

Abstract
Explicit modelling of metabolic networks relies on well-known mathematical tools and specialized computer programs. However, identifying and estimating the values of the very numerous enzyme parameters inherent to the models remain a tedious and difficult task, and the rate equations of the reactions are usually not known in sufficient detail. A way to circumvent this problem is to use ‘non-mechanistic’ models, which may account for the behaviour of the systems with a limited number of parameters. Working on the first part of glycolysis reconstituted in vitro, we showed how to derive, from titration experiments, values of effective enzyme activity parameters that do not include explicitly any of the classical kinetic constants. With a maximum of only two parameters per enzyme, this approach produced very good estimates for the flux values, and enabled us to determine the optimization conditions of the system, i.e. to calculate the set of enzyme concentrations that maximizes the flux. This fast and easy method should be valuable in the context of integrative biology or for metabolic engineering, where the challenge is to deal with the dramatic increase in the number of parameters when the systems become complex.
Keywords: biochemical pathway, flux computation, glycolysis, hyperbolic fitting, optimal enzyme concentration, systemic enzyme parameter
Abbreviations: BST, biochemical systems theory; CK, creatine kinase; cv, coefficients of variation; FBA, aldolase; G3PDH, glycerol-3-phosphate dehydrogenase; HK, hexokinase; MCA, metabolic control analysis; PFK, 6-phosphofructokinase; PGI, phosphoglucoisomerase; TPI, triose-phosphate isomerase
INTRODUCTION
The ‘omics’ approaches are challenging biochemical practice. The complete genome sequence data and microarray technologies reveal frequent occurrences of gene families with complex patterns of gene regulations, quantitative proteomics give expression data for hundreds of enzymes and isoenzymes, and metabolomics techniques give access to thousands of major metabolites. The way all these systemic variables are affected by development, environmental conditions and/or genetic background gives invaluable information to investigate the nature, organization and control of complex metabolic networks. Modelling metabolic systems relies on classical theoretical methods, such as differential equations, and on powerful computer programs. With these tools, extensive efforts have been made to achieve a comprehensive dynamic description of some essential pathways, such as glycolysis, especially in yeast [1,2], Trypanosoma brucei [3], erythrocytes [4–6] and muscle [7]. However, such approaches, which require a long-term effort of numerous laboratories for simple pathways, have severe limitations for more complex systems. The main obstacle for biochemical modelling is usually not the availability of mathematical and informatics tools, but the determination of the parameter values. Single rate equations may include more than ten parameters {as arises in the equation for PFK (6-phosphofructokinase; EC 2.7.1.11) [2]}, and the number of parameters increases dramatically with the size and complexity of the systems. Except for the concentration of the most abundant enzymes, which can be estimated with quantitative proteomics (e.g. [8]), there is no simple or direct way to measure essential parameters such as catalytic constants, Michaelis–Menten constants, or inhibition constants. For example, the robot-based platform recently developed to measure activities of approx. 20 enzymes in Arabidopsis is promising, but does not provide values for such essential parameters [9]. In vivo, it is questionable whether all the parameters can be estimated from perturbation experiments [10], in particular because the range of variation of metabolite levels is limited by homoeostasis constraints [11]. For some well-described pathways, computer simulations can be performed to estimate the parameter values by fitting the model with in vivo observations [12]. In any case, estimating the parameter values remains a tedious and time-consuming task, with multiple factors of inaccuracy, and no assurance that all the key parameters are identified.
Consequently, modelling efforts based on conceptual short-cuts are essential to simulate complex cellular behaviours from a limited amount of biological data. Flux balance analysis (see [13] for a review) represents a radical approach in this direction, since no kinetic information is included in the models. Flux balance analysis does not attempt to predict the exact behaviour of metabolic networks, but uses known constraints on the integrated function of multiple enzymes to separate the state that a system can reach from those it cannot. Constraints considered are the steady-state mass balance of metabolites, the effective irreversibility of reactions due to thermodynamic constraints, or the maximum flux capacity of enzymes or transport proteins. In the MCA (metabolic control analysis) [14,15] or in the BST (biochemical systems theory) [16], rate laws are not explicitly written, but kinetic parameters are not ignored. Both theoretical frameworks use systemic coefficients (control coefficients or logarithmic gains) and local coefficients (elasticity or kinetic order), which can be estimated experimentally, to quantify the behaviour of the systems. Derived from MCA or BST, various so-called ‘non-mechanistic’ models have been proposed that aim at using approximate equations to describe the behaviour of the systems with a reduced number of parameters. The power law representations, such as the S-systems, are obtained by specifying a rate constant (the multiplicative term) and writing one power term for each variable that directly influences the rate law (metabolites, enzymes and effectors) [17]. The (log)linear kinetic models rely on knowledge about the strength of interaction among the rates of enzyme-catalysed reactions and the concentrations of the various metabolites and regulators of the network [18]. The linlog kinetics proposes general expressions giving steady-state fluxes and metabolite levels as a function of enzyme concentration and response coefficients [11]. Finally, the ‘tendency modelling’ approach results in rate equations containing a minimal number of parameters, because it assumes that the behaviour of the metabolites is determined more by the stoichiometric structure of the network than by the exact enzyme mechanisms [19].
The present study is in the line of the modelling approaches using a limited number of parameters. We show how to use a simplified formalism based on MCA to derive global parameters that account for the kinetic behaviour of enzymes in their metabolic context. According to the structure of the pathway and to the position of the enzyme in the pathway, one or two parameters per enzyme are sufficient. We evaluated the validity of the method on a pathway reconstituted in vitro, an approach that just proved to be very efficient to study the systemic properties of the pathways, such as elasticities, flux control coefficients and transition time control coefficients [20–22], dynamic properties [23,24] and allosteric controls [25]. The upstream part of glycolysis, from HK (hexokinase; EC 2.7.1.1) to G3PDH (glycerol-3-phosphate dehydrogenase; EC 1.1.1.8), allowed us to measure the flux for a large number of different distributions of enzyme concentrations, and to compare the values with the theoretical predictions. We showed that the predicted flux values were tightly correlated to the measured flux values. In addition we derived equations to calculate the optimal distribution of enzyme concentrations for a given total enzyme content allocated to the network. Again the predicted values were fully consistent with observations. As the method does not require the estimation of any classical kinetic parameter such as Km, Vmax or Ki, the experimental effort for modelling and determining the conditions for optimization is thus significantly reduced. Application of the approach to metabolic engineering and to in vivo measurements is discussed.
THEORETICAL DEVELOPMENTS
Linear pathway of Michaelis–Menten enzymes
The flux J through a linear pathway of n unimolecular reversible reactions catalysed by Michaelis–Menten enzymes far from saturation is [14,15]:
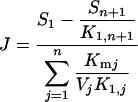 |
(1) |
where S1 and Sn+1 are respectively the concentrations of the initial and final substrates of the pathway, K1,n+1 is the product of the equilibrium constants of all the reactions, K1,j is the product of the equilibrium constants from reaction 1 to reaction Sj−1→Sj, Kmj is the Michaelis–Menten constant of enzyme j and Vj is the maximum velocity of enzyme j. Since the maximum velocity is given as Vj=Ejkcat j, with Ej the concentration of enzyme j and kcatj its catalytic constant, we have:
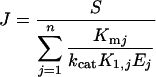 |
(2) |
where S=[S1]−[Sn+1]/K1,n+1.
For a given total enzyme concentration,
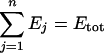 |
there is an optimal set of enzyme concentrations {E1*, E2*,…, En*}, that is, a distribution that maximizes the flux. At maximum flux, we have for all enzyme i [26,27]:
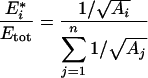 |
(3) |
where Aj=kcatjK1,j/Kmj.
Estimating systemic kinetic parameters
Equations (2) and (3) do not apply to pathways such as glycolysis that contain non-Michaelian–Menten and regulated enzymes. Nevertheless, we wanted to evaluate the extent to which a similar formalism could help in computing estimates of flux values and determining the conditions for optimizing metabolic systems of any complexity.
Consider a system converting a metabolite S1 into Sm+1 through a series of reactions catalysed by n reversible enzymes far from saturation. As branching is possible, n≥m. With a single input (S1) and output (Sm+1), the flux through the whole pathway at steady state is the rate of consumption of S1, which is equal to the rate of formation of Sm+1. On increasing the concentration Ei of enzyme i, the concentrations of other enzymes being fixed, the response of the flux is expected to display a quasi-hyperbolic ascending curve (saturation curve). This intuitive prediction is corroborated by an abundant theoretical and experimental literature (see the Discussion section). However, depending on the topology, the regulations and the constraints in the network, various mechanisms could result – at least theoretically – in a decrease in the flux when the concentration of certain enzymes increases (some examples are presented in the Discussion section). Moreover, non-linear behaviour, such as oscillations, can prevent a stable steady state being observed.
In the following developments, we considered only stable steady states, but for more generality we considered that the asymptotic response of the flux upon enzyme increase may be ascending or descending.
For fitting to experimental curves, we chose an expression of the flux J derived from eqn (2):
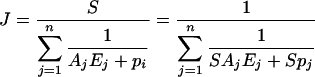 |
(4) |
where S and Ej have the same definition as above. The parameter Aj is a complex systemic kinetic parameter that depends on the type of enzyme and regulation, and takes the above value (Aj=kcatjK1,j/Kmj) only for Michaelis–Menten non-regulated enzymes in a linear pathway. Note that Aj may be negative, for example if the enzyme belongs to the backward branch of a substrate cycle. The dimension of Aj is μM−1·s−1. The parameter pj accounts for the ‘dispensability’ of enzyme j: if pj=0, then J=0 when Ej=0, and if |pj|≠0, then J≠0 when Ej=0. For example, in the system considered (see Scheme 1 in the Experimental section), removing the TPI (triose-phosphate isomerase; EC 5.3.1.1) does not bring the flux to zero because of the branching of the pathway at the reaction catalysed by the FBA (aldolase; EC 4.1.2.13). So the pTPI value is not null, unlike those of the other enzymes. The dimension of pj is s−1.
Scheme 1. Upstream part of glycolysis reconstructed in vitro.

All those parameters can be estimated with titration experiments, by varying in turn the concentration of each enzyme, the other concentrations being kept constant. For enzyme i, we have the following expression of the flux:
 |
(5) |
When Ei tends to infinity, the flux tends to the value:
 |
(6) |
Thus the flux may be given as:
 |
(7) |
The three parameters SAi, Spi and J∞ i of this function of Ei can be estimated by hyperbolic fitting of the titration curves [we used the nls package of the version 2.0.1 of the R software (http://www.r-project.org/)]. Note that the ordinate of the intercept for Ei=0, when Spi≠0, is:
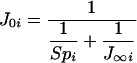 |
(8) |
Hence:
 |
(9) |
For indispensable enzymes, we have Spi=0, and the flux value tends to 0 when Ei tends to 0. With all the SAi and Spi estimates, the flux can be computed from eqn (4) for any set of Ei values.
Determination of the optimal distribution of enzyme concentrations
For a given total enzyme concentration, the optimal distribution of the concentrations {E1*, E2*,…, En*} for a flux expressed by eqn (4) can be calculated using the Lagrange multiplier method (see the Appendix). Writing ei*=Ei*/Etot, we get:
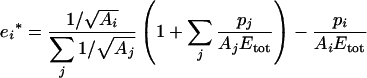 |
(10) |
This equation is valid only if Etot corresponds to a fixed total molarity. If Etot is a fixed total weight of enzymes (e.g. in g/l), as considered in the present study, the molecular masses of enzymes have to be included in the optimization equation, as shown in the Appendix. We get:
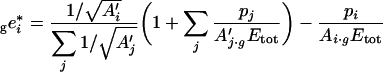 |
(11) |
where A′j=Aj/Mj, with Mj the molecular mass of enzyme j, and where the left subscript ‘g’ means that the enzyme concentrations are in g/l.
EXPERIMENTAL
Chemicals
D-Glucose, Pipes, ATP, NADH, phosphocreatine and all enzymes were obtained from Sigma. HK, PGI (phosphoglucoisomerase; EC 5.3.1.9) and TPI were purified from baker's yeast. FBA, G3PDH and CK (creatine kinase; EC 2.7.3.2) were purified from rabbit muscle, and PFK was from Bacillus stearothermophilus. All the purchased commercial enzymes were crystalline suspensions in ammonium sulphate. These proteins were desalted by using PD-10 desalting columns according to the manufacturer's recommendations (Amersham Biosciences). The protein concentrations were determined at λ=205 nm [28]. The molecular masses of the enzymes are: PGI, 61300 Da; PFK, 34000 Da; FBA, 39212 Da; and TPI, 26795 Da.
Measurements of kinetic parameters of isolated enzymes
All enzyme assays were carried out in a thermoregulated quartz cuvette (25 °C), in a buffer containing 50 mM Pipes (pH 7.5), 100 mM KCl and 5 mM magnesium acetate. HK activity was measured with 1 μM lactate dehydrogenase, 1 μM pyruvate kinase, 1 mM phosphoenolpyruvate and various concentrations of glucose. PGI activity was measured in the forward direction with 0.5 μM PFK, 5 μM FBA, 3 μM G3PDH and glucose 6-phosphate as substrate. PFK activity was measured with 0.25 μM FBA, 0.41 μM TPI, 0.9 μM G3PDH and various concentrations of fructose 6-phosphate. FBA activity was measured with 0.5 mM TPI and 1 mM G3PDH and fructose 1,6-bisphosphate as substrate. TPI activity was assayed with 1 μM G3PDH and glyceraldehyde 3-phosphate as substrate.
For estimating the kinetic parameters of isolated enzymes, we used the classical Eadie–Hofstee representation [29] where the maximal velocity V and the Michaelis–Menten constant Km were estimated by fitting the linear regression model
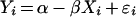 |
where Yi=vi/Si are experimental data points for the reaction rate vi at substrate concentration Si; Xi=vi; and ϵi is a random error term. In this representation, Km=1/β and V=α/β. Unbiased estimators Km and V, and their corresponding sampling variance, can be found by a Taylor series expansion of the ratios around the true values α and β (see for example [30]):
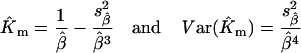 |
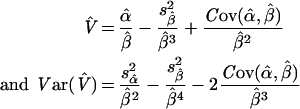 |
where 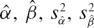 and
and 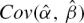 are estimated from the linear regression of vi/Si on vi.
are estimated from the linear regression of vi/Si on vi.
In vitro pathway and flux measurement
The system constructed in vitro was operated following Scheme 1.
The ATP pool is kept constant via the regeneration reaction catalysed by CK. HK is saturated due to the high glucose concentration (100 mM). The HK concentration was fixed in all experiments at 0.1 μM so that the glucose 6-phosphate input is the same in all the experiments. G3PDH, catalysing the oxidation of NADH, was kept constant, at high concentration (1 μM), in order to use the slope of the linear NADH decay as a measurement of the steady-state flux through the whole pathway. NADH consumption was monitored every 2 s with an Uvikon 850 spectrophotometer at 390 nm from 60 to 120 s. The assays were carried out at 25 °C in a buffer containing 50 mM Pipes (pH 7.5), 100 mM KCl, 20 mM phosphocreatine, 3 mM NADH and 5 mM magnesium acetate. Each reaction was started by addition of 1 mM ATP.
Titration curves
We varied in turn the concentrations of each enzyme, the concentration of the others being maintained at reference values computed on the basis of their physiological concentration estimated in the yeast strain S288C [8]. The reference values are 0.15 μM for PGI, 0.29 μM for PFK, 1.54 μM for FBA and 0.84 μM for TPI (respectively 9.1, 10.4, 60.1 and 22.3 mg/l). Hyperbolic fitting of the curves gave the relevant enzyme parameters that can be used for computing the predicted flux values (see the Theoretical developments subsection).
Selecting distributions of enzyme concentrations for flux measurement
To assess the reliability of the predicted flux values, and the validity of the model giving the optimal enzyme concentrations, we measured the flux values for a series of 121 distributions of enzyme concentrations, the total amount being kept constant. The sum of PGI, PFK, FBA and TPI concentrations was fixed at Etot=101.9 mg/l, chosen as the sum of the reference concentrations. In order to cover a large range of variation, we varied the FBA concentration (the most abundant enzyme) from 0 to Etot, taking ten values evenly distributed, and the proportions of the remaining enzymes (PGI, PFK and TPI) were drawn in β distributions with shape parameter α=1 and scale parameter
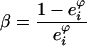 |
with eφi the reference proportion of enzyme i. In the 121 distributions obtained, enzyme concentrations ranged from 0 to 70 mg/l for PGI and PFK and from 1.66 to 66.9 mg/l for TPI.
Each flux measurement was repeated three times in three independent tubes. As the fluxes should be expressed in μM/s, the enzyme concentrations (in mg/l) were converted into μM using the molecular masses given by the manufacturer.
RESULTS
Estimation of the kinetic parameters of isolated enzymes
The kinetics of the commercial enzymes in the forward direction was studied under the same conditions as those used for the system constructed in vitro. The values of the enzyme kinetic parameters estimated using the Eadie–Hofstee representation were of the same order of magnitude as those indicated by the manufacturer (Table 1).
Table 1. Kinetic parameters of the enzymes.
Maximal rates (V) are expressed as units (i.e. μmol/min) per mg of total protein. Because V and Km are estimated from ratios in the Eadie–Hofstee representation, unbiased estimators and S.D. values of these parameters were calculated by Taylor series expansions (see the Materials and methods section).
| Experimental determination | Manufacturer values | |||||
|---|---|---|---|---|---|---|
| Enzyme | V±S.D. (units/mg of protein) | kcat (s−1) | Km±S.D. (mM) | V (units/mg of protein) | kcat (s−1) | Km (mM) |
| PGI | 1370±60 | 1410 | 1.65±0.15 | 504 | 514 | 1.4 |
| PFK | 70±1 | 41.7 | 0.033±0.001 | 164 | 150 | 0.029 |
| FBA | 42±2 | 27.7 | 0.012±0.002 | 14 | 9.3 | 0.05 |
| TPI | 14690±1900 | 6680 | 2.38±0.45 | 9500 | 4220 | 1.23 |
Titration experiments in the pathway constructed in vitro
Relationships between the flux and the concentration of each enzyme of the upstream part of glycolysis constructed in vitro, and the curves of the hyperbolic fittings, are shown in Figure 1. For each enzyme, the parameters J∞ i, SAi and Spi were estimated as indicated in the Theoretical developments subsection (Table 2). As expected, SAi values were poorly correlated with the ratio kcat iK1,i/Km i that can be estimated from isolated enzymes (results not shown). TPI alone is ‘dispensable’, since the flux with no TPI is estimated to be J0 TPI=10.47 μM/s, whereas J0 i for other enzymes is zero. So we have SpTPI=61.77 μM/s, and Spi=0 for PGI, PFK and FBA.
Figure 1. Titration curves.

(a) PGI, (b) PFK, (c) FBA and (d) TPI. The flux (y-axis) is in μM/s and the concentration of the enzymes (x-axis) is in μM.
Table 2. Systemic parameter values of the four enzymes estimated from titration curves.
J∞, J0, SA and Sp, see the text.
| Enzyme | J∞±S.D. (μM/s) | J0±S.D. (μM/s) | SA±S.D. (s−1) | Sp±S.D. (μM/s) |
|---|---|---|---|---|
| PGI | 13.22±0.5 | 0 | 499.4±49.6 | 0 |
| PFK | 17.61±1.0 | 0 | 115.5±15.1 | 0 |
| FBA | 18.24±0.7 | 0 | 22.5±2.4 | 0 |
| TPI | 12.60±0.1 | 10.5±0.1 | 22940±430 | 61.8±4.5 |
Comparing predicted and measured flux values
With the estimates of the SAi and Spi, we calculated a predicted flux value, Jpred, for any set of enzyme concentrations from eqn (4):
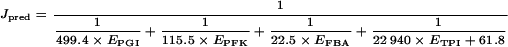 |
This predictor was used to compute the flux values for 121 distributions of enzyme concentrations, and these values were compared with the flux values measured in vitro, Jobs. Table 3 gives the Jpred and the Jobs values and the S.D. of the Jobs. The cv (coefficients of variation) never exceeded 0.14, and 84% of the cv were below 0.05. The linear correlation coefficient between the predicted and measured flux value was high (r=0.93) and the intercept of the regression line was not statistically different from zero (Figure 2). The slope of the regression line was statistically higher than 1 (a=1.38±0.18), which means that the predictor overestimates the observed flux by a constant factor. Thus Jpred/a appears to be a reliable predictor of the flux in the in vitro system.
Table 3. Predicted (Jpred) and measured flux (Jobs) for the 121 genotypes.
The enzyme concentrations are indicated in their respective columns. The last column (S.D.) indicates the standard deviation of the mean Jobs values computed with three replicates.
| PGI | PFK | FBA | TPI | Jpred | Jobs | S.D. | PGI | PFK | FBA | TPI | Jpred | Jobs | S.D. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 39.32 | 12.38 | 25.47 | 24.71 | 10.49 | 8.46 | 0.11 | 62 | 20.00 | 42.50 | 28.50 | 10.90 | 13.45 | 7.57 | 0.65 | |
| 2 | 51.97 | 14.22 | 25.47 | 10.23 | 10.92 | 8.45 | 0.23 | 63 | 35.00 | 32.50 | 8.50 | 25.90 | 4.59 | 2.39 | 0.21 | |
| 3 | 12.20 | 27.64 | 35.66 | 26.39 | 14.36 | 11.19 | 0.22 | 64 | 20.00 | 45.00 | 32.50 | 4.40 | 15.02 | 8.28 | 0.33 | |
| 4 | 32.45 | 28.74 | 35.66 | 5.03 | 15.84 | 9.68 | 0.14 | 65 | 55.00 | 7.50 | 22.50 | 16.90 | 8.40 | 4.53 | 0.65 | |
| 5 | 15.66 | 23.52 | 45.85 | 16.86 | 17.11 | 12.15 | 0.22 | 66 | 11.50 | 25.00 | 47.50 | 17.90 | 16.89 | 10.11 | 0.34 | |
| 6 | 15.55 | 30.53 | 45.85 | 9.96 | 17.96 | 10.40 | 0.22 | 67 | 22.50 | 30.00 | 8.50 | 40.90 | 4.54 | 2.34 | 0.10 | |
| 7 | 6.79 | 20.43 | 56.04 | 18.63 | 15.71 | 11.55 | 0.16 | 68 | 25.00 | 27.50 | 10.00 | 39.40 | 5.26 | 2.49 | 0.07 | |
| 8 | 3.33 | 5.75 | 66.23 | 26.58 | 8.74 | 9.24 | 0.04 | 69 | 45.00 | 37.50 | 8.50 | 10.90 | 4.63 | 2.32 | 0.06 | |
| 9 | 9.01 | 8.80 | 66.23 | 17.85 | 13.61 | 11.39 | 0.24 | 70 | 3.98 | 2.28 | 81.52 | 14.12 | 5.52 | 4.62 | 0.06 | |
| 10 | 4.24 | 25.75 | 66.23 | 5.67 | 14.94 | 11.70 | 0.30 | 71 | 3.81 | 2.71 | 81.52 | 13.85 | 6.17 | 6.12 | 0.12 | |
| 11 | 8.44 | 9.53 | 66.23 | 17.69 | 13.93 | 11.22 | 0.10 | 72 | 6.40 | 2.69 | 86.61 | 6.19 | 6.72 | 6.11 | 0.15 | |
| 12 | 5.92 | 5.25 | 76.42 | 14.30 | 10.03 | 8.90 | 0.06 | 73 | 4.75 | 2.63 | 81.52 | 13.00 | 6.27 | 5.13 | 0.19 | |
| 13 | 5.79 | 3.30 | 76.42 | 16.38 | 7.51 | 6.38 | 0.29 | 74 | 5.38 | 3.01 | 86.61 | 6.90 | 7.09 | 6.49 | 0.09 | |
| 14 | 4.23 | 2.62 | 76.42 | 18.62 | 6.09 | 5.46 | 0.10 | 75 | 6.37 | 4.17 | 76.42 | 14.94 | 8.86 | 7.71 | 0.34 | |
| 15 | 9.40 | 2.58 | 86.61 | 3.31 | 6.77 | 5.05 | 0.13 | 76 | 4.66 | 4.03 | 81.52 | 11.69 | 8.27 | 7.71 | 0.11 | |
| 16 | 3.72 | 1.95 | 86.61 | 9.61 | 4.90 | 3.99 | 0.13 | 77 | 24.36 | 7.48 | 56.04 | 14.01 | 13.23 | 9.73 | 0.11 | |
| 17 | 7.36 | 3.21 | 86.61 | 4.72 | 7.77 | 6.47 | 0.08 | 78 | 28.10 | 8.42 | 50.95 | 14.43 | 13.58 | 9.77 | 0.13 | |
| 18 | 37.90 | 12.60 | 35.66 | 15.73 | 13.24 | 9.74 | 0.05 | 79 | 8.76 | 5.41 | 66.23 | 21.49 | 10.55 | 8.96 | 0.08 | |
| 19 | 29.71 | 6.62 | 56.04 | 9.53 | 12.52 | 9.39 | 0.22 | 80 | 23.65 | 7.59 | 61.14 | 9.52 | 13.77 | 9.72 | 0.18 | |
| 20 | 13.29 | 8.21 | 56.04 | 24.36 | 13.11 | 10.08 | 0.05 | 81 | 6.82 | 14.17 | 76.42 | 4.49 | 16.17 | 11.33 | 0.38 | |
| 21 | 20.60 | 7.18 | 66.23 | 7.89 | 13.61 | 11.52 | 0.08 | 82 | 5.26 | 11.19 | 71.33 | 14.12 | 13.48 | 11.03 | 0.16 | |
| 22 | 9.88 | 3.73 | 86.61 | 1.67 | 8.92 | 7.31 | 0.04 | 83 | 9.64 | 16.24 | 66.23 | 9.79 | 17.45 | 11.56 | 0.23 | |
| 23 | 3.40 | 2.81 | 86.61 | 9.08 | 6.20 | 6.92 | 0.24 | 84 | 4.56 | 4.53 | 71.33 | 21.48 | 8.59 | 7.92 | 0.11 | |
| 24 | 70.00 | 5.00 | 5.00 | 21.90 | 2.44 | 1.22 | 0.08 | 85 | 8.62 | 3.47 | 86.61 | 3.19 | 8.36 | 7.25 | 0.11 | |
| 25 | 20.00 | 10.00 | 5.00 | 66.90 | 2.60 | 1.87 | 0.04 | 86 | 5.35 | 4.48 | 76.42 | 15.65 | 8.97 | 8.50 | 0.09 | |
| 26 | 40.00 | 10.00 | 40.00 | 11.90 | 13.13 | 9.70 | 0.55 | 87 | 22.56 | 7.84 | 50.95 | 20.55 | 12.94 | 9.86 | 0.05 | |
| 27 | 55.00 | 15.00 | 12.00 | 19.90 | 5.98 | 4.56 | 0.06 | 88 | 23.34 | 7.80 | 56.04 | 14.72 | 13.48 | 11.63 | 0.14 | |
| 28 | 15.00 | 15.00 | 55.00 | 16.90 | 16.79 | 12.90 | 0.53 | 89 | 27.65 | 9.99 | 45.85 | 18.40 | 13.89 | 10.05 | 0.09 | |
| 29 | 10.00 | 20.00 | 15.00 | 56.90 | 6.98 | 5.15 | 0.26 | 90 | 17.75 | 4.96 | 66.23 | 12.95 | 10.79 | 8.36 | 0.15 | |
| 30 | 40.00 | 35.00 | 5.00 | 21.90 | 2.78 | 1.72 | 0.02 | 91 | 18.53 | 4.91 | 71.33 | 7.12 | 10.97 | 9.08 | 0.35 | |
| 31 | 45.00 | 40.00 | 12.00 | 4.90 | 6.43 | 4.18 | 0.22 | 92 | 8.31 | 6.98 | 61.14 | 25.47 | 11.69 | 10.74 | 0.23 | |
| 32 | 15.00 | 40.00 | 45.00 | 1.90 | 18.22 | 12.07 | 0.81 | 93 | 15.24 | 5.45 | 76.42 | 4.78 | 11.75 | 9.76 | 0.03 | |
| 33 | 40.00 | 45.00 | 7.00 | 9.90 | 3.86 | 2.07 | 0.12 | 94 | 11.04 | 15.68 | 61.14 | 14.03 | 17.10 | 12.47 | 0.17 | |
| 34 | 3.00 | 45.00 | 50.00 | 3.90 | 12.10 | 10.50 | 0.35 | 95 | 7.06 | 14.74 | 76.42 | 3.67 | 16.53 | 12.23 | 0.13 | |
| 35 | 15.00 | 50.00 | 5.00 | 31.90 | 2.76 | 1.79 | 0.00 | 96 | 19.12 | 16.02 | 56.04 | 10.71 | 17.85 | 10.71 | 0.19 | |
| 36 | 25.00 | 50.00 | 12.00 | 14.90 | 6.40 | 4.18 | 0.15 | 97 | 11.51 | 13.37 | 66.23 | 10.79 | 16.91 | 12.63 | 0.15 | |
| 37 | 25.00 | 50.00 | 20.00 | 6.90 | 10.19 | 6.64 | 0.10 | 98 | 22.62 | 8.82 | 50.95 | 19.51 | 13.68 | 10.37 | 0.08 | |
| 38 | 35.00 | 60.00 | 5.00 | 1.90 | 2.80 | 1.89 | 0.01 | 99 | 45.65 | 13.30 | 25.47 | 17.47 | 10.72 | 7.65 | 0.32 | |
| 39 | 15.00 | 65.00 | 20.00 | 1.90 | 9.95 | 6.69 | 0.11 | 100 | 30.21 | 11.88 | 45.85 | 13.96 | 14.93 | 11.54 | 0.07 | |
| 40 | 25.00 | 70.00 | 2.00 | 4.90 | 1.14 | 0.74 | 0.08 | 101 | 9.06 | 16.44 | 56.04 | 20.35 | 15.97 | 11.49 | 0.10 | |
| 41 | 33.00 | 1.00 | 66.23 | 1.66 | 3.08 | 2.20 | 0.06 | 102 | 5.08 | 15.50 | 71.33 | 9.99 | 14.77 | 12.28 | 0.13 | |
| 42 | 1.00 | 33.00 | 66.23 | 1.66 | 6.30 | 7.23 | 0.01 | 103 | 19.19 | 17.00 | 56.04 | 9.67 | 18.20 | 11.05 | 0.29 | |
| 43 | 4.00 | 25.00 | 66.23 | 6.66 | 14.50 | 12.65 | 0.21 | 104 | 33.82 | 18.87 | 35.66 | 13.55 | 14.66 | 9.50 | 0.18 | |
| 44 | 7.00 | 12.00 | 66.23 | 16.66 | 14.61 | 12.35 | 0.21 | 105 | 21.50 | 7.41 | 56.04 | 16.94 | 13.05 | 10.52 | 0.07 | |
| 45 | 10.00 | 10.00 | 66.23 | 15.66 | 14.68 | 12.05 | 0.07 | 106 | 33.81 | 9.61 | 45.85 | 12.63 | 13.82 | 10.26 | 0.03 | |
| 46 | 13.00 | 13.00 | 66.23 | 9.66 | 17.09 | 11.90 | 0.14 | 107 | 11.15 | 8.50 | 61.14 | 21.11 | 13.48 | 10.82 | 0.19 | |
| 47 | 16.00 | 7.00 | 66.23 | 12.66 | 13.13 | 10.10 | 0.29 | 108 | 23.46 | 10.70 | 50.95 | 16.79 | 14.92 | 11.08 | 0.25 | |
| 48 | 19.00 | 7.00 | 66.23 | 9.66 | 13.34 | 10.11 | 0.27 | 109 | 16.40 | 17.41 | 50.95 | 17.14 | 17.04 | 11.75 | 0.10 | |
| 49 | 22.00 | 12.00 | 66.23 | 1.66 | 17.51 | 10.80 | n.d. | 110 | 10.61 | 18.22 | 50.95 | 22.12 | 16.13 | 11.57 | 0.29 | |
| 50 | 25.00 | 9.00 | 66.23 | 1.66 | 15.48 | 10.90 | 0.14 | 111 | 22.35 | 16.51 | 45.85 | 17.18 | 16.29 | 9.80 | 0.27 | |
| 51 | 28.00 | 6.00 | 66.23 | 1.66 | 12.43 | 9.35 | 0.46 | 112 | 18.13 | 15.35 | 56.04 | 12.38 | 17.50 | 11.79 | 0.08 | |
| 52 | 31.00 | 3.00 | 66.23 | 1.66 | 7.75 | 5.90 | 0.03 | 113 | 18.07 | 18.85 | 56.04 | 8.93 | 18.64 | 11.64 | 0.05 | |
| 53 | 6.00 | 18.00 | 66.23 | 11.66 | 15.81 | 9.31 | 0.10 | 114 | 12.34 | 16.16 | 56.04 | 17.36 | 16.85 | 11.58 | 0.06 | |
| 54 | 8.00 | 8.00 | 66.23 | 19.66 | 12.73 | 10.25 | 0.07 | 115 | 20.45 | 19.14 | 50.95 | 11.36 | 17.95 | 10.56 | 0.42 | |
| 55 | 10.00 | 15.00 | 66.23 | 10.66 | 17.14 | 11.45 | 0.49 | 116 | 12.00 | 20.03 | 56.04 | 13.82 | 17.82 | 10.79 | 0.24 | |
| 56 | 12.00 | 13.00 | 66.23 | 10.66 | 16.86 | 11.45 | 0.21 | 117 | 18.35 | 27.25 | 50.95 | 5.35 | 19.26 | 10.55 | 0.29 | |
| 57 | 14.00 | 18.00 | 66.23 | 3.66 | 19.32 | 11.60 | 0.00 | 118 | 9.90 | 28.14 | 56.04 | 7.82 | 18.48 | 10.95 | 0.26 | |
| 58 | 16.00 | 16.00 | 66.23 | 3.66 | 18.97 | 11.85 | 0.21 | 119 | 24.06 | 26.13 | 40.76 | 10.95 | 16.88 | 10.88 | 0.30 | |
| 59 | 47.50 | 37.50 | 3.50 | 13.40 | 1.97 | 1.10 | 0.03 | 120 | 11.23 | 21.97 | 50.95 | 17.75 | 17.06 | 11.11 | 0.07 | |
| 60 | 30.00 | 25.00 | 26.00 | 20.90 | 12.05 | 7.62 | 0.15 | 121 | 11.17 | 25.48 | 50.95 | 14.30 | 17.59 | 11.01 | 0.16 | |
| 61 | 37.50 | 47.50 | 5.00 | 11.90 | 2.79 | 1.62 | 0.05 |
Figure 2. Relationship between the measured flux Jobs and the predicted flux Jpred.

The regression line Jpred on Jobs is indicated.
Optimizing the metabolic system
As the total enzyme concentration was fixed, the relationship between the concentration of a particular enzyme and the flux can no longer be hyperbolic, since there is a global negative correlation between the concentrations. High concentration of one enzyme reduces that of the others, which may decrease the flux. Consistent with this prediction, plotting the observed flux as a function of the concentration of any one enzyme showed that the experimental points were distributed below a hump-shaped curve (Figure 3). For a given concentration of one of the enzymes, the flux may vary between 0 and a local maximum, depending on the distribution of the concentrations among the other enzymes. The distributions were randomly drawn, which explains the high extent of scatter of the points (some enzyme concentrations may be very low by chance).
Figure 3. Relationship between the concentration of each enzyme and the observed (A) and predicted (B) flux through the pathway.

The grey lozenges correspond to the theoretical optimal concentration of each enzyme, i.e. the concentration that leads to maximum flux. The error bars in (A) correspond to ±1 standard deviation.
The optimal proportion of each enzyme in the system was computed using eqn (11), which gave the proportions 0.155 for PGI, 0.240 for PFK, 0.590 for FBA and 0.015 for TPI. As the total enzyme concentration, Etot, was 101.9 mg/l, these values corresponded respectively to 15.8, 24.5, 60.1 and 1.5 mg/l. As expected, the flux value computed with these concentrations (Jpred max/a=14.62 μM/s) is higher than all the predicted or measured flux values, and the position of the optimal concentration for each enzyme corresponds to the top of the hump-shaped curve that ‘envelops’ all the points (Figure 3). For an enzyme concentration lower than the optimum, the local maximum flux increases with the enzyme concentration. For an enzyme concentration that exceeds the optimum, the local maximum flux decreases with the enzyme concentration.
DISCUSSION
We used the reconstituted upstream part of glycolysis to evaluate a method for estimating effective enzyme parameters that account for the behaviour of the enzyme within its metabolic pathway. The flux values predicted from these parameters proved to be quite close to the measured flux values. The systemic parameter Ai that we defined includes all the kinetic and regulation properties of the enzyme, but its estimation does not require any measurement of parameters such as Km, kcat or Ki. Thus the kinetic type of the enzyme does not interfere with the modelling. For instance, PFK, which is a regulated enzyme, was treated like PGI, a Michaelian enzyme. It is worth noting that Ai also contains information on the position of the catalysed reaction in the pathway, via the equilibrium constants. In linear pathways of Michaelis–Menten enzymes far from saturation, Ai includes merely the product of the equilibrium constants from the initial reaction to the reaction catalysed by enzyme i [14], but in the general case of complex networks there is no simple relationship between the equilibrium constants. The other parameter that we defined is the ‘dispensability’ (pi), which is different from zero whenever removing the enzyme from the system does not drive the flux to 0, a situation that occurs in the case of reticulation in the system. The parameters Ai and pi could not be estimated independently of S, a parameter that depends also on the equilibrium constants of the reactions and on the initial and final substrates of the pathway, but this had no incidence on the calculus of the predicted flux.
The visual inspection of the titration curves used to estimate these parameters revealed that for every enzyme, the experimental points follow a slightly more convex curve than the fitted hyperbola, so that the estimated J∞ i are higher than the apparent maximum experimental flux values. Thus the slope a of the regression line of Jpred on Jobs is higher than 1, so the right predictor of flux is Jpred/a. Other, more complex, functions could lead to better fittings. However, the quality of the prediction that we obtained with only two parameters does not justify making the model more complex. In addition, we think that the prediction could be even improved in a system in which the concentration of the first substrate could be maintained constant. Under our conditions, the first substrate of the system was glucose 6-phosphate; its input was constant because HK was saturated by glucose and did not vary, but its steady-state concentration depended on the concentration of the other enzymes. As a consequence, unlike what was assumed in the model, the parameter S could slightly vary, which inevitably increased the dispersion of the values.
The method for estimating global parameters can apply whether flux response towards enzyme variation is positive or negative, i.e. whether enzymes have positive or negative flux control coefficients. Here, the flux displayed a saturation curve for all enzymes, which means that the enzymes have a positive control coefficient for any concentration. This situation has been described for diverse pathways in many classical papers (e.g. [31–33]), and is theoretically expected in case of linear pathways [14] even if there are feedback or feedforward loops [34]. In this connection, it would be interesting to test the robustness of the method on the complete glycolysis, which would introduce strong feedback loops. In branched pathways, there are some particular situations where increasing an enzyme concentration could result in a decrease in the flux. (i) When the enzyme under study is located inside a feedback loop, negative control coefficients may be observed, depending on the parameter values of the network [34]. This is consistent with observations showing that increasing the trehalose-6-phosphate synthase may result in a decrease in trehalose synthesis, via the inhibition of HK by trehalose 6-phosphate [35]. (ii) When there is a substrate cycle (or ‘futile’ cycle), i.e. when a reaction, or a set of reactions, that converts metabolite Si into Sj, is opposed by a second set that reconverts Sj into Si, as observed for example with the cycle glucose–glucose 6-phosphate. Overexpression of glucose-6-phosphatase in rat hepatocytes decreases glucose 6-phosphate levels, with a negative control coefficient of this enzyme on glycogen synthesis [36]. In such cycles, increasing concentration of a backward enzyme is expected to decrease the whole flux to a minimal level, whereas a maximum flux value will be observed if the concentration of any one backward enzyme is null. (iii) When there is regulation by covalent modifications of enzymes, negative control coefficient may be observed. In the classical example of reversible phosphorylation of enzymes by protein kinases, the effects on the target enzymes can be activatory or inhibitory. Interestingly, when antagonistic enzymes in cycles are phosphorylated, usually one is activated and the other is inhibited [37]. Analogous, though more complex, mechanisms account for the high negative control coefficient of glycogen phosphorylase on glycogen synthesis [38]. All these situations necessarily result in negative values for SAi and Spi, which is allowed in our modelling since we made no assumption on the sign of those parameters. (It is worth noting that when the enzyme under study does not belong to the branch the flux of which is measured, negative flux control coefficients are expected [39,40], but this situation is not relevant to our approach, because we considered the flux through the whole pathway, with input flux equals output flux.)
The equations that we derived also allowed us to find the optimal distribution of enzyme concentrations in case of constraint on total enzyme. Despite the fact that the amount of enzyme the cell allocates to a given metabolic pathway is necessarily limited, biochemical modelling implicitly considers in most cases that enzyme concentrations are independent of each other, i.e. there is no limitation of resources (matter and energy) in the cell. However, some results clearly indicate that there are constraints on the variation of enzyme concentrations, particularly for the most abundant ones, due to competition for space, energy and cellular resources. Thus global negative correlations may occur between enzyme concentrations, resulting in the so-called ‘protein burden effect’ [41]. For example, it has been shown that increasing the expression of various glycolytic enzymes in Zymomonas mobilis causes a significant decrease in the glycolytic flux and growth rate [42]. In terms of economy of resources, and hence from the evolutionary point of view, it is important to achieve a given flux value with the minimum amount of total enzyme. Formally, this question is equivalent to the one that we addressed, namely how to achieve the maximum flux value with a given amount of total enzyme (for a general treatment of this question, see [26,27,43]). The equations that we derived allow this optimal distribution to be found.
Another noticeable interest of predicting optimal distribution of enzyme concentrations is for in vitro metabolic engineering. For instance, a process for the production of 2-oxo-L-gulonic acid, a precursor to vitamin C, implies two enzymatic reactions exterior to the non-living permeabilized Pantoea citrea cells used in the bioreactor [44]. The optimal concentrations of those extra enzymes could be determined following the method described in the present paper. Similarly, the approach could be applied to functional protein chips based on mRNA–enzyme fusion molecules hybridized on a DNA microarray, where immobilized enzymes retain activity proportional to the amount of capture DNA, allowing modulation of the relative activity of enzymes; this novel and elegant technique was used for the optimization of the trehalose synthesis pathway [35], but on an empirical base, while our theoretical treatment would make it rational.
The approach is not restricted to systems reconstituted in vitro, and can be applied to two experimental systems closer to in vivo conditions. With cell-free extracts, titration experiments using external enzymes are possible, as has been shown for characterizing control properties in glycolysis of mouse muscle [45] and rat liver [46]. Interestingly, it was shown that the kinetic parameters of commercial enzymes did not produce a reliable model of control properties of HK and PFK as compared with kinetic parameters determined in muscle extract [45]. In plants, isolated mesophyll cells from leaves of Digitaria sanguinalis keep functional plasmodesmata that allow the free exchange of low-molecular-mass compounds with the culture medium. This system has been used successfully to analyse the kinetic properties of the phosphoenolpyruvate carboxylase, with external malate dehydrogenase added to the medium [47]. Varying external enzymes in such experimental systems would allow the parameters we defined to be estimated ‘in cell’.
In conclusion, we developed a general method that allows us both to predict flux values from global enzyme parameters and to estimate optimal enzyme concentrations. This method is promising for integrative biology and could find applications in metabolic engineering.
Acknowledgments
We thank Professor Hans V. Westerhoff (Faculty of Earth and Life Sciences, Department of Molecular Cell Physiology, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands) for fruitful discussions, Professor David Fell (School of Biological and Molecular Sciences, Oxford Brookes University, Oxford, U.K.) for useful suggestions and Professor Olivier C. Martin (Laboratoire de Physique Théorique et Modèles Statistiques, Université Paris-Sud, Orsay Cedex, France) and Dr Delphine Sicard (UMR de Génétique Végétale, INRA/UPS/CNRS/INAPG, Gif-sur-Yvette, France) for careful reading of this paper and relevant remarks. J.B.F. was supported by a Thesis grant from the French Ministère de la Jeunesse, de l'Education Nationale et de la Recherche.
APPENDIX
Calculation of the optimal concentrations of the enzymes
We used the Lagrange multiplier method to find the distribution of enzyme concentrations that maximizes the flux for a given amount of total enzyme. At maximum flux, we have:
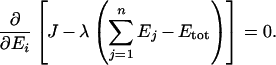 |
(A1) |
Taking the expression of the flux of the main text (eqn 4), we have:
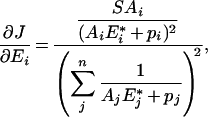 |
(A2) |
where E*i is the optimal concentration of enzyme i.
From eqn (A1), we see that all the derivatives are equal to λ, so we have:
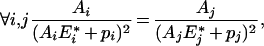 |
(A3) |
which is equivalent to:
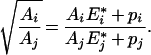 |
(A4) |
Hence:
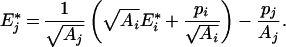 |
(A5) |
Introducing Etot, it yields:
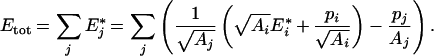 |
(A6) |
After rearrangements, we get:
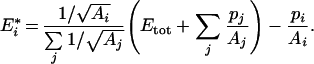 |
(A7) |
This equation leads to eqn (10) of the main text, with
 |
This general treatment, where implicitly the concentrations are expressed in mol/l, has to be adapted if the constraint on Etot is a fixed total weight of enzymes, as we did in the present study. In this case, the enzyme concentration, gEi (left subscript ‘g’ for g/l), can be written as:
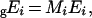 |
(A8) |
where Mi is the molecular mass of enzyme i. Thus the constraint is:
 |
(A9) |
and eqn (A1) becomes:
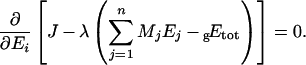 |
(A10) |
Here the derivatives of the flux are not equal, since we have:
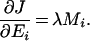 |
(A11) |
The flux equation can be rewritten to make the molecular masses apparent:
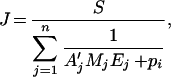 |
(A12) |
with A′j=Aj/Mj. Hence eqn (A11) can be written as:
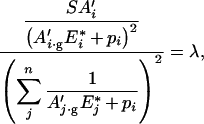 |
(A13) |
with gE*i the optimal concentration of enzyme i.
Thus replacing Ai (respectively Aj) by A′i (respectively A′j), and E*i (respectively E*j) by gE*i (respectively gE*j) in eqns (3)–(7), we obtain directly:
 |
(A14) |
This equation leads to eqn (11) of the main text, with
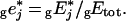 |
References
- 1.Theobald U., Mailinger W., Baltes M., Rizzi M., Reuss M. In vivo analysis of metabolic dynamics in Saccharomyces cerevisiae: II. Experimental observations. Biotechnol. Bioeng. 1997;55:305–316. doi: 10.1002/(SICI)1097-0290(19970720)55:2<305::AID-BIT8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 2.Teusink B., Passarge J., Reijenga C. A., Esgalhado E., van der Weijden C. C., Schepper M., Walsh M. C., Bakker B. M., van Dam K., Westerhoff H. V., Snoep J. L. Can yeast glycolysis be understood in terms of in vitro kinetics of the constituent enzymes? Testing biochemistry. Eur. J. Biochem. 2000;267:5313–5329. doi: 10.1046/j.1432-1327.2000.01527.x. [DOI] [PubMed] [Google Scholar]
- 3.Bakker B. M., Michels P. A., Opperdoes F. R., Westerhoff H. V. Glycolysis in bloodstream form Trypanosoma brucei can be understood in terms of the kinetics of the glycolytic enzymes. J. Biol. Chem. 1997;272:3207–3215. doi: 10.1074/jbc.272.6.3207. [DOI] [PubMed] [Google Scholar]
- 4.Bali M., Thomas S. R. A modelling study of feedforward activation in human erythrocyte glycolysis. C.R. Acad. Sci. III. 2001;324:185–199. doi: 10.1016/s0764-4469(00)01295-6. [DOI] [PubMed] [Google Scholar]
- 5.Mulquiney P. J., Kuchel P. W. Model of 2,3-bisphosphoglycerate metabolism in the human erythrocyte based on detailed enzyme kinetic equations: equations and parameter refinement. Biochem. J. 1999;342:581–596. [PMC free article] [PubMed] [Google Scholar]
- 6.Mulquiney P. J., Kuchel P. W. Model of 2,3-bisphosphoglycerate metabolism in the human erythrocyte based on detailed enzyme kinetic equations: computer simulation and metabolic control analysis. Biochem. J. 1999;342:597–604. [PMC free article] [PubMed] [Google Scholar]
- 7.Lambeth M. J., Kushmerick M. J. A computational model for glycogenolysis in skeletal muscle. Ann. Biomed. Eng. 2002;30:808–827. doi: 10.1114/1.1492813. [DOI] [PubMed] [Google Scholar]
- 8.Fiévet J., Dillmann C., Lagniel G., Davanture M., Negroni L., Labarre J., de Vienne D. Assessing factors for reliable quantitative proteomics based on two-dimensional electrophoresis. Proteomics. 2004;4:1939–1949. doi: 10.1002/pmic.200300731. [DOI] [PubMed] [Google Scholar]
- 9.Gibon Y., Blaesing O. E., Hannemann J., Carillo P., Hohne M., Hendriks J. H., Palacios N., Cross J., Selbig J., Stitt M. A robot-based platform to measure multiple enzyme activities in Arabidopsis using a set of cycling assays: comparison of changes of enzyme activities and transcript levels during diurnal cycles and in prolonged darkness. Plant Cell. 2004;16:3304–3325. doi: 10.1105/tpc.104.025973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizzi M., Baltes M., Theobald U., Reuss M. In vivo analysis of metabolic dynamics in Saccharomyces cerevisiae: II. Mathematical model. Biotechnol. Bioeng. 1997;55:592–608. doi: 10.1002/(SICI)1097-0290(19970820)55:4<592::AID-BIT2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 11.Visser D., Heijnen J. J. Dynamic simulation and metabolic re-design of a branched pathway using linlog kinetics. Metab. Eng. 2003;5:164–176. doi: 10.1016/s1096-7176(03)00025-9. [DOI] [PubMed] [Google Scholar]
- 12.Poolman M. G., Fell D. A., Thomas S. Modelling photosynthesis and its control. J. Exp. Bot. 2000;51:319–328. doi: 10.1093/jexbot/51.suppl_1.319. [DOI] [PubMed] [Google Scholar]
- 13.Edwards J. S., Covert M., Palsson B. Metabolic modelling of microbes: the flux-balance approach. Environ. Microbiol. 2002;4:133–140. doi: 10.1046/j.1462-2920.2002.00282.x. [DOI] [PubMed] [Google Scholar]
- 14.Kacser H., Burns J. A. The control of flux. Symp. Soc. Exp. Biol. 1973;27:65–104. [PubMed] [Google Scholar]
- 15.Heinrich R., Rapoport T. A. A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur. J. Biochem. 1974;42:89–95. doi: 10.1111/j.1432-1033.1974.tb03318.x. [DOI] [PubMed] [Google Scholar]
- 16.Savageau M. A. Concepts relating the behavior of biochemical systems to their underlying molecular properties. Arch. Biochem. Biophys. 1971;145:612–621. doi: 10.1016/s0003-9861(71)80021-8. [DOI] [PubMed] [Google Scholar]
- 17.Torres N. V., Voit E. O., Glez-Alcon C., Rodriguez F. An indirect optimisation method for biochemical systems: description of method and application to the maximization of the rate of ethanol, glycerol, and carbohydrate production in Saccharomyces cerevisiae. Biotechnol. Bioeng. 1997;55:758–772. doi: 10.1002/(SICI)1097-0290(19970905)55:5<758::AID-BIT6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 18.Hatzimanikatis V., Bailey J. E. Effects of spatiotemporal variations on metabolic control: approximate analysis using (log)linear kinetic models. Biotechnol. Bioeng. 1997;54:91–104. doi: 10.1002/(SICI)1097-0290(19970420)54:2<91::AID-BIT1>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 19.Visser D., van der Heijden R., Mauch K., Reuss M., Heijnen S. Tendency modeling: a new approach to obtain simplified kinetic models of metabolism applied to Saccharomyces cerevisiae. Metab. Eng. 2000;2:252–275. doi: 10.1006/mben.2000.0150. [DOI] [PubMed] [Google Scholar]
- 20.Giersch C. Determining elasticities from multiple measurements of flux rates and metabolite concentrations. Application of the multiple modulation method to a reconstituted pathway. Eur. J. Biochem. 1995;227:194–201. doi: 10.1111/j.1432-1033.1995.tb20376.x. [DOI] [PubMed] [Google Scholar]
- 21.Torres N. V., Souto R., Melendez-Hevia E. Study of the flux and transition time control coefficient profiles in a metabolic system in vitro and the effect of an external stimulator. Biochem. J. 1989;260:763–769. doi: 10.1042/bj2600763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saavedra E., Encalada R., Pineda E., Jasso-Chavez R., Moreno-Sanchez R. Glycolysis in Entamoeba histolytica. Biochemical characterization of recombinant glycolytic enzymes and flux control analysis. FEBS J. 2005;272:1767–1783. doi: 10.1111/j.1742-4658.2005.04610.x. [DOI] [PubMed] [Google Scholar]
- 23.Schellenberger W., Eschrich K., Hofmann E. Selforganization of a glycolytic reconstituted enzyme system: alternate stable stationary states, hysteretic transitions and stabilization of the energy charge. Adv. Enzyme Regul. 1980;19:257–284. doi: 10.1016/0065-2571(81)90019-4. [DOI] [PubMed] [Google Scholar]
- 24.Schellenberger W., Hervagault J. F. Irreversible transitions in the 6-phosphofructokinase/fructose 1,6-bisphosphatase cycle. Eur. J. Biochem. 1991;195:109–113. doi: 10.1111/j.1432-1033.1991.tb15682.x. [DOI] [PubMed] [Google Scholar]
- 25.Curien G., Ravanel S., Dumas R. A kinetic model of the branch-point between the methionine and threonine biosynthesis pathways in Arabidopsis thaliana. Eur. J. Biochem. 2003;270:4615–4627. doi: 10.1046/j.1432-1033.2003.03851.x. [DOI] [PubMed] [Google Scholar]
- 26.Kuchel P. W. Kinetic analysis of multienzyme systems in homogeneous solution. In: Welch G. R., editor. Organized Multienzyme Systems. New York: Academic Press; 1985. pp. 303–380. [Google Scholar]
- 27.Heinrich R., Schuster S., Holzhutter H. G. Mathematical analysis of enzymic reaction systems using optimization principles. Eur. J. Biochem. 1991;201:1–21. doi: 10.1111/j.1432-1033.1991.tb16251.x. [DOI] [PubMed] [Google Scholar]
- 28.Scopes R. K. Measurement of protein by spectrophotometry at 205 nm. Anal. Biochem. 1974;59:277–282. doi: 10.1016/0003-2697(74)90034-7. [DOI] [PubMed] [Google Scholar]
- 29.Cornish-Bowden A. London: Portland Press; 1995. Fundamentals of Enzyme Kinetics. [Google Scholar]
- 30.van Kempen G. M., van Vliet L. J. Mean and variance of ratio estimators used in fluorescence ratio imaging. Cytometry. 2000;39:300–305. doi: 10.1002/(sici)1097-0320(20000401)39:4<300::aid-cyto8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 31.Kacser H., Burns J. A. The molecular basis of dominance. Genetics. 1981;97:639–666. doi: 10.1093/genetics/97.3-4.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niederberger P., Prasad R., Miozzari G., Kacser H. A strategy for increasing an in vivo flux by genetic manipulations. The tryptophan system of yeast. Biochem. J. 1992;287:473–479. doi: 10.1042/bj2870473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rossignol R., Faustin B., Rocher C., Malgat M., Mazat J.-P., Letellier T. Mitochondrial threshold effects. Biochem. J. 2003;370:751–762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sen A. K. On the sign pattern of metabolic control coefficients. J. Theor. Biol. 1996;182:269–275. doi: 10.1006/jtbi.1996.0164. [DOI] [PubMed] [Google Scholar]
- 35.Jung G. Y., Stephanopoulos G. A functional protein chip for pathway optimization and in vitro metabolic engineering. Science. 2004;304:428–431. doi: 10.1126/science.1096920. [DOI] [PubMed] [Google Scholar]
- 36.Aiston S., Trinh K. Y., Lange A. J., Newgard C. B., Agius L. Glucose-6-phosphatase overexpression lowers glucose 6-phosphate and inhibits glycogen synthesis and glycolysis in hepatocytes without affecting glucokinase translocation. Evidence against feedback inhibition of glucokinase. J. Biol. Chem. 1999;274:24559–24566. doi: 10.1074/jbc.274.35.24559. [DOI] [PubMed] [Google Scholar]
- 37.Cohen P. The coordinated control of metabolic pathways by broad-specificity protein kinases and phosphatases. Curr. Top. Cell. Regul. 1985;27:23–37. doi: 10.1016/b978-0-12-152827-0.50010-4. [DOI] [PubMed] [Google Scholar]
- 38.Aiston S., Hampson L., Gomez-Foix A. M., Guinovart J. J., Agius L. Hepatic glycogen synthesis is highly sensitive to phosphorylase activity: evidence from metabolic control analysis. J. Biol. Chem. 2001;276:23858–23866. doi: 10.1074/jbc.M101454200. [DOI] [PubMed] [Google Scholar]
- 39.LaPorte D. C., Walsh K., Koshland D. E., Jr The branch point effect. Ultrasensitivity and subsensitivity to metabolic control. J. Biol. Chem. 1984;259:14068–14075. [PubMed] [Google Scholar]
- 40.Kruckeberg A. L., Neuhaus H. E., Feil R., Gottlieb L. D., Stitt M. Decreased-activity mutants of phosphoglucose isomerase in the cytosol and chloroplast of Clarkia xantiana. Impact on mass-action ratios and fluxes to sucrose and starch, and estimation of flux control coefficients and elasticity coefficients. Biochem. J. 1989;261:457–467. doi: 10.1042/bj2610457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peter Smits H., Hauf J., Muller S., Hobley T. J., Zimmermann F. K., Hahn-Hagerdal B., Nielsen J., Olsson L. Simultaneous overexpression of enzymes of the lower part of glycolysis can enhance the fermentative capacity of Saccharomyces cerevisiae. Yeast. 2000;16:1325–1334. doi: 10.1002/1097-0061(200010)16:14<1325::AID-YEA627>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 42.Snoep J. L., Yomano L. P., Westerhoff H. V., Ingram L. O. Protein burden in Zymomonas mobilis: negative flux and growth control due to overproduction of glycolytic enzymes. Microbiology. 1995;141:2329–2337. [Google Scholar]
- 43.Lion S., Gabriel F., Bost B., Fievet J., Dillmann C., de Vienne D. An extension to the metabolic control theory taking into account correlations between enzyme concentrations. Eur. J. Biochem. 2004;271:4375–4391. doi: 10.1111/j.1432-1033.2004.04375.x. [DOI] [PubMed] [Google Scholar]
- 44.Banta S., Boston M., Jarnagin A., Anderson S. Mathematical modeling of in vitro enzymatic production of 2-keto-L-gulonic acid using NAD(H) or NADP(H) as cofactors. Metab. Eng. 2002;4:273–284. doi: 10.1006/mben.2002.0231. [DOI] [PubMed] [Google Scholar]
- 45.Puigjaner J., Rais B., Burgos M., Comin B., Ovadi J., Cascante M. Comparison of control analysis data using different approaches: modelling and experiments with muscle extract. FEBS Lett. 1997;418:47–52. doi: 10.1016/s0014-5793(97)01347-1. [DOI] [PubMed] [Google Scholar]
- 46.Torres N. V., Mateo F., Melendez-Hevia E., Kacser H. Kinetics of metabolic pathways. A system in vitro to study the control of flux. Biochem. J. 1986;234:169–174. doi: 10.1042/bj2340169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pierre J. N., Prieto J. L., Gadal P., Vidal J. In situ C4 phosphoenolpyruvate carboxylase activity and kinetic properties in isolated Digitaria sanguinalis mesophyll cells. Photosynth. Res. 2004;79:349–355. doi: 10.1023/B:PRES.0000017179.31351.f0. [DOI] [PubMed] [Google Scholar]