls FerH and EGFP expression in the kidneys of Dox-treated LAPFerH double transgenic mice.

Mice of the LAPFerH model were treated with Dox for 2 weeks in drinking water. At necropsy, a LAPFerH double transgenic mouse was killed and the contents of the thorax were viewed under normal (A, C) and epifluorescent (B, D) light. Organs are denoted by the single letters C (colon), K (kidney), L (liver) and S (spleen). Note the position of the spleen in (A), and its lack of fluorescence in (B). The kidneys were strongly fluorescent (B), with prominent fluorescence in the cortex (C, D), while fluorescence in the liver was relatively much weaker, though evident. In (E), kidney tissues of mice were analysed by Western blotting for expression of FerH, TfR1 and GAPDH (loading control). Lanes 1–3 represent three single transgenic FerHmIRE/TetO7/EGFP mice, while lanes 4–8 represent five double transgenic LAPFerH mice. In (F), kidney tissues from Tet-ON double transgenic LAPFerH mice treated with Dox (lanes 1–4) or with sucrose vehicle (lanes 5–7) were evaluated by Western blotting.
As seen in Figure 4(E), when ferritin protein expression was evaluated by Western blotting, FerH was increased 14.2±4.8-fold (mean±S.D.) in kidneys of Dox-fed LAPFerH mice as compared with single transgenic mice. A robust expression of EGFP always accompanied FerH induction. As depicted in Figure 4(F), transgenic ferritin overexpression was strictly dependent on Dox, demonstrating both spatial and temporal control of transgene expression in this model. Western blotting demonstrated no expression of EGFP in the absence of Dox, confirming tight regulation by the Tet-ON rtTA2S-S2 transgene (results not shown).
Assessment of the potential for toxicity of FerH overexpression in vivo
FerH overexpression has been reported to induce cell death in selected cell culture systems [29]. We therefore evaluated ferritin-overexpressing mice for signs of toxicity or apoptosis. Double transgenic LAPFerH mice that express Dox-inducible FerH in the kidney and control mice were treated with Dox for 8 weeks. No behavioural alterations, changes in weight, or other signs of systemic toxicity were noted. We performed histological evaluation of fixed tissues collected at the 8 week endpoint. H/E sections of the kidney, spleen, liver, heart and small intestine were all normal, with no signs of injury. We also assessed kidney function by measuring creatinine and protein/creatinine ratios in urine. Creatinine levels in urine were unaffected by genotype [creatinine in urine was 495±164 μg/ml in LAPFerH ferritin overexpressers and 554±325 μg/ml in controls (means±S.D., n=5–8; P>0.717 by unpaired t test); protein/creatinine ratios in urine were likewise not different between groups (results not shown)]. Together with histological data, this suggests that overexpression of ferritin does not lead to renal toxicity in this model system.
Evaluation of the impact of FerH induction in kidney tissue on systemic iron parameters
Because ferritin sequesters iron, it was important to test whether its induction in the kidney caused systemic iron depletion. Since most of the iron in the body is found in haemoglobin, several blood iron parameters were measured to determine if mice expressing the FerH transgene were anaemic. As shown in Table 1, haematocrits and plasma iron parameters were not significantly different between double transgenic and control mice. In addition, to confirm that Dox treatment was not toxic to tissues of iron absorption, mice receiving a standard Dox regimen were necropsied and evaluated histologically. No signs of toxicity were detected either grossly or in H/E sections of the colon and small intestine (results not shown).
Table 1. Blood iron parameters of ferritin-overexpressing and -non-overexpressing mice.
Blood parameters were measured as described in the Experimental section. Results are means±S.E.M. The number of observations (n) is given in parentheses. None of these parameters were significantly different between groups (P>0.05, unpaired Student's t test).
| Ferritin expression | ||
|---|---|---|
| Induced | Normal | |
| Haematocrit | 0.52±0.1 (n=8) | 0.51 (n=2) |
| Plasma iron (μM) | 34±2 (n=10) | 31±3 (n=4) |
| Total iron binding capacity (μM) | 60±4 (n=10) | 64±5 (n=4) |
| Transferrin saturation (μM) | 58±5 (n=10) | 49±4 (n=4) |
Impact of FerH induction in kidney tissue on cellular iron proteins
To determine if transgenic FerH expression caused a significant change in tissue iron metabolism, we examined expression of TfR1, the principal protein responsible for the uptake of transferrin-bound iron into cells. The results (Figure 4) indicate that overexpression of FerH in transgenic mice results in an increase in TfR1 that is dependent on both the transactivator (Figure 4E) and Dox (Figure 4F). Thus, in Dox-treated double transgenic LAPFerH mice, the increase in FerH was associated with a 4.5±2.1-fold (mean±S.D.) increase in TfR1 expression.
IRP binding increases when iron is depleted and decreases when iron is in excess. Extracts were prepared at the time of killing [23] from kidneys of the same animals evaluated by Western blotting for ferritin and TfR1. The gel shift (see Figure 5) indicates that IRP activity is increased in kidneys of double transgenic Dox-treated LAPFerH mice by 2.3±0.9-fold (mean±S.D.) over the single transgenic FerHmIRE/TetO7/EGFP mice, a response consistent with decreased labile iron, and the observed increase in TfR1 expression.
Figure 5. Impact of FerH induction on IRP activity in vivo.

Double transgenic LAPFerH mice were treated with Dox for 2 weeks and IRP assays were performed. Extracts assessed in lanes 5–9 and 13–17 are from the Dox-fed double transgenic mice depicted in the Western blots of Figure 4(E). Lanes 1–3 and 10–12 are from the Dox-fed single transgenic controls depicted in the same Western-blot analyses. Extracts in lanes 10–17 were analysed after the addition of 2-mercaptoethanol (BME), as described in the Experimental section.
Evaluation of effects of Dox in controls
Dox is a weak iron chelator [30], and therefore had the potential to exert its own influence on cellular iron levels in the kidney. To evaluate the impact of Dox, mice bearing no transgenes were treated with a standard Dox regimen or sucrose vehicle, and tissues of the kidney were analysed by Western blotting. No changes in ferritin or TfR were seen (results not shown). This finding indicates that the changes in FerH that we observed in the kidney were due to Dox's stimulation of transgene expression and not to a chelation effect of Dox.
DISCUSSION
We show in the present study that FerH can be conditionally overexpressed and temporally regulated in a tissue-specific manner in transgenic mice using a tetracycline-regulated transgene. The tetracycline-regulated transgene approach was chosen for two reasons. First, it was formally possible that excessive FerH expression might be toxic to cells or tissues, since prolonged overexpression of FerH in tissue culture has been reported to result in growth inhibition [6] or apoptosis [29]. By using a conditional expression system, such a potential toxicity could be avoided. Secondly, the availability of tissue-specific tetracycline-regulated transactivator mouse strains enables assessment of the effect of increased FerH expression on multiple target tissues. In experiments described here, we crossed mice containing the FerH transgene with mice carrying a tet transactivator driven by the broadly active CMV promoter, yielding progeny with elevated expression of FerH in a variety of tissues, particularly skeletal muscle and splenic omentum (Figure 3). Crosses with mice carrying a transactivator driven by a LAP promoter with a more restricted tissue expression resulted in overexpression of FerH in the kidney, and to a lesser extent in the liver, of double transgenic offspring (Figure 4). In the Tet-ON system, the expression of transgenic FerH was tightly regulated by Dox (Figure 4F). This new reverse transactivator has been demonstrated to have a lower background and thus a 10-fold greater range of induction than the original reverse tet transactivator [28]. Thus FerH expression in this model can be controlled spatially and temporally. FerH expression was not associated with demonstrable toxicity in either model system.
To provide a convenient marker of transgenic FerH expression, we used a bicistronic promoter that simultaneously drives expression of EGFP and FerH. Increased FerH expression and the presence of EGFP correlated well (Figure 3C), indicating that EGFP can be used as a convenient surrogate marker of transgenic FerH expression. Given the ubiquitous nature of endogenous ferritin expression, we relied on EGFP expression to determine how tightly regulated the Dox-controlled transgenes were. Our observations, both visually and using Western blots, were that Dox administration was a requirement for transgene expression, indicating that the rtTA2S-S2LAP transgene is not leaky. Although in our experiments we measured EGFP only at necropsy, EGFP can be detected in living animals [27] and in our experiments was readily apparent in double transgenic carriers in the kidneys (LapFerH model) of shaved, Dox-treated mice, or in the eyes (CMVFerH model) of adults (results not shown). Thus, in addition to its utility in the rapid identification of double transgenic progeny, EGFP expression may be potentially useful in highlighting tissues that express transgenic ferritin, as well as in following changes in expression over time in living animals.
We had previously observed in vitro that TNF (tumour necrosis factor) and oxidants induce ferritin via a transcriptional mechanism that is distinct from the translational IRP-dependent mechanism by which iron induces ferritin [13,31]. We postulate that this may represent an alternative pathway for modulation of iron homoeostasis that becomes important under conditions of chronic inflammation or oxidative stress. Thus in vitro studies have demonstrated that in response to chronic exposure to stimuli such as TNFα [7,31,32] oxidants [13,33] or chemopreventive agents [13,34,35] that induce ferritin, the depletion of intracellular iron resulting from ferritin overexpression may out-pace the ability of IRP-dependent induction of TfR to restore iron homoeostasis. In collaboration with other mechanisms (such as cytokine-dependent induction of hepcidin, a peptide involved in regulating iron efflux [36]), such an induction of ferritin may contribute to a re-setting of the cellular ‘ferro-stat’ to a state of chronic iron depletion in certain pathophysiological settings [37].
With this model we have specifically overexpressed FerH, thus enabling us to examine its ability to drive changes in iron homoeostasis in vivo. To prevent feedback repression of the FerH transgene by the IRPs, we rendered the ferritin mRNA non-responsive to IRP-dependent regulation by introducing a single base-pair deletion into the IRE [16]. This strategy enabled robust FerH overexpression in double transgenic mice (Figure 4E). In the case of FerH/LAP double transgenics, FerH expression was sufficient to alter parameters of overall tissue iron homoeostasis, resulting in activation of IRP1 (Figure 5) and induction of TfR1 (Figure 4E) in the kidney. Thus we found that overexpression of FerH is sufficient to elicit a phenotype of iron depletion in vivo. Elevation of TfR in the kidney was not an indirect result of changes in systemic iron levels, since transferrin levels, transferrin saturation and haematocrit were not statistically different in controls and double transgenic mice (Table 1). Notably, despite a 2 week induction of ferritin, IRP activity was elevated (Figure 5) in FerH overexpressers, indicating that FerH modulation need not simply be a response to changes in tissue iron (its classic role), but can itself drive overall tissue iron balance. The increase in IRP activity and resultant increase in TfR1 expression that we report provide strong mechanistic evidence in vivo that altered ferritin expression can adjust the cellular ‘ferro-stat’.
Several studies support the concept that FerH manipulation can alter iron homoeostasis in vivo. Ferreira et al. [38] reported that heterozygous FerH knockout mice accumulate FerL, a result consistent with an increase in the labile iron pool engendered by reduced expression of FerH. An increase in FerL was also observed in the brains of an independently derived FerH heterozygous (+/−) mouse, although not all findings were consistent with an increase in intracellular iron in the brains of these mice [39]. In another study, overexpression of a human FerH gene in dopaminergic SN neurons of transgenic mice led to an increase in Fe(III) and a decrease in Fe(II) in the SN of the brain [15], consistent with increased sequestration of iron in ferritin in these mice. Families with rare gain-of-function mutations in the FerH IRE that increase the sensitivity of ferritin to IRP-mediated repression exhibit iron overload [40], further suggesting that FerH levels can regulate tissue iron homoeostasis in humans.
Extensive in vitro experiments have demonstrated the critical importance of FerH in regulating both iron homoeostasis and stress mediated by cytokines, oxidants and chemopreventive agents. Experiments presented here demonstrate that FerH is also a potent regulator of tissue iron homoeostasis in vivo.
Acknowledgments
We acknowledge the excellent advice of Dr Tim Kute (Department of Pathology, Wake Forest University School of Medicine, Winston-Salem, NC, U.S.A.), and the technical assistance of Dr Rong Ma, Mr Donald Floyd and Ms Julie Brown. We thank Dr Vicki Davis (Burns and Allen Research Institute, Los Angeles, CA, U.S.A.) for help in the initial stages of preparation of the transgenic construct. This work was supported by an NIDDK (National Institute of Diabetes and Digestive and Kidney Diseases) MERIT award R37 DK42412-15 (F. M. T.) and a K01 award 1 K01 DK065876 (J. W.).
References
- 1.Lawson D. M., Treffry A., Artymiuk P. J., Harrison P. M., Yewdall S. J., Luzzago A., Cesareni G., Levi S., Arosio P. Identification of the ferroxidase centre in ferritin. FEBS Lett. 1989;254:207–210. doi: 10.1016/0014-5793(89)81040-3. [DOI] [PubMed] [Google Scholar]
- 2.Santambrogio P., Levi S., Arosio P., Palagi L., Vecchio G., Lawson D. M., Yewdall S. J., Artymiuk P. J., Harrison P. M., Jappelli R., et al. Evidence that a salt bridge in the light chain contributes to the physical stability difference between heavy and light human ferritins. J. Biol. Chem. 1992;267:14077–14083. [PubMed] [Google Scholar]
- 3.Levi S., Santambrogio P., Cozzi A., Rovida E., Corsi B., Tamborini E., Spada S., Albertini A., Arosio P. The role of the L-chain in ferritin iron incorporation. Studies of homo and heteropolymers. J. Mol. Biol. 1994;238:649–654. doi: 10.1006/jmbi.1994.1325. [DOI] [PubMed] [Google Scholar]
- 4.Picard V., Epsztejn S., Santambrogio P., Cabantchik Z. I., Beaumont C. Role of ferritin in the control of the labile iron pool in murine erythroleukemia cells. J. Biol. Chem. 1998;273:15382–15386. doi: 10.1074/jbc.273.25.15382. [DOI] [PubMed] [Google Scholar]
- 5.Theil E. C. Ferritin: at the crossroads of iron and oxygen metabolism. J. Nutr. 2003;133:1549S–1553S. doi: 10.1093/jn/133.5.1549S. [DOI] [PubMed] [Google Scholar]
- 6.Cozzi A., Corsi B., Levi S., Santambrogio P., Albertini A., Arosio P. Overexpression of wild type and mutated human ferritin H-chain in HeLa cells: in vivo role of ferritin ferroxidase activity. J. Biol. Chem. 2000;275:25122–25129. doi: 10.1074/jbc.M003797200. [DOI] [PubMed] [Google Scholar]
- 7.Kwak E. L., Larochelle D. A., Beaumont C., Torti S. V., Torti F. M. Role for NF-κB in the regulation of ferritin H by tumor necrosis factor-α. J. Biol. Chem. 1995;270:15285–15293. doi: 10.1074/jbc.270.25.15285. [DOI] [PubMed] [Google Scholar]
- 8.Epsztejn S., Glickstein H., Picard V., Slotki I. N., Breuer W., Beaumont C., Cabantchik Z. I. H-ferritin subunit overexpression in erythroid cells reduces the oxidative stress response and induces multidrug resistance properties. Blood. 1999;94:3593–3603. [PubMed] [Google Scholar]
- 9.Hentze M. W., Kuhn L. C. Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8175–8182. doi: 10.1073/pnas.93.16.8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rouault T. A. Post-transcriptional regulation of human iron metabolism by iron regulatory proteins. Blood Cells Mol. Dis. 2002;29:309–314. doi: 10.1006/bcmd.2002.0571. [DOI] [PubMed] [Google Scholar]
- 11.Arosio P., Levi S. Ferritin, iron homeostasis, and oxidative damage. Free Radical Biol. Med. 2002;33:457–463. doi: 10.1016/s0891-5849(02)00842-0. [DOI] [PubMed] [Google Scholar]
- 11a.Wilkinson J., IV, Pietsch E. C., Torti S. V., Torti F. M. Ferritin regulation by oxidants and chemopreventive xenobiotics. Adv. Enzyme Regul. 2003;43:135–151. doi: 10.1016/s0065-2571(02)00037-7. [DOI] [PubMed] [Google Scholar]
- 12.Orino K., Lehman L., Tsuji Y., Ayaki H., Torti S. V., Torti F. M. Ferritin and the response to oxidative stress. Biochem. J. 2001;357:241–247. doi: 10.1042/0264-6021:3570241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuji Y., Ayaki H., Whitman S. P., Morrow C. S., Torti S. V., Torti F. M. Coordinate transcriptional and translational regulation of ferritin in response to oxidative stress. Mol. Cell. Biol. 2000;20:5818–5827. doi: 10.1128/mcb.20.16.5818-5827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konjin A., Hershko C. New York: John Wiley and Sons; 1988. Iron in Immunity, Cancer and Inflammation. [Google Scholar]
- 15.Kaur D., Yantiri F., Rajagopalan S., Kumar J., Mo J. Q., Boonplueang R., Viswanath V., Jacobs R., Yang L., Beal M. F., et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron. 2003;37:899–909. doi: 10.1016/s0896-6273(03)00126-0. [DOI] [PubMed] [Google Scholar]
- 16.Jaffrey S. R., Haile D. J., Klausner R. D., Harford J. B. The interaction between the iron-responsive element binding protein and its cognate RNA is highly dependent upon both RNA sequence and structure. Nucleic Acids Res. 1993;21:4627–4631. doi: 10.1093/nar/21.19.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagy A., Gertsensten M., Vintersten K., Behringer R. 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Press; 2003. Manipulating the Mouse Embryo: A Laboratory Manual. [Google Scholar]
- 18.Kistner A., Gossen M., Zimmermann F., Jerecic J., Ullmer C., Lubbert H., Bujard H. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10933–10938. doi: 10.1073/pnas.93.20.10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallagher A. R., Schonig K., Brown N., Bujard H., Witzgall R. Use of the tetracycline system for inducible protein synthesis in the kidney. J. Am. Soc. Nephrol. 2003;14:2042–2051. doi: 10.1097/01.asn.0000079615.38843.4a. [DOI] [PubMed] [Google Scholar]
- 20. Reference deleted.
- 21.Schonig K., Schwenk F., Rajewsky K., Bujard H. Stringent doxycycline dependent control of CRE recombinase in vivo. Nucleic Acids Res. 2002;30:e134. doi: 10.1093/nar/gnf134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Persijn J. P., van der Slik W., Riethorst A. Determination of serum iron and latent iron-binding capacity (LIBC) Clin. Chim. Acta. 1971;35:91–98. doi: 10.1016/0009-8981(71)90298-1. [DOI] [PubMed] [Google Scholar]
- 23.Festa M., Colonna A., Pietropaolo C., Ruffo A. Oxalomalate, a competitive inhibitor of aconitase, modulates the RNA-binding activity of iron-regulatory proteins. Biochem. J. 2000;348:315–320. [PMC free article] [PubMed] [Google Scholar]
- 24.Mullner E. W., Neupert B., Kuhn L. C. A specific mRNA binding factor regulates the iron-dependent stability of cytoplasmic transferrin receptor mRNA. Cell. 1989;58:373–382. doi: 10.1016/0092-8674(89)90851-9. [DOI] [PubMed] [Google Scholar]
- 25.Gossen M., Bujard H. Studying gene function in eukaryotes by conditional gene inactivation. Annu. Rev. Genet. 2002;36:153–173. doi: 10.1146/annurev.genet.36.041002.120114. [DOI] [PubMed] [Google Scholar]
- 26.Schonig K., Bujard H. Generating conditional mouse mutants via tetracycline-controlled gene expression. Methods Mol. Biol. 2003;209:69–104. doi: 10.1385/1-59259-340-2:69. [DOI] [PubMed] [Google Scholar]
- 27.Okabe M., Ikawa M., Kominami K., Nakanishi T., Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 28.Urlinger S., Baron U., Thellmann M., Hasan M. T., Bujard H., Hillen W. Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc. Natl. Acad. Sci. U.S.A. 2000;97:7963–7968. doi: 10.1073/pnas.130192197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cozzi A., Levi S., Corsi B., Santambrogio P., Campanella A., Gerardi G., Arosio P. Role of iron and ferritin in TNFα-induced apoptosis in HeLa cells. FEBS Lett. 2003;537:187–192. doi: 10.1016/s0014-5793(03)00114-5. [DOI] [PubMed] [Google Scholar]
- 30.Grenier D., Huot M. P., Mayrand D. Iron-chelating activity of tetracyclines and its impact on the susceptibility of Actinobacillus actinomycetemcomitans to these antibiotics. Antimicrob. Agents Chemother. 2000;44:763–766. doi: 10.1128/aac.44.3.763-766.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torti S. V., Kwak E. L., Miller S. C., Miller L. L., Ringold G. M., Myambo K. B., Young A. P., Torti F. M. The molecular cloning and characterization of murine ferritin heavy chain, a tumor necrosis factor-inducible gene. J. Biol. Chem. 1988;263:12638–12644. [PubMed] [Google Scholar]
- 32.Pham C. G., Bubici C., Zazzeroni F., Papa S., Jones J., Alvarez K., Jayawardena S., De Smaele E., Cong R., Beaumont C., et al. Ferritin heavy chain upregulation by NF-κB inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119:529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 33.Cairo G., Tacchini L., Pogliaghi G., Anzon E., Tomasi A., Bernelli-Zazzera A. Induction of ferritin synthesis by oxidative stress. Transcriptional and post-transcriptional regulation by expansion of the ‘free’ iron pool. J. Biol. Chem. 1995;270:700–703. doi: 10.1074/jbc.270.2.700. [DOI] [PubMed] [Google Scholar]
- 34.Wasserman W. W., Fahl W. E. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. U.S.A. 1997;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pietsch E. C., Chan J. Y., Torti F. M., Torti S. V. Nrf2 mediates the induction of ferritin H in response to xenobiotics and cancer chemopreventive dithiolethiones. J. Biol. Chem. 2003;278:2361–2369. doi: 10.1074/jbc.M210664200. [DOI] [PubMed] [Google Scholar]
- 36.Nemeth E., Tuttle M. S., Powelson J., Vaughn M. B., Donovan A., Ward D. M., Ganz T., Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 37.Torti F. M., Torti S. V. Regulation of ferritin genes and protein. Blood. 2002;99:3505–3516. doi: 10.1182/blood.v99.10.3505. [DOI] [PubMed] [Google Scholar]
- 38.Ferreira C., Santambrogio P., Martin M. E., Andrieu V., Feldmann G., Henin D., Beaumont C. H ferritin knockout mice: a model of hyperferritinemia in the absence of iron overload. Blood. 2001;98:525–532. doi: 10.1182/blood.v98.3.525. [DOI] [PubMed] [Google Scholar]
- 39.Thompson K., Menzies S., Muckenthaler M., Torti F. M., Wood T., Torti S. V., Hentze M. W., Beard J., Connor J. Mouse brains deficient in H-ferritin have normal iron concentration but a protein profile of iron deficiency and increased evidence of oxidative stress. J. Neurosci. Res. 2003;71:46–63. doi: 10.1002/jnr.10463. [DOI] [PubMed] [Google Scholar]
- 40.Kato J., Fujikawa K., Kanda M., Fukuda N., Sasaki K., Takayama T., Kobune M., Takada K., Takimoto R., Hamada H., et al. A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am. J. Hum. Genet. 2001;69:191–197. doi: 10.1086/321261. [DOI] [PMC free article] [PubMed] [Google Scholar]